

Abstract
Juvenile hormone (JH) regulates insect development by a poorly understood mechanism. Application of JH agonist insecticides to Drosophila melanogaster during the ecdysone-driven onset of metamorphosis results in lethality and specific morphogenetic defects, some of which resemble those in mutants of the ecdysone-regulated Broad-Complex (BR-C). The Methoprene-tolerant (Met) bHLH–PAS gene mediates JH action, and Met mutations protect against the lethality and defects. To explore relationships among these two genes and JH, double mutants were constructed between Met alleles and alleles of each of the BR-C complementation groups: broad (br), reduced bristles on palpus (rbp), and 2Bc. Defects in viability and oogenesis were consistently more severe in rbp Met or br Met double mutants than would be expected if these genes act independently. Additionally, complementation between BR-C mutant alleles often failed when MET was absent. Patterns of BRC protein accumulation during metamorphosis revealed essentially no difference between wild-type and Met-null individuals. JH agonist treatment did not block accumulation of BRC proteins. We propose that MET and BRC interact to control transcription of one or more downstream effector genes, which can be disrupted either by mutations in Met or BR-C or by application of JH/JH agonist, which alters MET interaction with BRC.
HORMONAL regulation of insect development involves the relatively well-understood steroid 20-hydroxyecdysone (20E) (Henrich and Brown 1995; Riddiford et al. 2000; Spindler et al. 2001; Thummel and Chory 2002) and the more enigmatic sesquiterpenoid juvenile hormone (JH). 20E orchestrates molting and metamorphosis and regulates reproduction (Wyatt and Davey 1996; Riddiford et al. 2000). Its dimeric receptor ECR/USP is a 20E-dependent transcription factor that regulates the expression of target genes, including both primary response (early) genes, such as Broad-Complex (BR-C), that are directly and rapidly induced by 20E and secondary response (late) genes that are regulated by the primary response gene products (Ashburner et al. 1974; Richards 1997). JH action during larval development, at least in lepidopteran and hemimetabolous insects, is to maintain the “status quo,” probably to allow proper larval molting and prevent premature metamorphosis (Williams 1961; Zhou and Riddiford 2002). At the end of larval development, circulating JH is degraded, enabling 20E to trigger metamorphosis (Riddiford 1996). JH reappears in many adult insects to control both oogenesis and male accessory gland function (Wyatt and Davey 1996). Neither the JH receptor nor its molecular mechanism of action is well understood (Gilbert et al. 2000), although JH, as well as JH agonists (JHA), can regulate gene expression (Jones 1995; Dubrovsky et al. 2000; Sempere et al. 2002). The 20E and JH signaling pathways interact during development. In hemipteran and lepidopteran larvae, withdrawal of JH resulted in 20E induction of precocious metamorphosis, whereas application of JH during the last larval instar resulted in a supernumerary instar or larval-pupal intermediate (Riddiford 1994; Gilbert et al. 2000). These two hormones also interact in adults to control oogenesis (Soller et al. 1999).
In Drosophila melanogaster, metamorphosis is controlled by several waves of 20E secretion: the first at the end of the third larval instar driving pupararium formation, the second 10–12 hr after pupararium formation (APF) triggering pupation, and finally a large wave beginning 25–30 hr APF (Handler 1982; Riddiford 1993). JH has been detected in Drosophila larvae but not pupae (Bownes and Rembold 1987; Sliter et al. 1987) when its absence is thought to permit 20E control of metamorphosis. Application of exogenous JH or JH analog insecticides, such as methoprene and pyriproxyfen which act as JHA (Staal 1975; Wilson 2004), does not block pupararium formation or pupation. Rather, it results in disruptions of the adult abdominal epidermis, male genitalia orientation, and many internal tissues, resulting in death during the late pupal (pharate adult) stage (Ashburner 1970; Madhavan 1973; Postlethwait 1974; Wilson and Fabian 1986; Riddiford and Ashburner 1991; Restifo and Wilson 1998).
The primary response gene BR-C is composed of three lethal complementation groups: broad (br), reduced bristles on the palpus (rbp), and 2Bc, mutations of which cause death during prepupal or pupal stages (Belyaeva et al. 1980; Kiss et al. 1988). Null alleles of the entire gene, called nonpupariating1 (npr1), cause death in late third-instar larvae (Kiss et al. 1988; Gonzy et al. 2002), demonstrating that BR-C function is essential for metamorphic onset. BR-C uses alternative splicing to encode a small family of transcription factors with amino-terminal BTB–POZ domains linked to one of four possible C2H2 zinc-finger domains: BRC–Z1, BRC–Z2, BRC–Z3, and BRC–Z4 (Dibello et al. 1991; Bayer et al. 1996). During metamorphosis BRC isoforms regulate numerous downstream effector genes (Guay and Guild 1991; Karim et al. 1993; von Kalm et al. 1994; Hodgetts et al. 1995; Bayer et al. 1996, 1997; Crossgrove et al. 1996; Liu and Restifo 1998; Mugat et al. 2000; Dubrovsky et al. 2001; Dunne et al. 2002; Sempere et al. 2003). Although there is some functional redundancy among the isoforms, in general, BRC–Z1 provides the function for rbp, BRC–Z2 for br, and BRC–Z3 for 2Bc (Crossgrove et al. 1996; Bayer et al. 1997; Sandstrom et al. 1997; Liu and Restifo 1998; Consoulas et al. 2005).
BR-C metamorphosis phenotypes include failures of larval tissue histolysis (Lee and Baehrecke 2001; Kucharova-Mahmood et al. 2002), epidermal morphogenesis (Kiss et al. 1988), and internal tissue remodeling (Restifo and White 1991, 1992; Sandstrom et al. 1997; Consoulas et al. 2005). Several phenotypes, such as a split-brain abnormality (Restifo and White 1991) were common to all BR-C mutants, while others, such as failure of thoracic muscle attachment or persistence of larval salivary glands (Restifo and White 1992) were restricted to one or two complementation groups. This suggested that BRC proteins operate in two pathways, “common,” requiring all three functions and “restricted,” requiring a subset of them (Restifo and White 1991, 1992; Restifo and Wilson 1998). Following metamorphosis BR-C is expressed in egg chamber follicle cells to function during oogenesis (Huang and Orr 1992; Deng and Bownes 1997; Tzolovsky et al. 1999).
Our previous work showed that lethal pharate adults developing from wild-type methoprene-treated larvae showed a selective BR-C phenocopy, including disruptions of the central nervous system and salivary glands (Restifo and Wilson 1998). The methoprene syndrome was striking in that it included none of the restricted-pathway defects, which is inconsistent with methoprene simply blocking the 20E induction of BR-C expression during the larval-to-pupal transition. We interpreted the pathological effects of methoprene to reflect dysfunction of BR-C and probably additional primary response genes during metamorphosis, resulting in aberrant expression of secondary response genes. In contrast, other investigators found that the JHA pyriproxyfen caused abnormal pupal cuticle gene expression, apparently due to abnormal BR-C expression (Zhou and Riddiford 2002).
Methoprene-tolerant (Met) is essential for the manifestation of the toxic and morphogenetic effects of JH/JHA in D. melanogaster (Wilson and Fabian 1986; Riddiford and Ashburner 1991; Wilson 1996; Restifo and Wilson 1998). Met mutants are resistant to these effects of methoprene (Wilson and Fabian 1986). MET can bind JH III with specificity and nanomolar affinity (Shemshedini and Wilson 1990; Miura et al. 2005), suggesting that it is a component of a JH receptor. Met encodes a bHLH–PAS transcriptional regulator family member (Ashok et al. 1998) and MET can activate a reporter gene in transfected Drosophila S-2 cells (Miura et al. 2005).
We hypothesize that BR-C and Met function together in one or more aspects of development. If methoprene disrupts 20E-mediated metamorphic development or oogenesis by acting on BR-C or its downstream genes, one would expect double mutants to show synergistic genetic interaction, such as synthetic lethality of viable alleles, shifts in lethal phase, or enhanced oogenesis defects. We found that animals carrying both Met and BR-C mutations showed just such synergistic interactions. However, we did not detect any major disruption in BRC protein accumulation following methoprene treatment, suggesting that the MET and BRC interact to regulate expression of downstream effector gene(s).
MATERIALS AND METHODS
Stocks:
BR-C mutant alleles used in this study were previously used to investigate internal tissue metamorphosis (Restifo and White 1991, 1992; Restifo and Merrill 1994; Sandstrom et al. 1997; Consoulas et al. 2005). BR-C mutant progeny were identified by visible markers yellow, white, and/or singed. Wild-type BR-C function was provided by second- and/or third-chromosome BRC–Z1, BRC–Z2, or BRC–Z3 cDNA transgenes with hsp70 promoters (Bayer et al. 1997). The third-chromosome hsBRC-Z3 transgene, provided by C. Bayer (University of Central Florida, Orlando, FL) is leaky in a temperature-sensitive manner (R. Spokony, H. J. Clark, and L. L. Restifo, unpublished data). Survival to adult eclosion of 2Bc1/Y; hsBRC-Z3/+ varied with rearing temperature: 9% at 22–23°, 12% at 25°, and 29% at 29°. Most of the Met alleles used in this study were isolated from a methoprene-susceptible isogenic vermilion (v) strain as previously described (Wilson and Fabian 1987). After isolation, each Met allele was backcrossed to v for 5–7 generations to minimize background genome differences that might impact comparison of phenotypes. A functional copy of Met+ was provided by a second-chromosome genomic transgene (Ashok et al. 1998; Wilson and Ashok 1998). The Y-borne duplications y2Y67g 19.1 (hereafter termed y2Y67g), bearing BR-C+ (Belyaeva et al. 1980; Lindsley and Zimm 1992) and y+Yv+BS− (hereafter termed Y-Met+), bearing Met+ (Lindsley and Zimm 1992) were used to cover BR-C and Met mutations, respectively.
Double mutants were constructed by genetic recombination, and each was maintained heterozygous with an FM7 balancer chromosome. Each double-mutant chromosome was tested to verify the presence of each mutation and the absence of inadvertently introduced lethal mutations.
D. melanogaster culture and methoprene treatment:
Stocks and crosses were cultured on one of three standard media with mold inhibitors, at 25° with a 12:12 L:D photoperiod unless otherwise specified. For experiments comparing phenotypes, the same culture medium and other conditions were used for all genotypes. For progeny phenotype analysis, cultures were performed in glass vials (Capital Vial) without larval crowding, and all progeny were censused and examined, either as adults or as uneclosed pupae.
Cultures were tested for methoprene resistance on diagnostic doses given in Table 5 of ZR-2008, the biologically active isomer of methoprene [isopropyl-(2E,4E)-11-methoxy-3,7,11-trimethyl-2,4-dodecadienonate], as previously described (Wilson 1996; Wilson and Ashok 1998). At lethal concentrations, mortality occurs in Met+ typically during the pharate adult stage. Eclosing survivors were examined for methoprene-induced morphogenetic defects of malrotated male genitalia and defective sternal bristles, particularly on the posterior sternites. For analysis of BRC proteins, 100–120 OreRC or v Met27 eggs were transferred to glass bottles containing Drosophila Instant Food (Carolina Biologicals) with either a high-lethal dose of methoprene or vehicle (acetone) alone, as described in Restifo and Wilson (1998). Control and methoprene-treated cultures were reared in parallel.
TABLE 5.
Survival and morphological defects in various BR-C and Met flies following treatment with methoprene
| Methoprene dose
|
||||
|---|---|---|---|---|
| Genotype | 0.05 | 0.01 | 0.005 | 0.001 |
| Oregon-RC | 0 | 0 | 11 (100) | 62 (69) |
| rbp2/rbp2/Y | 0 | 0 | 7.6 (100) | 70 (63) |
| br1/br1/Y | 0 | 0 | 13 (89) | 75 (74) |
| 2Bc1/FM7 | 0 | 0 | 3.1 (100) | 58 (48) |
| npr13/FM7 | ND | 0 | 14 (100) | 67 (72) |
| Met3/Met3/Y | 45 (0) | 67 (0) | 92 (0) | ND |
| br1 Met3/br1 Met3/Y | 40 (0) | 74 (0) | 81 (0) | ND |
Mean N = 63, range 40–79, individuals were evaluated at each methoprene dose applied to at least triplicate cultures of 30 individual larvae. Survival is expressed as percentage of individuals of the indicated genotype surviving to adulthood. Numbers in parentheses represent the percentage of survivors having abnormal sternite bristle/male genitalia. Met3 was used as the Met allele because of high viability with br1. ND, not determined.
BR-C phenotype examination:
Well-described BR-C phenotypes, including lethality and epidermal defects of wing and maxillary palpus morphology (Kiss et al. 1988) were examined. Lethality was assigned to the prepupal, pupal, or pharate adult stage on the basis of external appearance (Bainbridge and Bownes 1981). Homozygous or hemizygous Met27 pupae can be identified by a slightly elongated pupal case (T. G. Wilson, unpublished data). For quantitative lethal-phase analysis, white or very young brown prepupae were transferred to moistened ashless filter paper (Whatman no. 42) in small glass petri dishes and allowed to continue developing in a humid chamber. Developmental stage was monitored daily until eclosion or death was evident. We found some variability (<5%) in the survival rates from pupariation to eclosion of Met br1 and Met rbp2 in different genetic backgrounds. To accurately compare the phenotypes of the various allele combinations, crosses were standardized using FM7-balanced mothers whenever possible.
Ovipositional rate and ovary examination:
Females isolated within 4–6 hr after eclosion were provided with wild-type (OreRC) males in food vials sprinkled with baker's yeast. Egg counts were made at 2-day intervals when the medium was changed, and fertility of the eggs was noted. Oogenesis was assessed by dissecting ovaries from females at several times after eclosion and examining for the presence of stages 8–14 vitellogenic oocytes as previously described (Wilson and Ashok 1998).
Analysis of BRC protein accumulation by immunoblotting:
White prepupae (WPP) were collected and either homogenized immediately or placed in humid chambers for further development. In experiments with late-pupal stages, animals were resynchronized at head eversion. Protein extraction was based on the method of Emery et al. (1994). For each time point, 5–10 animals were homogenized in 50–100 μl sample buffer with a Teflon pestle in a microcentrifuge tube. Sample buffer consisted of 75 mm Tris-HCl, pH 6.8, 6% SDS, 15% glycerol, 10% β-mercaptoethanol and protease inhibitors (0.1 μg/μl pepstatin A, 0.5 μg/μl leupeptin, and 10 mm PMSF, Sigma). Following centrifugation for 10 min at 14,000 rpm (Eppendorf 5415C), the supernatant was used immediately for electrophoresis or stored at −80° for up to 3 weeks, which did not compromise BRC protein stability (data not shown).
Extracts representing 0.25- or 0.5-animal equivalents were heated for 5 min at 90°, quick chilled on ice for 10 min, and separated by SDS–PAGE (Towbin et al. 1979). For optimal band separation and size assessment, we used large (16 × 18 cm) 10% acrylamide gels on a Hoefer SE 600 Ruby electrophoresis apparatus at constant current (30 mA) for 5 hr. Otherwise, 12% acrylamide gels (7 × 8 cm) were run on a Bio-Rad mini-PROTEAN II apparatus at constant voltage (195 V) for 50 min. Proteins were transferred to nitrocellulose membranes (Protran, 0.45 μm, Schleicher & Schuell) by electroblotting at 4° overnight. Overall protein pattern was detected by staining the membrane with 0.5% Ponceau-S (Sigma, St. Louis).
Nonspecific binding sites were blocked with phosphate-buffered saline (PBS) plus 0.1% Tween 20 (TPBS) and 5% w/v nonfat dry milk powder (Bio-Rad) for 90 min at room temperature. Blots were probed with either anti-BRcore (mAb25E9) or anti-Z1 (mAb3C11) mouse monoclonal antibodies (Emery et al. 1994), diluted 1:2,500 or 1:100, respectively, in PBS + 5% w/v milk powder, for 2 hr at room temperature or overnight at 4°. After three 10-min washes in TPBS, the blots were incubated with HRP-conjugated goat anti-mouse IgG (Cappel, ICN) at 1:10,000 (with anti-BRcore) or 1:2,500 (with anti-Z1) for 1 hr at room temperature. After another three 10-min washes, the signal was revealed by chemilumiscent detection of HRP (ECL detection kit; Amersham-Pharmacia) and exposure to X-ray film (Kodak X-OMAT AR). To evaluate lane loading, the blot was stripped (by serial 10-min washes in water, 0.2N NaOH, water, and TPBS), reblocked, and reprobed with anti-actin (mAb1501; Chemicon) at 1:20,000 (Mackler and Reist 2001).
RESULTS
Interaction between Met and rbp alleles:
We first investigated the impact of reduced Met function on rbp mutants with marginal viability. Heteroallelic mutants carrying the viable allele rbp2 with the BRC-null allele npr13 showed ∼60% eclosion (Figure 1). Careful examination of rbp2/npr13 mutants, starting at puparium formation and continuing throughout metamorphosis, showed that 100% of them were able to pupate, after which ∼15% died as early pupae (Figure 1). Eighty percent of the original cohort survived to late pharate adult stage, with ∼20% failing to eclose. In contrast, when the hypomorphic-viable allele Met3 was crossed onto each of the BR-C mutant chromosomes, the survival curve of the resulting heteroallelic double mutants was shifted (Figure 1), showing a dramatic reduction in viability.
Figure 1.

BRC and Met mutations interact to cause synergistic enhancement of lethality during metamorphosis. Survival curves were obtained by selecting individuals as very young prepupae, culturing them at 25° in a humid chamber, and evaluating their developmental progress, with staging based on Bainbridge and Bownes (1981). The x-axis is not strictly proportional to time because the stages are of unequal duration and mutants of different genotypes develop at different rates. Single-mutant BRC progeny, y rbp2 w/y npr13 w sn3 (n = 156) were generated by crossing y npr13 w sn3/Binsn females to y rbp2 w/y2Y67g males. Control siblings from the same cross, y rbp2 w/Binsn (n = 157) showed 100% eclosion. Double mutants, y rbp2 w v Met3/y npr13 w v Met3 (n = 115) were generated by crossing y npr13 w v Met3/FM7, y31d B v females to y rbp2 w v Met3/Y males. Sibling controls from that cross, y rbp2 w v Met3/FM7, y31d B v (n = 114) had 100% eclosion. Data for v Met3 (n = 284) and v Met27 (n = 125) include similar numbers of hemizygous male and homozygous female progeny from the respective inter-sib matings.
We next examined survival in double mutants carrying Met27, a bono fide null allele (Wilson and Ashok 1998), with each of two alleles of rbp: rbp2, a weak allele, and rbp1, a severe allele (Belyaeva et al. 1980; Kiss et al. 1988). Met27 flies show good survival, with ≤15% mortality during pupal development (Figure 1). Flies homozygous for rbp2 and carrying one copy of Met27 showed good survival to adults (Table 1). In contrast, survival of rbp2 Met27 homozygotes to the adult stage was poor, dying typically during the pharate adult stage (Table 1).
TABLE 1.
Survival to adulthood of females carrying various combinations of BR-C and Met alleles
| Mutant progeny of interest | N | Estimated survival of mutant (%) |
|---|---|---|
| rbp alleles | ||
| rbp2 Met27/rbp2 Met27 | 189 | <2 |
| rbp2 Met27/rbp2 | 498 | 86 |
| rbp2 Met27/rbp2 Met27; p[Met+]/+ | 298 | 84 |
| rbp1 Met27/rbp1 Met27 | 204 | 0 |
| rbp1 Met27/rbp1 | 350 | 0 |
| rbp1 Met27/rbp2 Met27 | 278 | 0 |
| rbp2 Met27/rbp1 | 265 | 100 |
| rbp2 Met27/rbp1 Met27; p[Met+]/+ | 384 | 80 |
| br alleles | ||
| br1 Met27 br1 Met27 | 238 | <2 |
| br1/br1 Met27 | 195 | 84 |
| br1 Met27/br1Met27; p[Met+] | 277 | 64 |
| br5 Met27/br1 Met27 | 135 | 0 |
| br5 Met27/br1 | 247 | 54 |
| br5 Met27/rbp1 Met27 | 239 | 0 |
| br5 Met27/rbp1 | 271 | 38 |
| br5 Met27/rbp2 Met27 | 294 | 10 |
| br5 Met27/rbp2 | 125 | 82 |
| br1 Met27/rbp1 Met27 | 182 | 8 |
| br1 Met27/rbp1 | 242 | 88 |
| 2Bc alleles | ||
| 2Bc1 Met27/2Bc1 Met27 | 119 | 0 |
| 2Bc1 Met27/2Bc1 | 247 | 0 |
| 2Bc1 Met27/rbp2 Met27 | 360 | 78 |
| 2Bc1 Met27/rbp1 Met27 | 133 | 0 |
| 2Bc1/rbp1 Met27 | 238 | 92 |
| 2Bc1 Met27/br1 Met27 | 350 | 82 |
| 2Bc1 Met27/br5 Met27 | 208 | 24 |
| 2Bc1/br5 Met27 | 178 | 104 |
Flies were generated from FM7-bearing females carrying the first chromosome listed in the above genotypes. N is the total female progeny examined from a particular cross, and estimated survival is the percentage of non-FM7 females of interest divided by 0.5. Homozygous Met27 female survival is presented first, followed by the corresponding heterozygous Met27 female survival, and finally representative p[Met+] transgenic survival.
Double mutants homozygous for Met27 and rbp1 did not survive to adulthood, as expected (Table 1). The lethal phase was shifted from the readily discernable pharate adult stage seen in rbp1 pupae (Belyaeva et al. 1980; Kiss et al. 1988) to an earlier, less well-defined prepupal/early pupal stage, judging from visual observation of the pupae. Heteroallelic rbp2 Met27/rbp1 Met27 individuals, in contrast to surviving rbp2 Met27/rbp1 individuals, were also prepupal/early pupal lethals (Table 1). Therefore, loss of Met+ gene product resulted in pharate adult lethality in rbp2 and shifted the lethal phase of rbp1 to an earlier stage.
Lethality in rbp2 Met27 homozygotes or rbp2 Met27/rbp1 Met27 heteroallelic pupae could be rescued by one copy of Met+ as the p[EN71] transgene (Table 1).
Interaction between Met and br alleles:
We next tested for interaction between Met and br. A viable br allele, br1, exists (Morgan et al. 1925; Kiss et al. 1988). Individuals homozygous for br1 Met27 were usually lethal, but escaper adults eclosed in small numbers (Table 1), and hemizygotes showed slightly higher viability (Table 3) and were fertile. Examination of br1 Met27 pupae showed lethality in both pupal and especially pharate adult stages. Transgenic br1 Met27; p[EN71]/+ flies readily survived (Table 1). Individuals homozygous or hemizygous for the severe br5 allele were lethal in early pupal development (Kiss et al. 1988) and double mutants of Met27 with br5 were likewise lethal in prepupal/early pupal development. Heteroallelic br5 Met27/br1 pupae survived well, but br5 Met27/br1 Met27 heteroallelic individuals were lethal in prepupal/early pupal development (Table 1), an effect of Met27 similar to that seen in rbp2 Met27/rbp1 Met27.
TABLE 3.
Oviposition by females of various BR-C and Met genotypes
| Eggs laid/female/2-day period: day after eclosion
|
|||||
|---|---|---|---|---|---|
| Genotype | N | 2 | 4 | 6 | 8 |
| br1 v Met27/br1 v Met27 | 63 | 0 | 1.1 | <1 | <1 |
| br1 v Met27/br1 | 20 | 6.6 | 30.9 | 42.6 | 38.0 |
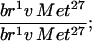 p[Met+]/+ p[Met+]/+
|
20 | 1.0 | 13.0 | 14.0 | 14.6 |
| v/v | 30 | 9.3 | 76.6 | 129 | 136 |
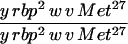 |
43 | 0 | 0 | <1 | <1 |
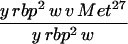 |
20 | 4.8 | 17.8 | 17.6 | 16.6 |
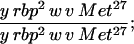 p[Met+]/+ p[Met+]/+
|
30 | 4.1 | 29.8 | 34.0 | 32.3 |
Each value is the mean of egg counts from females of the indicated genotypes isolated from at least two cultures. Due to low preadult survival, 5–7 cultures were required to produce the indicated numbers of the exceptional double mutant homozygotes.
Interaction between br1 and other Met alleles:
It is possible that the effects of Met27 in the double mutants are allele specific. To determine if other alleles of Met also show an interaction with br1, double mutants were constructed and the resultant hemizygotes examined. Table 2 shows hemizygote survival values for each allele combination. Examination of the pupae showed the lethal phase generally to be pharate adult, as found for br1 Met27. The only Met-weak allele recovered to date is MetE1, and the double mutant showed good survival. The remaining alleles resulted from mutagenesis screens employing a variety of mutagens (Wilson and Fabian 1987; Ashok et al. 1998) and generally show poorer survival with br1. Therefore, the interaction between Met and br1 is not allele specific for Met27.
TABLE 2.
Survival of br1 hemizygotes and oviposition by homozygotes carrying various Met alleles
| Genotype of X chromosome | Hemizygotes (% of F1 adults) | Oviposition (eggs/female/10 days) |
|---|---|---|
| br1 | 27 | 880 ± 50 |
| Met3 | 25 | 773 ± 81 |
| br1 Met | 19 | 179 ± 25 |
| br1 Met3 | 25 | 134 ± 9 |
| br1 MetA3 | 5.4 | 16 ± 7 |
| br1 MetE1 | 26 | 987 ± 79 |
| br1 Met128 | 14 | 26 ± 8 |
| br1 Met253 | 15 | 12 ± 3 |
| br1 MetW3 | 7.8 | 27 ± 7 |
| br1 Met27 | 2.8 | 3.4 ± 0.8 |
| br1 MetD29 | 21 | 32 ± 5 |
For each chromosome, F1 progeny were generated from at least three separate cultures of FM7-balanced females × FM7/Y or br1 Met/y2Y67g males, the latter to generate homozygous females for oviposition determination. Survival is expressed as the percentage of adult hemizygotes for the indicated X chromosome present among the F1 progeny. Oviposition (eggs laid ± SEM) was determined over a 10-day period beginning at 2–4 day post eclosion for females that were homozygous for the indicated genotype. When >100 eggs were laid during a 2-day period on the food surface, the total oviposition was estimated by extrapolating from one counted quadrant of the food surface. The oviposition rate of Met3 was similar to that of the other Met alleles, except for the lower rate of Met27 Wilson and Ashok (1998).
Interaction between Met and 2Bc alleles:
Finally, we tested for interaction between Met and 2Bc. Both alleles of 2Bc, 2Bc1 and 2Bc2, have a similar phenotype of prepupal lethality (Belyaeva et al. 1980; Kiss et al. 1988). Individuals homozygous for 2Bc1 Met27 (Table 1) or 2Bc2 Met27 (data not shown) were also lethal as expected, and visual examination of the pupae showed the stage of prepupal lethality to be indistinguishable from that of 2Bc individuals. Therefore, either Met does not interact with 2Bc or the prepupal/early pupal-lethal phase cannot be shifted to an earlier stage in individuals carrying Met27.
Altered BR-C complementation patterns in the absence of MET:
BR-C complementation group mutants complement one another to varying extents, with rbp partially complementing br, and 2BC fully complementing both rbp and br (Belyaeva et al. 1980; Kiss et al. 1988). To determine if Met27 influences complementation for viability, double mutants for both the weak and severe alleles of br and rbp and for 2Bc were crossed and survival of the F1 examined. The absence of Met+ resulted in failure of br5 and rbp1 to complement one another as well as reduced complementation between br5 and rbp2 (Table 1). Complementation was reduced between 2Bc1 and either br5 or rbp1. However, complementation in the Met27 double mutants continued to be robust between either of the weak alleles br1 or rbp2 with 2Bc1 (Table 1). Therefore, an absence of MET reduced complementation to varying degrees, depending on the alleles examined, being less evident with the weak alleles and with the 2Bc1 mutation.
Oogenesis in BR-C Met double mutants:
Both of these genes are involved in oogenesis in Drosophila (Huang and Orr 1992; Wilson and Ashok 1998; Tzolovsky et al. 1999). Normally, vitellogenic oocyte development begins within 12 hr following eclosion, and mature eggs begin to be oviposited within 48 hr (King 1970; Wilson and Ashok 1998). BR-C homozygotes for the weak alleles br1 and rbp2 showed good oviposition, although below that of v/v females (Table 3). Met27 females show oogenesis reduced to ∼20% of wild type (Wilson and Ashok 1998). To determine if interaction between BR-C and Met is apparent during this process in adults, oviposition was examined in BR-C Met homozygotes at 2-day intervals following eclosion. Homozygous br1 Met27 and rbp2 Met27 females occasionally survived to adulthood, and survivors showed strong reductions in oviposition: both br1 Met27 and rbp2 Met27 homozygotes laid only a few eggs during an 8-day examination period, and casual examination of these females for another week showed no change in the ovipositional pattern.
To determine if the ovipositional failure was due to a defect in oogenesis or in oviposition, both br1 Met27 and rbp2 Met27 females were dissected at 5–7 days following eclosion and their vitellogenic oocytes staged and censused. Despite having access to ample yeast and courting wild-type males, there were few (usually 0–3) vitellogenic oocytes in the ovaries of these females, showing that the defect resides in oogenesis, not oviposition. However, no degenerating oocytes were present, a condition suggestive of hormonal disruption (Wilson 1982; Soller et al. 1999).
Oviposition was also measured in the br1 Met double-mutant combinations of the various Met alleles. Some of the allele combinations resulted in severe disruption of oviposition (Table 2), presumably due to defects in oogenesis, as seen for Met27. Other combinations resulted in ovipositional rates higher than those of br1 Met27. In summary, the severe depression in oviposition seen for some Met allele combinations represents a strong allele interaction with br1 and is not specific for the Met27 allele.
The link between BRC and methoprene:
We addressed the enigmatic relationship between BR-C and methoprene in three contexts: (i) the overlap between methoprene-induced defects and BR-C common phenotypes, (ii) the influence of BR-C function on sensitivity to methoprene, and (iii) the effect of methoprene on BR-C expression.
The BR-C common phenotypes include malrotation of male genitalia:
In our previous study, we showed that methoprene treatment of wild type causes a specific partial phenocopy of BRC-associated internal defects (Restifo and Wilson 1998). Here, we tested whether BR-C mutants of each complementation group show the well-known methoprene-induced malrotation of the male genitalia (Postlethwait 1974; Wilson and Fabian 1986). In some cases, this required combining strong mutations with moderate wild-type transgene activity, an established method for revealing late developmental functions (Hall and Thummel 1998). The malrotation phenotype is of particular interest because a genetic interaction between Met and spin, a Fas2 mutation with a malrotation phenotype, has recently been described (Adam et al. 2003).
For rbp, we found that 100% of rbp1/Y hemizygotes have malrotated genitalia (Table 4). This fully penetrant phenotype was rescued by a BR-C+ Y-borne duplication y2Y67g and uncovered by the Y-borne duplication with an interstitial deletion y2YSz280 that lacks all BR-C sequences, confirming that it maps to the BR-C region. To generate br mutant males that die as pharate adults (when the genitalia are pigmented), we partially rescued br-null mutants br5/Y using two transgenic copies of heat-shock-inducible BRC–Z2. Optimal heat-shock protocols rescue lethality (Bayer et al. 1997), gene expression (Liu and Restifo 1998), and CNS morphogenesis (R. F. Spokony and L. L. Restifo, unpublished data). To obtain partial rescue, we heat-shocked unsynchronized third-instar larvae once (37°, 1 hr), and then twice more 18 and 23 hr later. This resulted in very small numbers of pharate adults, 93% of which (14/15) had malrotated genitalia (Table 4). For 2Bc, we first observed malrotation in two very rare, late-dying 2Bc2/Y mutants (Restifo and White 1991, 1992; Consoulas et al. 2005). To examine larger numbers, we used a “leaky” BRC–Z3 transgene whose expression is dependent on temperature (see materials and methods). We found malrotated genitalia, inversely related to rearing temperature: 14% at 25° and 40% at 22–23° (Table 4).
TABLE 4.
Malrotation of the male genitalia is a developmental phenotype of all BR-C complementation groups
| Genotype | Culture conditions | Phenotype: % with malrotation |
|---|---|---|
| reduced bristles on palps | ||
| rbp1/Y | 25° standard | 100 (n = 31) |
| rbp1/y2Y67g | 25° standard | 0 (n = 128) |
| rbp1/y2YSz280 | 25° standard | 100 (n = 23) |
| broad | ||
| br5/Y; hsZ2/+; hsZ2/+ | 37° heat shocks; partial rescue | 93 (n = 15) |
| y w sn3/Y; hsZ2/+; hsZ2/+ | 37° heat shocks | 0 (n = 16) |
| br5/y2Y67g | 37° heat shocks | 4 (n = 48) |
| lethal(1)2Bc | ||
| 2Bc2/Y | 25° standard | 100 (n = 2) |
| 2Bc1/Y; hsZ3/+ | 25°; modest partial rescue | 14 (n = 28) |
| 2Bc1/Y; hsZ3/+ | 22–23°; modest partial rescue | 40 (n =53) |
| 2Bc1/y2Y67g | 22–23° | 0.8 (n = 261) |
| yw/Y; hsZ3/+ | 22–23° | 0 (n =582) |
Neither ubiquitous expression of BRC–Z2 nor BRC–Z3 in wild type caused malrotation. Genetic controls, br5/y2Y67g or 2Bc1/y2Y67g, exposed to the corresponding temperature protocol showed only very rare malrotation (4 or <1%, respectively), confirming the mapping of the phenotype to BR-C and suggesting the possibility of a very small heat-shock effect. In summary, BR-C mutants of all three complementation groups have malrotated male genitalia, which adds this methoprene-induced defect to the list of BR-C common phenotypes.
Sensitivity of BR-C mutants to methoprene:
Met mutations confer semidominant resistance to both the toxic and morphogenetic effects of methoprene (Wilson and Fabian 1986; Restifo and Wilson 1998). To determine if BR-C mutations, either singly or in double-mutant combination with Met, affect the response to methoprene treatment, larvae were raised in the presence of one of four diagnostic concentrations of methoprene and evaluated for survival and the external morphology of surviving adults. Viable BR-C mutations do not change the susceptibility to methoprene from that of wild type (Table 5). Similarly, reducing the dose of 2Bc+ or BR-C+ by 50% (2Bc1/FM7 or npr13/FM7, respectively) did not shift the sensitivity to methoprene. Moreover, in the double mutant, br1 did not affect the resistance conferred by Met3. Hence, BR-C+ function does not appear to impact the MET-dependent methoprene-sensitivity mechanism.
Effect of Met and methoprene on BRC protein accumulation:
We investigated BRC protein expression for two reasons. First, a plausible mechanistic explanation for the genetic interaction between Met and BR-C would be that Met+ upregulates BR-C expression, and that reduced BRC levels in Met mutants would enhance the lethality of partial-loss-of-function BR-C genotypes. Second, reported effects on BR-C transcript levels caused by JH/JHA treatment (Zhou et al. 1998; Zhou and Riddiford 2002) have failed to provide an explanation for the methoprene phenocopy of BR-C common defects, especially those involving internal structures of the head and thorax (Restifo and Wilson 1998).
The BRC family of proteins, which migrate as three size groups (Emery et al.,1994; Mugat et al. 2000), is readily detected by immunoblotting of proteins extracted from whole white prepupae (Figure 2A). The largest group, Emery's p118, is thought to represent BRC–Z4; the middle group, Emery's p91 and p81 contains BRC–Z1 and –Z3; the smallest group, Emery's p64 and p57 contains BRC–Z2. Over the subsequent 24 hr, especially after head eversion (∼12 hr APF), BRC protein levels declined (Figure 2B). In Met27 mutants, the pattern and relative abundance of BRC isoforms detected over this first day of metamorphosis was indistinguishable from those of wild type (Figure 2B). Likewise, methoprene treatment of wild-type animals did not change the overall quantities and isoform patterns of BRC proteins (Figure 2C).
Figure 2.

BRC protein accumulation during the first day of metamorphosis is not affected by lack of MET or by treatment with methoprene. Immunoblotting of whole-body protein extracts with anti-BRcore; 0.5-animal equivalents per lane. (A) Wild-type (OreRC) white prepupae. The migration positions of molecular weight markers are shown on the far left. The individual boxes on the right show optimized images of each group of BRC proteins, obtained by changing exposure times or amounts of protein loaded. The indicated molecular weight estimates are averages based on three or more independent experiments. (B and C) Time course of BRC accumulation in animals collected at puparium formation and sampled every 8 hr. The rightmost lanes contain protein from BR-C-null mutant (npr13/Y) wandering third-instar larvae as a negative control to verify the specificity of the antibody. Each blot was reprobed for actin as an indicator of protein loading. hAPF, hours after puparium formation. (B) Wild-type (OreRC) and Met-null mutant (v Met27). The BRC protein profiles are qualitatively and quantitatively indistinguishable. (C) Control and methoprene-treated OreRC. The BRC protein profiles are very similar.
In independent experiments, BRC proteins were evaluated over a 3-day interval, representing ∼75% of metamorphosis, during which animals were resynchronized at head eversion. At 24 hr APF (12 hr after head evesion), BRC proteins were detectable in the methoprene-treated animals, but not in the controls (Figure 3A). Similarly, methoprene-treated animals showed mild persistence of BRC–Z1 during midpupal stages, following a peak in accumulation at the normal time, 8 hr APF. There was no reappearance of BRC–Z1 or any other BRC isoforms during mid-to-late pupal stages (Figure 3A), even on very long exposures of the immunoblots (data not shown). In summary, chronic larval treatment with lethal doses of the JH agonist and mimic methoprene does not block BRC protein accumulation during the first day of metamorphosis, a developmental window in which BR-C function is essential.
Figure 3.

BRC protein accumulation is mildly prolonged by methoprene treatment. Animals were collected at puparium formation, sampled at 0 and 8 hr, resynchronized at head eversion (∼12 hr APF in control wild-type animals), and then sampled 4 hr after head eversion (= 16 hr APF) and at 8-hr intervals thereafter. Immunoblotting with anti-BRcore and anti-Z1; 0.5-animal equivalents per lane. In each experiment, control and methoprene-treated animals were reared and processed in parallel, the gels were run simultaneously in the same apparatus, and the antibody labeling and detection were performed in parallel. Each of the blots was reprobed for actin as an indicator of protein loading. (A) Wild type (OreRC). Two independent immunoblotting experiments are shown. Treatment with a lethal dose of methoprene causes a mild prolongation of BRC protein accumulation, especially of the abundant 75–85-kDa bands, which contain primarily BRC–Z1. Methoprene does not cause reappearance of BRC in the midpupal period (40– 64 hr APF). (B) Met-null mutant (Met27). Sequential immunoblotting for BRC–Z1 and BRcore. As in wild type, BRC accumulation is mildly enhanced by treatment with a lethal dose of methoprene.
Over the 3-day interval, BRC protein profiles in Met27 mutants were normal, in both the presence and absence of methoprene (Figure 3B). These data demonstrate that the accumulation of BRC proteins at the onset of metamorphosis is MET independent. Hence, altered BRC expression, at least at the level of the whole-body protein accumulation, cannot explain the genetic interactions between BR-C and Met mutations.
DISCUSSION
In this work, we have examined two genes required for signaling by 20E (BR-C) and JH (Met) to probe for interaction between these pathways. We have found evidence for interaction between Met and BR-C as reflected by synergistically reduced viability and oogenesis seen in double mutants. Consistent results were seen with different combinations of Met and br or Met and rbp alleles (Figure 1; Tables 1 and 2), indicating that the interactions are not allele specific in either direction.
Met interacted with both the weak-viable alleles br1 and rbp2 and with the severe alleles br5 and rbp1 (Table 1) during pupal development. Each of the weak alleles possesses sufficiently functional gene product to permit completion of pupal development; but this amount is insufficient when MET is absent or defective. The more severe rbp1 homogygotes are pupal lethal, but only at late metamorphosis, in the pharate adult stage (Belyaeva et al. 1980). Lethality was shifted in rbp1 Met27 pupae to prepupal/early pupal development, suggesting that MET absence causes the residual rbp1 function to be insufficient during these earlier stages in pupal development. Homozygotes of br5 and 2Bc die in the early and late prepupal stage, respectively (Kiss et al. 1988), and the double mutants with Met27 showed a similar phenotype, demonstrating that the interaction cannot shift lethality to an earlier stage, late third-instar larvae. Our observations are consistent with the interaction between BR-C and Met beginning in prepupal or early pupal development. While we interpret the Met–BR-C interaction as enhancing the lethality of br and rbp mutations, it is also possible that Met becomes an essential gene when BR-C function is reduced, or that the interaction is mutual, such that both mutations become more severe in phenotype when they are present together.
Genetic interaction became strikingly evident when complementation failures between mutant alleles from different BR-C complementation groups occurred in the presence of Met27 (Table 1). Without MET, developing animals may be less able to make use of the partial functional redundancy among BRC isoforms that has been documented previously (Bayer et al. 1997).
The interaction between mutant alleles of BR-C and Met was also evident in the adult stage when oogenesis was examined. Both the rate of oviposition and the paucity of vitellogenic oocytes in ovaries of br1 Met27 and rbp2 Met27 females reflects almost complete failure of oogenesis, with only a few eggs oviposited during the lifetime of the female.
Previous studies have also detected BR-C interaction with other genes. Double mutants of BR-C with another primary response gene E74 show interaction for some but not all of the phenotypic characters (Fletcher and Thummel 1995). In addition to interactions among transcription regulators of the ecdysone cascade, br alleles interact with genes involved in imaginal disc morphogenesis, including those encoding an atypical serine protease, Stubble-stubboid (Beaton et al. 1988; Appel et al. 1993), nonmuscle myosin II heavy chain (zipper), the Drosophila serum response factor transcription factor [blistered: (Gotwals and Fristrom 1991)], the small GTPase Rho1, cytoplasmic tropomyosin, and 22 others (Ward et al. 2003).
Although BR-C expression and function overlap the JH/JHA-sensitive period, data presented above demonstrate that methoprene treatment does not block BRC expression in either wild-type or Met-null mutants (Figures 2 and 3). Furthermore, the methoprene phenocopy, which excludes complementation group-specific defects (e.g., larval salivary gland persistence, which is rbp-restricted), is not consistent with methoprene simply reducing BRC expression (Restifo and Wilson 1998). We propose that JH application results in abnormal function of BRC proteins, thus phenocopying certain characteristics common to all BR-C mutants. Therefore, the link between BR-C mutant phenotypes and JH-induced defects could be abnormal regulation of target genes, resulting in the phenotypic characteristics observed (Figure 4). Previously, we suggested several possibilities to explain methoprene pathology and BR-C phenocopy, including BRC interaction with an unidentified partner, perhaps MET (Restifo and Wilson 1998). We believe that the Met–BR-C genetic interaction reported in this work can be explained best by this hypothesized protein–protein interaction between MET and BRC to regulate one or more target genes. Supporting our hypothesis are the following findings: first, both proteins are located in the nucleus (Emery et al. 1994; Restifo and Hauglum 1998; Pursley et al. 2000; Renault et al. 2001), so there is no compartmental barrier to interaction. Second, both proteins appear to be transcription factors: BRC isoforms bind specific DNA sequences (von Kalm et al. 1994; Dubrovsky et al. 2001) and regulate transcription (Dubrovsky et al. 1994; Hodgetts et al. 1995; Crossgrove et al. 1996; Mugat et al. 2000). BR-C mutants have misexpressed secondary-response and other target genes (Guay and Guild 1991; Karim et al. 1993; Mugat et al. 2000; Renault et al. 2001; Dunne et al. 2002; Sempere et al. 2003). MET is a member of the bHLH–PAS family of transcription factors (Ashok et al. 1998) and was recently shown to act as one (Miura et al. 2005). Third, both are found at common times during development, such as prepupae (Karim et al. 1993; Ashok et al. 1998) and during vitellogenic oocyte development (Tzolovsky et al. 1999; Pursley et al. 2000). Finally, PAS domains in bHLH–PAS proteins are thought to promote protein–protein interaction (Heery et al. 1997), either with other PAS proteins or as coactivators with nuclear receptor proteins (Xu et al. 1999), and the BTB/POZ domain of BRC has been implicated in protein–protein interaction (Zollman et al. 1994; Melnick et al. 2002).
Figure 4.

Proposed scheme for regulation by MET and BRC–Z1 of a target gene necessary for pupal viability. Illustrated are three hypothesized transcriptional situations for rbp: top, wild type; center, in the presence of wild-type MET, strong hypomorphic rbp alleles cause lethality in the pharate adult stage; bottom, in rbp Met double mutants, lethality is shifted to the prepupal stage. No interaction between MET and Z1 is shown, but formation of heterodimers is possible. Likewise, each is shown binding DNA when the protein is wild type but not when mutant, although DNA binding by lesioned protein is possible. The presumed level of target-gene transcription is reflected by arrow thickness.
In Met27 mutants, BRC protein accumulation profiles are normal (Figures 2 and 3). Since metamorphosis is not derailed in Met27 pupae, BRC+ function in these pupae does not seem to be adversely affected. The fly may be protected from absence of MET by functional redundancy (Wilson and Ashok 1998). A candidate for the redundant substitute is the PAS gene germ cell expressed (gce), a gene with high (∼70% amino acid identity) homology to Met (Moore et al. 2000) that could substitute for MET to rescue larval and/or pupal development. However, this substitute does not appear to be satisfactory if BR-C is mutant. When a gce mutant becomes available, its phenotype could help evaluate this hypothesis.
How does the application of exogenous JH act to phenocopy BR-C? It is clear that the action of these compounds occurs through MET, probably acting as a JH receptor component (Wilson and Fabian 1986; Shemshedini et al. 1990; Shemshedini and Wilson 1990; Ashok et al. 1998; Miura et al. 2005). JH is present during larval development when it presumably acts to prevent premature metamorphosis resulting from each wave of 20E secretion that triggers a molt. This failsafe mechanism may occur by JH binding by and conformational change of MET, resulting in regulation of genes necessary for molting or perhaps simply blocking expression of metamorphic genes. Studies with Drosophila S-2 cells have implicated the transcription factor E75A in promoting JH regulation of larval development (Dubrovsky et al. 2004). At metamorphosis, when little or no JH is present (Bownes and Rembold 1987; Sliter et al. 1987), BR-C is expressed, and we propose that BRC dimerizes with the nonliganded MET protein to regulate a different set of target genes, promoting the initiation of metamorphosis. If exogenous JH is present during this time, it binds to MET and results in a more larval conformation, resulting in inappropriate binding to BRC and leading to a change in target-gene expression patterns consequently seen as defects characteristic of BR-C mutants.
Other work has implicated BR-C in the action of the JH agonist pyriproxyfen during metamorphic disruption. Zhou and Riddiford (2002) showed that application of this compound to white prepupae resulted in re-expression of BRC–Z1 in the abdomen during late pupal development, which in turn caused abnormal development of abdominal epidermis, including bristle disturbances. Those findings differ from ours with methoprene in two significant ways. First, a lethal dose of methoprene caused a mild enhancement and prolongation of BRC protein accumulation in young pupae, but no re-expression at later times (Figure 3A). Second, the modest effect of methoprene on BRC protein profiles cannot mediate the developmental effects of this JHA because the same mild persistence of BRC was seen in Met27 mutants (Figure 3B), which are protected against methoprene-induced defects. It is not clear what underlies the difference in response of BR-C to methoprene and pyriproxyfen. We note that pyriproxyfen is a more powerful JH agonist than methoprene (Riddiford and Ashburner 1991), but qualitative differences in the actions of the two compounds may exist as well.
In summary, our results provide genetic evidence that supports other studies implicating BR-C as a focal point for interaction of JH and 20E signaling pathways, and they suggest that BRC and MET interact to regulate expression of one or more effector genes involved in metamorphic development.
Acknowledgments
We thank Cynthia Bayer for stocks and for pointing out the malrotated genitalia in partially rescued br males. T.G.W. carried out some of this work in the laboratory of Mary Bownes at the University of Edinburgh. L.L.R. thanks Hannah V. Foster and H. Jolene Clark for help with crosses, lethal phase determination, and male genitalia phenotype scoring. This work was supported by grants to T.G.W. (National Science Foundation grant IBN 0322136 and National Institutes of Health grant AI052290) and L.L.R. (Flinn Foundation Interdisciplinary Genetics Research Grant and National Institutes of Health HD038363). D.M.D. was supported by a Flinn Foundation Genetics Training Grant.
References
- Adam, G., N. Perrimon and S. Noselli, 2003. The retinoic-like juvenile hormone controls the looping of left-right asymmetric organs in Drosophila. Development 130: 2397–2406. [DOI] [PubMed] [Google Scholar]
- Appel, L. F., M. Prout, R. Abu-Shumays, A. Hammond, J. C. Garbe et al., 1993. The Drosophila Stubble-stubboid gene encodes an apparent transmembrane serine protease required for epithelial morphogenesis. Proc. Natl. Acad. Sci. USA 90: 4937–4941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner, M., 1970. Effects of juvenile hormone on adult differentiation of Drosophila melanogaster. Nature 227: 187–189. [DOI] [PubMed] [Google Scholar]
- Ashburner, M., C. Chihara, P. Meltzer and G. Richards, 1974. Temporal control of puffing activity in polytene chromosomes. Cold Spring Harbor Symp. Quant. Biol. 38: 655–662. [DOI] [PubMed] [Google Scholar]
- Ashok, M., C. Turner and T. G. Wilson, 1998. Insect juvenile hormone resistance gene homology with the bHLH-PAS family of transcriptional regulators. Proc. Natl. Acad. Sci. USA 95: 2761–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bainbridge, S. P., and M. Bownes, 1981. Staging the metamorphosis of Drosophila melanogaster. J. Embryol. Exp. Morphol. 66: 57–80. [PubMed] [Google Scholar]
- Bayer, C. A., B. Holley and J. W. Fristrom, 1996. A switch in broad-complex zinc-finger isoform expression is regulated posttranscriptionally during the metamorphosis of Drosophila imaginal discs. Dev. Biol. 177: 1–14. [DOI] [PubMed] [Google Scholar]
- Bayer, C. A., L. von Kalm and J. W. Fristrom, 1997. Relationships between protein isoforms and genetic functions demonstrate functional redundancy at the Broad-Complex during Drosophila metamorphosis. Dev. Biol. 187: 267–282. [DOI] [PubMed] [Google Scholar]
- Beaton, A. H., I. Kiss, D. Fristrom and J. W. Fristrom, 1988. Interaction of the Stubble-stubboid locus and the Broad-Complex of Drosophila melanogaster. Genetics 120: 453–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belyaeva, E. S., M. G. Aizenzon, V. F. Semeshin, I. Kiss, K. Koczya et al., 1980. Cytogenetic analysis of the 2B3–4-2B11 region of the X-chromosome of Drosophila melanogaster. I. Cytology of the region and mutant complementation groups. Chromosoma 81: 281–306. [DOI] [PubMed] [Google Scholar]
- Bownes, M., and H. Rembold, 1987. The titre of juvenile hormone during the pupal and adult stages of the life cycle of Drosophila melanogaster. Eur. J. Biochem. 164: 709–712. [DOI] [PubMed] [Google Scholar]
- Consoulas, C., R. B. Levine and L. L. Restifo, 2005. The steroid hormone-regulated gene Broad-Complex is required for dendritic growth of motorneurons during metamorphosis of Drosophila. J. Comp. Neurol. 485: 321–337. [DOI] [PubMed] [Google Scholar]
- Crossgrove, K., C. A. Bayer, J. W. Fristrom and G. M. Guild, 1996. The Drosophila Broad-Complex early gene directly regulates late gene transcription during the ecdysone-induced puffing cascade. Dev. Biol. 180: 745–758. [DOI] [PubMed] [Google Scholar]
- Deng, W.-M., and M. Bownes, 1997. Two signalling pathways specify localised expression of the Broad-Complex in Drosophila eggshell patterning and morphogenesis. Development 124: 4639–4647. [DOI] [PubMed] [Google Scholar]
- DiBello, P. R., D. A. Withers, C. A. Bayer, J. W. Fristrom and G. M. Guild, 1991. The Drosophila Broad-Complex encodes a family of related, zinc finger-containing proteins. Genetics 129: 385–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubrovsky, E. B., G. Dretzen and M. Bellard, 1994. The Drosophila Broad-Complex regulates developmental changes in transcription and chromatin structure of the 67B heat-shock gene cluster. J. Mol. Biol. 241: 353–362. [DOI] [PubMed] [Google Scholar]
- Dubrovsky, E. B., V. A. Dubrovskaya, A. L. Bilderback and E. M. Berger, 2000. The isolation of two juvenile hormone-inducible genes in Drosophila melanogaster. Dev. Biol. 224: 486–495. [DOI] [PubMed] [Google Scholar]
- Dubrovsky, E. B., V. A. Dubrovskaya and E. M. Berger, 2001. Selective binding of Drosophila BR-C isoforms to a distal regulatory element in the hsp23 promoter. Insect Biochem. Mol. Biol. 31: 1231–1239. [DOI] [PubMed] [Google Scholar]
- Dubrovsky, E. B., V. A. Dubrovskaya and E. M. Berger, 2004. Hormonal regulation and functional role of Drosophila E75A orphan nuclear receptor in the juvenile hormone signaling pathway. Dev. Biol. 268: 258–270. [DOI] [PubMed] [Google Scholar]
- Dunne, J. C., V. Kondylis and C. Rabouille, 2002. Ecdysone triggers the expression of Golgi genes in Drosophila imaginal discs via Broad-Complex. Dev. Biol. 245: 172–186. [DOI] [PubMed] [Google Scholar]
- Emery, I. F., V. Bedian and G. M. Guild, 1994. Differential expression of Broad-Complex transcription factors may forecast distinct developmental tissue fates during Drosophila metamorphosis. Development 120: 3275–3287. [DOI] [PubMed] [Google Scholar]
- Fletcher, J. C., and C. S. Thummel, 1995. The ecdysone-inducible Broad-Complex and E75 early genes interact to regulate target gene transcription and Drosophila metamorphosis. Genetics 141: 1025–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert, L. I., N. A. Granger and R. M. Roe, 2000. The juvenile hormones: historical facts and speculations on future research directions. Insect Biochem. Mol. Biol. 30: 617–644. [DOI] [PubMed] [Google Scholar]
- Gonzy, G., G. V. Pokholkova, F. Peronnet, B. Mugat, O. V. Demakova et al., 2002. Isolation and characterization of novel mutations of the Broad-Complex, a key regulatory gene of ecdysone induction in Drosophila melanogaster. Insect Biochem. Mol. Biol. 32: 121–132. [DOI] [PubMed] [Google Scholar]
- Gotwals, P. J., and J. W. Fristrom, 1991. Three neighboring genes interact with the Broad-Complex and the Stubble-stubbloid locus to affect imaginal disc morphogenesis in Drosophila. Genetics 127: 747–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guay, P. S., and G. M. Guild, 1991. The ecdysone-induced puffing cascade in Drosophila salivary glands: a Broad-Complex early gene regulates intermolt and late gene transcription. Genetics 129: 169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, B. L., and C. S. Thummel, 1998. The RXR homolog Ultraspiracle is an essential component of the Drosophila ecdysone receptor. Development 125: 4709–4717. [DOI] [PubMed] [Google Scholar]
- Handler, A. M., 1982. Ecdysteroid titers during pupal and adult development in Drosophila melanogaster. Dev. Biol. 93: 73–82. [DOI] [PubMed] [Google Scholar]
- Heery, D. M., E. Kalkhoven, S. Hoare and M. G. Parker, 1997. A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature 387: 733–736. [DOI] [PubMed] [Google Scholar]
- Henrich, V. C., and N. E. Brown, 1995. Insect nuclear receptors: A developmental and comparative perspective. Insect Biochem. Mol. Biol. 25: 881–897. [DOI] [PubMed] [Google Scholar]
- Hodgetts, R., W. C. Clark, S. O'Keefe, M. Schouls, K. Crossgrove et al., 1995. Hormonal induction of dopa decarboxylase in the epidermis of Drosophila is mediated by the Broad-Complex. Development 121: 3913–3922. [DOI] [PubMed] [Google Scholar]
- Huang, R.-Y., and W. C. Orr, 1992. Broad-Complex function during oogenesis in Drosophila melanogaster. Dev. Genet. 13: 277–288. [DOI] [PubMed] [Google Scholar]
- Jones, G., 1995. Molecular mechanisms of action of juvenile hormone. Annu. Rev. Entomol. 40: 147–169. [DOI] [PubMed] [Google Scholar]
- Karim, F. D., G. M. Guild and C. S. Thummel, 1993. The Drosophila Broad-Complex plays a key role in controlling ecdysone-regulated gene expression at the onset of metamorphosis. Development 118: 977–988. [DOI] [PubMed] [Google Scholar]
- King, R. C., 1970. Ovarian Development in Drosophila melanogaster. Academic Press, New York.
- Kiss, I., A. H. Beaton, J. Tardiff, D. Fristrom and J. W. Fristrom, 1988. Interactions and developmental effects of mutations in the Broad-Complex of Drosophila melanogaster. Genetics 118: 247–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucharova-Mahmood, S., I. Raska, B. M. Mechler and R. Farkas, 2002. Temporal regulation of Drosophila salivary gland degeneration by the Broad-Complex transcription factors. J. Struct. Biol. 140: 67–78. [DOI] [PubMed] [Google Scholar]
- Lee, C.-Y., and E. H. Baehrecke, 2001. Steroid regulation of autophagic programmed cell death during development. Development 128: 1443–1455. [DOI] [PubMed] [Google Scholar]
- Lindsley, D. L., and G. G. Zimm, 1992. The Genome of Drosophila melanogaster. Academic Press, New York.
- Liu, E., and L. L. Restifo, 1998. Identification of a Broad Complex-regulated enhancer in the developing visual system of Drosophila. J. Neurobiol. 34: 253–270. [DOI] [PubMed] [Google Scholar]
- Mackler, J. M., and N. E. Reist, 2001. Mutations in the second C2 domain of synaptotagmin disrupt synaptic transmission at Drosophila neuromuscular junctions. J. Comp. Neurol. 436: 4–16. [PubMed] [Google Scholar]
- Madhavan, K., 1973. Morphogenetic effects of juvenile hormone and juvenile hormone mimics on adult development of Drosophila. J. Insect Physiol. 19: 441–453. [DOI] [PubMed] [Google Scholar]
- Melnick, A., G. Carlile, K. F. Ahmad, C.-L. Kiang, C. Corcoran et al., 2002. Critical residues within the BTB domain of PLZF and Bcl-6 modulate interaction with corepressors. Mol. Cell. Biol. 22: 1804–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura, K., M. Oda, S. Makita and Y. Chinzei, 2005. Characterization of the Drosophila Methoprene-tolerant gene product. FEBS J. 272: 1169–1178. [DOI] [PubMed] [Google Scholar]
- Moore, A. W., S. Barbel, L. Y. Jan and Y. N. Jan, 2000. A genomewide survey of basic helix-loop-helix factors in Drosophila. Proc. Natl. Acad. Sci. USA 97: 10436–10441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan, T. H., C. Bridges and A. H. Sturtevant, 1925. The genetics of Drosophila. Bibliogr. Genet. 2: 145. [Google Scholar]
- Mugat, B., V. Brodu, J. Kejzlarova-Lepesant, C. Antoniewski, C. A. Bayer et al., 2000. Dynamic expression of broad-complex isoforms mediates temporal control of an ecdysteroid target gene at the onset of Drosophila metamorphosis. Dev. Biol. 227: 104–117. [DOI] [PubMed] [Google Scholar]
- Postlethwait, J. H., 1974. Juvenile hormone and the adult development of Drosophila. Biol. Bull. 147: 119–135. [DOI] [PubMed] [Google Scholar]
- Pursley, S., M. Ashok and T. G. Wilson, 2000. Intracellular localization and tissue specificity of the Methoprene-tolerant (Met) gene product in Drosophila melanogaster. Insect Biochem. Mol. Biol. 30: 839–845. [DOI] [PubMed] [Google Scholar]
- Renault, N., K. King-Jones and M. Lehmann, 2001. Downregulation of the tissue-specific transcription factor Fork head by Broad-Complex mediates a stage-specfic response. Development 128: 3729–3737. [DOI] [PubMed] [Google Scholar]
- Restifo, L. L., and W. Hauglum, 1998. Parallel molecular genetic pathways operate during CNS metamorphosis in Drosophila. Mol. Cell. Neurosci. 11: 134–148. [DOI] [PubMed] [Google Scholar]
- Restifo, L. L., and V. K. Merrill, 1994. Two Drosophila regulatory genes, Deformed and the Broad Complex, share common functions in development of adult CNS, head, and salivary glands. Dev. Biol. 162: 465–485. [DOI] [PubMed] [Google Scholar]
- Restifo, L. L., and K. White, 1991. Mutations in a steroid hormone-regulated gene disrupt the metamorphosis of the central nervous system in Drosophila. Dev. Biol. 148: 174–194. [DOI] [PubMed] [Google Scholar]
- Restifo, L. L., and K. White, 1992. Mutations in a steroid hormone-regulated gene disrupt the metamorphosis of internal tissues in Drosophila: salivary glands, muscle, and gut. Roux's Arch. Dev. Biol. 201: 221–234. [DOI] [PubMed] [Google Scholar]
- Restifo, L. L., and T. G. Wilson, 1998. A juvenile hormone agonist reveals distinct developmental pathways mediated by ecdysone-inducible Broad Complex transcription factors. Develop. Genet. 22: 141–159. [DOI] [PubMed] [Google Scholar]
- Richards, G., 1997. The ecdysone regulatory cascades in Drosophila. Adv. Dev. Biol. 5: 81–135. [Google Scholar]
- Riddiford, L. M., 1993. Hormones and Drosophila development, pp. 899–939 in The Development of Drosophila melanogaster, edited by M. Bate and A. Martinez Arias. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Riddiford, L. M., 1994. Cellular and molecular actions of juvenile hormone I. General considerations and premetamorphic actions. Adv. Insect Physiol. 24: 213–274. [Google Scholar]
- Riddiford, L. M., 1996. Molecular aspects of juvenile hormone action in insect metamorphosis, pp. 223–251 in Metamorphosis: Postembryonic Reprogramming of Gene Expression in Amphibian and Insect Cells, edited by L. I. Gilbert, J. R. Tata and B. G. Atkinson. Academic Press, San Diego.
- Riddiford, L. M., and M. Ashburner, 1991. Effects of juvenile hormone mimics on larval development and metamorphosis of Drosophila melanogaster. Gen. Comp. Endocrinol. 82: 172–183. [DOI] [PubMed] [Google Scholar]
- Riddiford, L. M., J. W. Truman and P. Cherbas, 2000. Ecdysone receptors and their biological actions. Vitam. Horm. 60: 1–73. [DOI] [PubMed] [Google Scholar]
- Sandstrom, D. J., C. A. Bayer, J. W. Fristrom and L. L. Restifo, 1997. Broad-Complex transcription factors regulate thoracic muscle attachment in Drosophila. Dev. Biol. 181: 168–185. [DOI] [PubMed] [Google Scholar]
- Sempere, L. F., E. B. Dubrovsky, V. A. Dubrovskaya, E. M. Berger and V. Ambros, 2002. The expression of the let-7 small regulatory RNA is controlled by ecdysone during metamorphosis in Drosophila melanogaster. Dev. Biol. 244: 170–179. [DOI] [PubMed] [Google Scholar]
- Sempere, L. F., N. S. Sokol, E. B. Dubrovsky, E. M. Berger and V. Ambros, 2003. Temporal regulation of microRNA expression in Drosophila melanogaster mediated by hormonal signals and Broad-Complex gene activity. Dev. Biol. 259: 9–18. [DOI] [PubMed] [Google Scholar]
- Shemshedini, L., and T. G. Wilson, 1990. Resistance to juvenile hormone and an insect growth regulator in Drosophila is associated with an altered cytosolic juvenile hormone binding protein. Proc. Natl. Acad. Sci. USA 87: 2072–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shemshedini, L., M. Lanoue and T. G. Wilson, 1990. Evidence for a juvenile hormone receptor involved in protein synthesis in Drosophila melanogaster. J. Biol. Chem. 265: 1913–1918. [PubMed] [Google Scholar]
- Sliter, T. J., B. J. Sedlak, F. C. Baker and P. A. Schooley, 1987. Juvenile hormone in Drosophila melanogaster. Identification and titer determination during development. Insect Biochem. 17: 161–165. [Google Scholar]
- Soller, M., M. Bownes and E. Kubli, 1999. Control of oocyte maturation in sexually mature Drosophila females. Dev. Biol. 208: 337–351. [DOI] [PubMed] [Google Scholar]
- Spindler, K.-D., S. Przibilla and M. Spindler-Barth, 2001. Moulting hormones of arthropods: molecular mechanisms. Zoology 103: 189–201. [Google Scholar]
- Staal, G. B., 1975. Insect growth regulators with juvenile hormone activity. Annu. Rev. Entomol. 20: 417–460. [DOI] [PubMed] [Google Scholar]
- Thummel, C. S., and J. Chory, 2002. Steroid signaling in plants and insects-common themes, different pathways. Genes Dev. 16: 3113–3129. [DOI] [PubMed] [Google Scholar]
- Towbin, H., T. Staehelin and J. Gordon, 1979. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. USA 76: 4350–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzolovsky, G., W. M. Deng, T. Schlitt and M. Bownes, 1999. The function of the Broad-Complex during Drosophila melanogaster oogenesis. Genetics 153: 1371–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Kalm, L., K. Crossgrove, D. von Seggern, G. M. Guild and S. K. Beckendorf, 1994. The Broad-Complex directly controls a tissue-specific response to the steroid hormone ecdysone at the onset of Drosophila metamorphosis. EMBO J. 13: 3505–3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward, R. E., J. Evans and C. S. Thummel, 2003. Genetic modifer screens in Drosophila demonstrate a role for Rho1 signaling in ecdysone-triggered imaginal disc morphogenesis. Genetics 165: 1397–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, C. M., 1961. The juvenile hormone. II. Its role in the endocrine control of molting, pupation, and adult development in the Cecropia silkworm. Biol. Bull. 121: 572–585. [Google Scholar]
- Wilson, T. G., 1982. A correlation between juvenile hormone deficiency and vitellogenic oocyte degeneration in Drosophila melanogaster. Wilhelm Roux Arch. Entwicklungsmech. Org. 191: 257–263. [DOI] [PubMed] [Google Scholar]
- Wilson, T. G., 1996. Genetic evidence that mutants of the Methoprene-tolerant gene of Drosophila melanogaster are null mutants. Arch. Insect Biochem. Physiol. 32: 641–649. [DOI] [PubMed] [Google Scholar]
- Wilson, T. G., 2004. The molecular site of action of juvenile hormone and juvenile hormone insecticides during metamorphosis: how these compounds kill insects. J. Insect Physiol. 50: 111–121. [DOI] [PubMed] [Google Scholar]
- Wilson, T. G., and M. Ashok, 1998. Insecticide resistance resulting from an absence of target-site gene product. Proc. Natl. Acad. Sci. USA 95: 14040–14044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, T. G., and J. Fabian, 1986. A Drosophila melanogaster mutant resistant to a chemical analog of juvenile hormone. Develop. Biol. 118: 190–201. [DOI] [PubMed] [Google Scholar]
- Wilson, T. G., and J. Fabian, 1987. Selection of methoprene-resistant mutants of Drosophila melanogaster, pp. 179–188 in Molecular Entomology, edited by J. Law. UCLA Symposia on Molecular and Cellular Biology, New Series, Los Angeles.
- Wyatt, G. R., and K. G. Davey, 1996. Cellular and molecular actions of juvenile hormone II. Roles of juvenile hormone in adult insects. Adv. Insect Physiol. 26: 1–155. [Google Scholar]
- Xu, L., C. K. Glass and M. G. Rosenfeld, 1999. Coactivator and corepressor complexes in nuclear receptor function. Curr. Opin. Genet. Dev. 9: 140–147. [DOI] [PubMed] [Google Scholar]
- Zhou, B., K. Hiruma, T. Shinoda and L. M. Riddiford, 1998. Juvenile hormone prevents ecdysteroid-induced expression of broad complex RNAs in the epidermis of the tobacco hornworm, Manduca sexta. Dev. Biol. 203: 233–244. [DOI] [PubMed] [Google Scholar]
- Zhou, X., and L. M. Riddiford, 2002. Broad specifies pupal development and mediates the ‘status quo’ action of juvenile hormone on the pupal-adult transformation in Drosophila and Manduca. Development 129: 2259–2269. [DOI] [PubMed] [Google Scholar]
- Zollman, S., D. Godt, G. G. Prive, J.-L. Couderc and F. A. Laski, 1994. The BTB domain, found primarily in zinc finger proteins, defines an evolutionarily conserved family that includes several developmentally regulated genes in Drosophila. Proc. Natl. Acad. Sci. USA 91: 10717–10721. [DOI] [PMC free article] [PubMed] [Google Scholar]