

Abstract
Most models of positive directional selection assume codominance of the beneficial allele. We examine the importance of this assumption by implementing a coalescent model of positive directional selection with arbitrary dominance. We find that, for a given mean fixation time, a beneficial allele has a much weaker effect on diversity at linked neutral sites when the allele is recessive.
THE fixation of a beneficial allele leaves a signature in patterns of genetic variation at linked neutral sites. If this signature is well characterized, it can be used to identify recent adaptations from polymorphism data. To date, most models developed to characterize the effects of positive directional selection (termed “selective sweep”) have assumed that the favored allele is codominant. In other words, if the fitnesses of the three genotypes are given by 1, 1 + sh, and 1 + s (where s is the selection coefficient), then 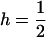 . While the dominance coefficients of advantageous mutations are largely unknown, this assumption is likely to be unrealistic (Jimenez-Sanchez et al. 2001; Kondrashov and Koonin 2004). The heterozygote effect is known to be a crucial parameter governing the rate of evolution, especially in the context of X–autosome comparisons (Orr and Betancourt 2001; Betancourt et al. 2004).
. While the dominance coefficients of advantageous mutations are largely unknown, this assumption is likely to be unrealistic (Jimenez-Sanchez et al. 2001; Kondrashov and Koonin 2004). The heterozygote effect is known to be a crucial parameter governing the rate of evolution, especially in the context of X–autosome comparisons (Orr and Betancourt 2001; Betancourt et al. 2004).
The parameter h influences the trajectory of the favored allele from introduction to fixation and hence may be an important determinant of the signature of directional selection in polymorphism data. Analytic results demonstrate that when Ns is large, the mean fixation time of the favored allele is approximately the same for h and (1 − h) (N is the diploid effective population size) (van Herwaarden and van der Wal 2002). This approximation is highly accurate as long as N and Ns are large. This result might be taken to imply that the effects on polymorphism of the fixation event are very similar. However, as we show below, even when the mean fixation time is the same, the effect on polymorphism is not.
To examine this, we implement a general model of positive directional selection, allowing for weak selection (i.e., small Ns) as well as arbitrary dominance to be modeled. We use a coalescent approach introduced by Kaplan et al. (1989) and developed in Griffiths (2003) and Coop and Griffiths (2004). This approach allows us to generate polymorphism data from a neutral locus linked to a site at which a favorable allele has recently reached fixation in the population. The program implementing the algorithm produces output in the format of ms (Hudson 2002) and is available upon request to K.T.
COALESCENT MODEL OF POSITIVE DIRECTIONAL SELECTION
We focus on a neutrally evolving, autosomal region and assume the standard neutral model of a random-mating population of constant size. At one site within this region, a favorable allele arises and eventually reaches fixation in the population. Genotype fitnesses are given as above and the scaled selection parameter is 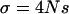 . Since we consider models of directional selection, h is constrained to be between 0 and 1. There are two steps involved in generating a sample from the neutral locus: (1) generation of the trajectory of a favored allele from introduction to fixation and (2) generation of an ancestral recombination graph for the neutral locus, conditional on this trajectory.
. Since we consider models of directional selection, h is constrained to be between 0 and 1. There are two steps involved in generating a sample from the neutral locus: (1) generation of the trajectory of a favored allele from introduction to fixation and (2) generation of an ancestral recombination graph for the neutral locus, conditional on this trajectory.
The first step is accomplished by using a variable-sized-jump random walk to approximate to the diffusion process, conditional on fixation (for details see Przeworski et al. 2005). Briefly, the trajectory frequency of the favored allele, x, changes after a small time interval, Δt, by
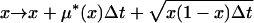 |
or
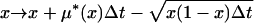 |
with equal probability, where 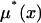 is the mean allele frequency change conditional on eventual fixation, and
is the mean allele frequency change conditional on eventual fixation, and
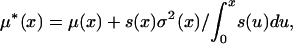 |
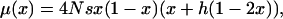 |
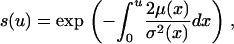 |
and
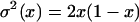 |
(see, e.g., Griffiths 2003; Ewens 2004). The integral was obtained numerically. We set Δt = 1/400N and error checked our program by comparison to results from SelSim (Spencer and Coop 2004; Przeworski et al. 2005).
To generate the ancestral recombination graph, we start at the present and proceed backward in time. Recombination occurs at a constant rate per base pair and is specified by the population recombination parameter 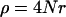 , where r is the recombination rate per site per generation. All recombination events are crossovers with no associated gene conversion. The beneficial allele fixes at time 0. While the selected site is polymorphic in the population, there are three possible events: coalescent events within either allelic class, with probability
, where r is the recombination rate per site per generation. All recombination events are crossovers with no associated gene conversion. The beneficial allele fixes at time 0. While the selected site is polymorphic in the population, there are three possible events: coalescent events within either allelic class, with probability 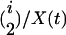 or
or 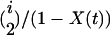 [where i and j are the numbers of ancestral lineages of the favored and unfavored alleles and X(t) is the frequency of the favored allele at time t], and recombination events within classes or between classes, which occur with probability
[where i and j are the numbers of ancestral lineages of the favored and unfavored alleles and X(t) is the frequency of the favored allele at time t], and recombination events within classes or between classes, which occur with probability  . At the time of the last event, z, the time to the next event, τ, is given by solving
. At the time of the last event, z, the time to the next event, τ, is given by solving 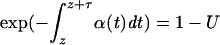 , where U is a uniform random number. The next event at time
, where U is a uniform random number. The next event at time 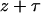 is chosen randomly with probability
is chosen randomly with probability  , where
, where  is the instantaneous rate of event k (e.g., recombination within the favored class), and
is the instantaneous rate of event k (e.g., recombination within the favored class), and  is the rate of any event, at time
is the rate of any event, at time 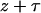 . Once the time is reached when the favored allele first arose, the process is given by the standard coalescent (Hudson 1990). After generation of the ancestral recombination graph, mutations are superimposed on the genealogy. We assume that they occur according to the infinite-site mutation model. The population mutation parameter is
. Once the time is reached when the favored allele first arose, the process is given by the standard coalescent (Hudson 1990). After generation of the ancestral recombination graph, mutations are superimposed on the genealogy. We assume that they occur according to the infinite-site mutation model. The population mutation parameter is 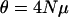 , where μ is the mutation rate per site per generation.
, where μ is the mutation rate per site per generation.
Trajectories of the beneficial allele:
Figure 1 presents the average sojourn time of the favorable allele, conditional on fixation. When selection is strong, the mean fixation time is approximately the same for h and (1 − h), a reversibility property established by van Herwaarden and van der Wal (2002). In this case, the fixation time is the shortest when h = 0.5. When instead selection is weak (i.e., when 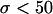 in Figure 1), the approximation becomes worse and the average fixation time increases with h. These observations can be understood by examining the trajectory of the allele conditional on fixation.
in Figure 1), the approximation becomes worse and the average fixation time increases with h. These observations can be understood by examining the trajectory of the allele conditional on fixation.
Figure 1.

The average fixation time of a favorable allele for different dominance coefficients. The scaled selection coefficient 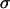 is on the x-axis and the fixation time in generations is on the y-axis. N is the diploid effective population size. The mean fixation time of a neutral allele is 4N generations. The solid line (green) is for h = 0.1, the long dashed line (red) is for h = 0.5, and the short dashed line (blue) is for h = 0.9. The inset is an enlarged view of the trajectories when selection is strong.
is on the x-axis and the fixation time in generations is on the y-axis. N is the diploid effective population size. The mean fixation time of a neutral allele is 4N generations. The solid line (green) is for h = 0.1, the long dashed line (red) is for h = 0.5, and the short dashed line (blue) is for h = 0.9. The inset is an enlarged view of the trajectories when selection is strong.
For strong selection, an example is provided in Figure 2a. When the allele is rare, it is found almost exclusively in heterozygotes. Thus, if it is recessive (e.g., h = 0.1), it will be hidden from selection in the early phases and take longer to reach appreciable frequency. Once it increases in frequency and is also found in homozygotes, the allele spreads rapidly across the population until fixation. If instead the derived allele is dominant (e.g., h = 0.9), the allele is immediately visible to selection and so initially increases in frequency more rapidly. However, once the beneficial allele is at high frequency, the unfavorable allele tends to be hidden from selection in heterozygotes and is therefore delayed in its rise from high frequency to fixation. For strong selection, the trajectories of the favored allele for h and (1 − h) therefore become symmetric and the mean fixation times become the same.
Figure 2.

Examples of the trajectory of the favored allele from introduction to fixation. (a) σ = 4000; (b) σ = 50. Time scaled by the population size is on the x-axis and the frequency of the favored allele is on the y-axis. The solid line (green) is for h = 0.1, the long dashed line (red) is for h = 0.5, and the short dashed line (blue) is for h = 0.9.
An example of a trajectory of the favorable allele under weak selection is shown in Figure 2b. Conditional on fixation, the favored allele rapidly increases in frequency in the initial stages—otherwise, it would be eliminated from the population by drift. Given the rapid ascent in frequency at this early stage, recessive alleles fix more rapidly than dominant ones, whose rise in frequency is relatively slower at high frequencies. As a result, the mean fixation time increases with h.
Effect of the fixation event on polymorphism:
How are these differences in trajectories reflected in polymorphism data? To examine this, we consider the effect of a fixation event on diversity levels at linked neutral sites, summarizing the data by 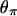 (Tajima 1983),
(Tajima 1983), 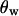 (Watterson 1975), and
(Watterson 1975), and 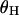 (Fay and Wu 2000). Averages of the three statistics are plotted against the distance from the selected site in Figure 3, a, b, and c, respectively. Parameters
(Fay and Wu 2000). Averages of the three statistics are plotted against the distance from the selected site in Figure 3, a, b, and c, respectively. Parameters 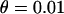 ,
, 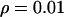 ,
, 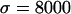 , and
, and  are chosen to be plausible for strong selection in Drosophila melanogaster (Andolfatto and Przeworski 2000). The effect of h on diversity levels is most obviously seen in
are chosen to be plausible for strong selection in Drosophila melanogaster (Andolfatto and Przeworski 2000). The effect of h on diversity levels is most obviously seen in 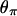 (Figure 3a), so that we focus on this case. Two observations emerge:
(Figure 3a), so that we focus on this case. Two observations emerge:
Close to the selected site, the fixation event has a stronger effect for smaller h; i.e., diversity levels decrease with h.
However, diversity levels recover to their neutral expectations faster for smaller h. For these parameters, for example, the diversity level recovers to half of its neutral expectation (i.e., 10/kb) by 8 kb for a recessive allele vs. 21 kb for a dominant allele.
Figure 3.

The effect of strong directional selection. Average (a) θπ, (b) θW, and (c) θH are shown as functions of the distance (in kilobases) from the selected site, for different dominance coefficients. (d) Mean, 25th, and 75th percentiles of Tajima's D; (e) mean, 25th, and 75th percentiles of Fu and Li's D. Parameters are 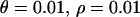 ,
, 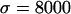 . The solid line (green) is for h = 0.1, the long dashed line (red) is for h = 0.5, and the short dashed line (blue) is for h = 0.9.
. The solid line (green) is for h = 0.1, the long dashed line (red) is for h = 0.5, and the short dashed line (blue) is for h = 0.9.
The first observation can be understood as follows: close to the selected site, there will be little or no recombination during the selective phase. Thus, most ancestral lineages will coalesce when the favored allele first reaches low frequency (going backward in time). For a given fixation time, this happens more rapidly for recessive alleles. As a result, the genealogy is shallower for smaller h. This effect on the genealogy is most notable in the value of 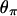 rather than
rather than 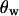 and
and 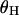 because this statistic is most sensitive to the height of the genealogy (Tajima 1989b).
because this statistic is most sensitive to the height of the genealogy (Tajima 1989b).
The second result stems from the difference in the shape of the trajectory. As shown in Figure 2a, when h is small, most of the sojourn time is when the allele is at low frequency in the population. During this phase, the allele will have the opportunity to recombine onto other backgrounds. In other words, the favored allele will tend to increase in frequency on multiple backgrounds, preserving more of the diversity that existed when it first arose. In contrast, for dominant alleles, most of the sojourn time is spent at higher frequency, when there is less opportunity for the favored allele to recombine onto other backgrounds. This results in a wider signature of a fixation event for larger h-values.
The behavior of  for different h-values (Figure 3c) can be understood in the same way. Large values of
for different h-values (Figure 3c) can be understood in the same way. Large values of 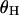 reflect a lopsided genealogy (i.e., one with a long internal branch leading to most of the gene copies in the sample) because of rare recombination events that occur while the favored allele is at intermediate frequency in the population (Barton 1998; Fay and Wu 2000; Przeworski 2002). If instead the beneficial allele recombines while it is at low frequency, the genealogy is more likely to be balanced and therefore
reflect a lopsided genealogy (i.e., one with a long internal branch leading to most of the gene copies in the sample) because of rare recombination events that occur while the favored allele is at intermediate frequency in the population (Barton 1998; Fay and Wu 2000; Przeworski 2002). If instead the beneficial allele recombines while it is at low frequency, the genealogy is more likely to be balanced and therefore  tends to be lower.
tends to be lower.
We also present the average, 25th, and 75th percentiles of Tajima's (1989a) D and Fu and Li's (1993) D, two widely used summaries of the allele frequency spectrum (Figure 3). Tajima's D is the (approximately normalized) difference between π and θW while Fu and Li's D considers the (approximately normalized) difference between θW and another unbiased estimator of θ, on the basis of the number of singletons in the sample (Fu and Li 1993). The neutral expectation of both statistics is ∼0 under the neutral equilibrium model. Figure 3, d and e, presents the two statistics as a function of distance from the selected site for different h-values. As can be seen, both reach 0 faster for smaller h. For example, for these parameters, the means of these statistics 18 kb from the selected site are ∼0 when h = 0.1, but they are still negative 40 kb away for h = 0.9. This finding suggests that, all else being equal, it will be more difficult to detect a selective sweep if the beneficial allele was recessive.
Finally, we compare the effect of a beneficial substitution for different h-values when selection is weak (e.g., 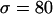 in Figure 4). For a given fixation time, the trajectories of a beneficial allele are similar to each other for different h-values (Figure 2b), so there is little difference in the effect on polymorphism data. Moreover, given that for all h-values the sojourn time of the beneficial allele is not much shorter than that of a neutral allele (Figure 1), its fixation does not distort polymorphism levels much relative to the neutral case (Figure 4).
in Figure 4). For a given fixation time, the trajectories of a beneficial allele are similar to each other for different h-values (Figure 2b), so there is little difference in the effect on polymorphism data. Moreover, given that for all h-values the sojourn time of the beneficial allele is not much shorter than that of a neutral allele (Figure 1), its fixation does not distort polymorphism levels much relative to the neutral case (Figure 4).
Figure 4.

The effect of weak directional selection. Average (a) θπ, (b) θW, and (c) θH are shown as functions of the distance (in kilobases) from the selected site, for different dominance coefficients. (d) Mean, 25th, and 75th percentiles of Tajima's D; (e) mean, 25th, and 75th percentiles of Fu and Li's D. Parameters are 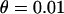 ,
, 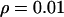 ,
, 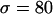 . The solid line (green) is for h = 0.1, the long dashed line (red) is for h = 0.5, and the short dashed line (blue) is for h = 0.9.
. The solid line (green) is for h = 0.1, the long dashed line (red) is for h = 0.5, and the short dashed line (blue) is for h = 0.9.
Implications:
Using an approximation to the fixation process of advantageous mutations, we find that the dominance coefficient, h, of a favored allele can have a marked influence on the signature of directional selection.
First, as selection becomes weaker, the mean sojourn times for alleles with dominance coefficient h and (1 − h) are no longer the same. This finding may have few practical implications, however, as we can only hope to detect strong selective sweeps (see Figure 4). But h also has a marked effect on the shape of the trajectory for strong selection. Even though the mean fixation time is the same for h and (1 − h), the time spent at low frequency differs substantially. This difference produces distinct genealogies and hence distinct patterns of polymorphism after the fixation of a beneficial mutation. In particular, our simulations show that the fixation of dominant alleles influences a larger genomic region, suggesting that this type of favorable substitution may be easiest to detect from polymorphism data.
The prevalence of positive selection on dominant alleles is unknown. Comparisons of X and autosomal diversity and divergence have suggested that a substantial fraction of advantageous alleles may be recessive (Begun and Whitley 2000; Schofl and Schlotterer 2004; Lu and Wu 2005). In humans, there is at least one example of a selective sweep in which the beneficial allele is thought to be recessive: the fixation of the null allele at the Duffy locus in sub-Saharan populations that experience vivax malarial pressures (Hamblin and Di Rienzo 2000). This said, there are also anecdotal examples of dominant beneficial mutations, such as those underlying lactose tolerance (Jobling et al. 2003). Moreover, Haldane's sieve—the idea that a dominant allele has a greater chance of fixation—suggests that most fixation events on autosomes may involve dominant alleles, unless mutations to recessive alleles are much more common.
It may be possible to gain some insight into heterozygote effects on the basis of the protein product of the gene. For example, mutations in enzymes are thought to be more likely to be recessive, while those in transcription factors may be more likely to be dominant (Jimenez-Sanchez et al. 2001). However, most of these observations stem from mutations to disease alleles that are deleterious and it is unclear whether the same can be expected of new mutations that confer a fitness advantage. In any case, our results suggest that, when available, information about dominance coefficients should be integrated into models of directional selection.
Acknowledgments
We thank Graham Coop for helpful discussions and comments on the manuscript. This work was supported by National Institutes of Health grant GM072861 and by an Alfred P. Sloan research fellowship to M.P. in Computational Molecular Biology.
References
- Andolfatto, P., and M. Przeworski, 2000. A genome-wide departure from the standard neutral model in natural populations in Drosophila. Genetics 156: 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton, N. H., 1998. The effect of hitch-hiking on neutral genealogies. Genet. Res. 72: 123–133. [Google Scholar]
- Begun, D. J., and P. Whitley, 2000. Reduced X-linked nucleotide polymorphism in Drosophila simulans. Proc. Natl. Acad. Sci. USA 97: 5960–5965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betancourt, A. J., Y. Kim and H. A. Orr, 2004. A pseudo-hitchhiking model of X vs. autosomal diversity. Genetics 168: 2261–2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coop, G., and R. C. Griffiths, 2004. Ancestral inference on gene trees under selection. Theor. Popul. Biol. 66: 219–232. [DOI] [PubMed] [Google Scholar]
- Ewens, W. J., 2004. Mathematical Population Genetics. Springer, New York.
- Fay, J. C., and C.-I Wu, 2000. Hitchhiking under positive Darwinian selection. Genetics 155: 1405–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y. X., and W. H. Li, 1993. Statistical test of neutrality of mutations. Genetics 133: 693–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths, R. C., 2003. The frequency spectrum of a mutation, and its age, in a general diffusion model. Theor. Popul. Biol. 64: 241–251. [DOI] [PubMed] [Google Scholar]
- Hamblin, M. T., and A. Di Rienzo, 2000. Detection of the signature of natural selection in humans: evidence from the Duffy blood group locus. Am. J. Hum. Genet. 66: 1669–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., 1990. Gene genealogy and the coalescent process, pp. 1–14 in Oxford Surveys in Evolutionary Biology, Vol. 7, edited by D. Futuyma and J. Antonovics. Oxford University Press, Oxford.
- Hudson, R. R., 2002. Generating samples under a Wright–Fisher neutral model of genetic variation. Bioinformatics 18: 337–338. [DOI] [PubMed] [Google Scholar]
- Jimenez-Sanchez, G., B. Childs and D. Valle, 2001. Human disease genes. Nature 409: 853–855. [DOI] [PubMed] [Google Scholar]
- Jobling, M. A., M. Hurles and C. Tyler-Smith, 2003. Human Evolutionary Genetics: Origins, Peoples and Disease, p. 419. Garland Science, London/New York.
- Kaplan, N. L., R. R. Hudson and C. H. Langley, 1989. The “hitchhiking effect” revisited. Genetics 123: 887–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov, F. A., and E. V. Koonin, 2004. A common framework for understanding the origin of genetic dominance and evolutionary fates of gene duplications. Trends Genet. 20: 287–291. [DOI] [PubMed] [Google Scholar]
- Lu, J., and C.-I Wu, 2005. Weak selection revealed by the whole-genome comparison of the X chromosome and autosomes of human and chimpanzee. Proc. Natl. Acad. Sci. USA 102: 4063–4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr, H. A., and A. J. Betancourt, 2001. Haldane's sieve and adaptation from the standing genetic variation. Genetics 157: 875–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przeworski, M., 2002. The signature of positive selection at randomly chosen loci. Genetics 160: 1179–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przeworski, M., G. Coop and J. D. Wall, 2005. The signature of positive selection on standing genetic variation. Evolution 59: 2312–2323. [PubMed] [Google Scholar]
- Schofl, G., and C. Schlotterer, 2004. Patterns of microsatellite variability among X chromosomes and autosomes indicate a high frequency of beneficial mutations in non-African D. simulans. Mol. Biol. Evol. 21: 1384–1390. [DOI] [PubMed] [Google Scholar]
- Spencer, C. C. A., and G. Coop, 2004. SelSim: a program to simulate population genetic data with natural selection and recombination. Bioinformatics 20: 3673–3675. [DOI] [PubMed] [Google Scholar]
- Tajima, F., 1983. Evolutionary relationship of DNA sequences in finite populations. Genetics 105: 437–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, F., 1989. a Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, F., 1989. b The effect of change in population size on DNA polymorphism. Genetics 123: 597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Herwaarden, O. A., and N. J. van der Wal, 2002. Extinction time and age of an allele in a large finite population. Theor. Popul. Biol 61: 311–318. [DOI] [PubMed] [Google Scholar]
- Watterson, G. A., 1975. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7: 256–276. [DOI] [PubMed] [Google Scholar]