

Abstract
Genetic association studies are rapidly becoming the experimental approach of choice to dissect complex traits, including tolerance to drought stress, which is the most common cause of mortality and yield losses in forest trees. Optimization of association mapping requires knowledge of the patterns of nucleotide diversity and linkage disequilibrium and the selection of suitable polymorphisms for genotyping. Moreover, standard neutrality tests applied to DNA sequence variation data can be used to select candidate genes or amino acid sites that are putatively under selection for association mapping. In this article, we study the pattern of polymorphism of 18 candidate genes for drought-stress response in Pinus taeda L., an important tree crop. Data analyses based on a set of 21 putatively neutral nuclear microsatellites did not show population genetic structure or genomewide departures from neutrality. Candidate genes had moderate average nucleotide diversity at silent sites (πsil = 0.00853), varying 100-fold among single genes. The level of within-gene LD was low, with an average pairwise r2 of 0.30, decaying rapidly from ∼0.50 to ∼0.20 at 800 bp. No apparent LD among genes was found. A selective sweep may have occurred at the early-response-to-drought-3 (erd3) gene, although population expansion can also explain our results and evidence for selection was not conclusive. One other gene, ccoaomt-1, a methylating enzyme involved in lignification, showed dimorphism (i.e., two highly divergent haplotype lineages at equal frequency), which is commonly associated with the long-term action of balancing selection. Finally, a set of haplotype-tagging SNPs (htSNPs) was selected. Using htSNPs, a reduction of genotyping effort of ∼30–40%, while sampling most common allelic variants, can be gained in our ongoing association studies for drought tolerance in pine.
THE neutral theory of molecular evolution states that nucleotide diversity is governed by the population mutation parameter 4Neμ, where μ is the per-generation, per-site mutation rate. Over the past 2 decades, identification of candidate genes under selection in natural populations has relied on the analysis of nucleotide diversity patterns within and between species and departures of allele (haplotype) distributions from neutral expectations (i.e., neutrality tests; see reviews in Kreitman 2000; Ford 2002; Rosenberg and Nordborg 2002). Two major patterns emerged from these analyses in a wide range of genes and organisms. One type of loci showed an excess of intermediate-frequency haplotypes, frequently arranged around two highly divergent lineages (e.g., Filatov and Charlesworth 1999; Tian et al. 2002), and the other was characterized by an excess of rare haplotypes (e.g., Olsen et al. 2002; see Pot et al. 2005 for pine). These departures of the site-frequency spectrum from the neutral expectation, as long as they were not due to demography or population structure, were associated with balancing selection and with purifying selection or selective sweeps caused by positive selection, respectively.
Genetic association between allelic variants and trait differences on a population scale is a powerful, and relatively recent, approach to identifying genes or alleles that contribute to variation in adaptive traits (Long and Langley 1999; see Neale and Savolainen 2004 for conifers). Population stratification is the most common source of systematic bias in association studies (Buckler and Thornsberry 2002; Hirschhorn and Daly 2005). Putatively neutral molecular markers, such as nuclear microsatellites, are generally used to detect population structure and other population and demographic processes that might produce false positives in association studies (Rosenberg et al. 2002). Optimization of association mapping requires knowledge of the patterns of nucleotide diversity and linkage disequilibrium for each particular species and candidate gene set. In addition, standard neutrality tests applied to DNA sequences of a single or a few gene(s) can be used in selecting candidate genes or amino acid sites that are putatively under selection for association mapping.
Forest trees play a crucial role in terrestrial ecosystems, offering major ecological benefits in terms of climate control, carbon fixation, and wildlife maintenance. Drought stress is the most common cause of tree mortality and is responsible for severe annual yield losses in commercial species (up to ∼65% in Pinus taeda L.; Burns and Honkala 1990). Understanding the physiological mechanisms and the genetic basis of drought-stress tolerance has been a long-standing interest for plant biologists (e.g., Ingram and Bartels 1996; Seki et al. 2003; see Newton et al. 1991 for forest trees). However, progress on identification of drought-related genes and development of expressional studies in forest trees are relatively recent (Chang et al. 1996; Dubos and Plomion 2003; Watkinson et al. 2003). The molecular basis of dehydration tolerance in trees is extremely complex and a wide variety of expressional candidate genes has been suggested. Increased expression of dehydrins has been found in different conifer trees during both seed development (Jarvis et al. 1996) and drought stress (Richard et al. 2000; Watkinson et al. 2003). Chang et al. (1996), using a subtractive hybridization approach, identified four cDNA clones with drought-induced expression in P. taeda: lp2, with a high homology to S-adenosylmethionine synthetase (sams), an intermediate in the synthesis of ethylene; lp3, expressed predominantly in roots and later found to belong to a small family of ABA-inducible genes (Padmanabhan et al. 1997); lp4, similar to a type I copper-containing glycoprotein; and lp5, expressed almost exclusively in roots and coding for a glycine-rich protein similar to cell wall proteins. Other major expressional candidate genes for drought-stress response identified in trees encode protein kinases (Dubos and Plomion 2003; Dubos et al. 2003), cysteine proteases (Tranbarger and Misra 1996), iron storage proteins (Li et al. 1998), antioxidants (Li et al. 1998; Karpinska et al. 2001), and pathogenesis-related proteins (Dubos and Plomion 2001; Dubos et al. 2003).
Conifers are long-lived, widely distributed organisms that, in general, exhibit high levels of heterozygosity and large effective population sizes. Therefore, it has been suggested that conifers may show high levels of nucleotide variation (Dvornyk et al. 2002). However, the first results on DNA sequence variation for conifers showed, at best, moderate estimates of nucleotide diversity (e.g., Kado et al. 2003; Brown et al. 2004; Pot et al. 2005). Average population differentiation was also moderate in conifers (Kado et al. 2003; but see Pot et al. 2005 for korrigan and pp1 genes), even when extreme phenotypes were sampled (García-Gil et al. 2003). For example, García-Gil et al. (2003) did not find any functional differentiation at the photosensory domains of two phytochrome loci among populations sampled along a latitudinal cline that was associated with marked differences in growth phenology (as shown by common garden experiments). Patterns of nucleotide diversity and/or population differentiation that deviate from the neutral expectation, potentially indicating the action of natural selection, have been described only for a few genes and tree species [acl5 in Cryptomeria japonica (L. f.) D. Don (Kado et al. 2003); f3h1, 4cl1, and mt-like in Pseudotsuga menziesii (Mirb.) Franco (Krutovsky and Neale 2005); and pp1, korrigan, and CesA3 in pines (Pot et al. 2005)]. Large effective population sizes in conifers would result in low linkage disequilibrium (LD) due to high recombination rates at the population level. This prediction agrees with empirical data in conifers, where lack of LD among genes and relatively rapid decay of LD within genes (200–1500 bp) have been observed (Brown et al. 2004; Rafalski and Morgante 2004). However, it is possible but currently unknown if more extensive LD exists in particular tree species or populations that experienced historical bottlenecks in Pleistocene glacial refugia, both in Europe and in America.
The standing variation in natural populations is patterned as a consequence of the interplay among genetic drift, demography, population structure, and natural selection. In this article, we used a data set of 21 nuclear microsatellites for detecting population structure and demographic processes that might cause spurious associations in association studies and bias neutrality tests, and sequenced all or portions of 18 candidate genes for drought-stress response in P. taeda, an important tree crop. Our sample covered the southeastern native range of P. taeda, including Florida, a putative Pleistocene glacial refugium of this species (Schmidtling et al. 1999; Al-Rabab'ah and Williams 2002), which was not extensively sampled in our previous studies (see Brown et al. 2004). We have used DNA sequences to estimate levels of nucleotide diversity and linkage disequilibrium, to identify candidate genes under selection (by means of neutrality tests), and to select haplotype-tagging single-nucleotide polymorphisms (htSNPs) for our current genetic association studies.
MATERIALS AND METHODS
Plant material:
A sample of 32 seed megagametophytes (the haploid, maternally derived nutritive tissue of conifer seeds) of P. taeda (1 from each of 30 trees and 2 from 1 tree) was used for SNP discovery. Seed donors included 22 unrelated, first-generation selections (plus trees) from undisturbed natural stands covering the southeastern range of P. taeda [Atlantic Coastal Plain (ACP), central Florida, northern Florida, Marion County, and Gulf Coast provenances; see supplemental Table S1 at http://www.genetics.org/supplemental/] and nine second-generation selections produced by controlled crosses among first-generation selections within the Atlantic Coastal Plain provenance. These trees are currently part of the Forest Biology Research Cooperative (FBRC) Tree Improvement Program(University of Florida, Gainesville, FL). The second-generation trees may introduce a slight bias, due to the inclusion of four pairs of half-sibs and three trees that have first-generation selections as parents (see supplemental Table S1). However, because of the high levels of heterozygosity in this species and meiotic segregation, the bias is considered negligible.
Candidate gene selection:
Candidate genes for drought-stress response were selected on the basis of (1) homology of contig assemblies of P. taeda expressed sequence tags (ESTs) in public databases (DDBJ/EMBL/GenBank) with drought-stress response genes in model species; (2) homology of sequences from the unigene set (∼20,500 nonredundant genes) assembled at North Carolina State University on the basis of six xylem EST libraries (accessed through http://pinetree.ccgb.umn.edu/) with drought-stress response genes in model species; and (3) the overabundance of ESTs in root libraries from P. taeda trees under drought stress compared to control trees as indicated by “electronic” Northerns using the MAGIC Gene Discovery tool (University of Georgia, http://fungen.org/Projects/Pine/Pine.htm). Two other genes, ppap12 and lp3-3, were also selected because they showed differential expression under drought treatments as shown by reverse Northerns in P. pinaster (Dubos et al. 2003) and P. taeda (Padmanabhan et al. 1997), respectively.
DNA isolation, amplification, and sequencing:
Haploid genomic DNA was extracted from megagametophytes, using the Plant DNeasy kit (QIAGEN, Valencia, CA) after seed germination. PCR primers were designed to amplify a 400- to 1000-bp fragment in nine nuclear loci and previously published primers were used for an additional nine genes (see supplemental Table S2 at http://www.genetics.org/supplemental/). Primers were designed to amplify full-length genes for lp3-3, dhn-1, and dhn-2. Sequence data were obtained directly from PCR products on an ABI 377 automated sequencer, using the BigDye Terminator v. 3.1 cycle sequencing kit (Applied Biosystems, Foster City, CA). All samples were sequenced from both ends. Base calling and assembly of forward and reverse reads were done using phred and phrap programs (Ewing et al. 1998; Gordon et al. 1998; http://bozeman.mbt.washington.edu/phredphrapconsed.html) under a Unix environment. Multiple alleles from a locus were aligned in the multiple-alignment consed extensions (MACE) program (B. Gilliland and C. Langley, University of California, Davis, CA). All chromatograms were checked visually and a putative sequence variant was accepted only when the phred scores for all sequences exceeded 25 at that site. Resequencing was performed as needed to maintain this quality criterion. Since the DNA samples were haploid, the identification of haplotypes (i.e., alleles) was unambiguous.
Mapping of candidate genes:
Six of the 18 candidate genes were mapped previously (Brown et al. 2003). Mapping of the remaining 12 loci was attempted using two reference mapping populations of P. taeda, the qtl and base pedigrees (details in Brown et al. 2001). Five candidate genes (lp3-1, dhn-1, rd21A-like, cpk3, and ppap12) were mapped using denaturing gradient gels (DGGE) according to Temesgen et al. (2001) and 1 (lp3-3) was mapped using a template-directed dye-termination incorporation assay (TDI) with fluorescence polarization (FP) detection (TDI 5′–3′ primer: TTGCCAGTAGCATACACATCTG). FP–TDI was done using the AcycloPrime-FP SNP detection kit and a Wallac VICTOR2 fluorescence plate reader (Perkin-Elmer Life and Analytical Sciences, Torrance, CA). The other 6 candidate genes either were unlinked (sod-chl) or lacked suitable polymorphisms (i.e., parents of the pedigrees did not segregate for any SNP or primers for FP–TDI could not be designed due to the existence of repetitive regions near SNPs: ferritin, erd-3, dhn-2, lp5-like, and ug-2_498). A consensus map was obtained together with other markers following Brown et al. (2001).
Population structure and demographic processes:
Population stratification is the most common systematic bias producing false-positive associations in association studies (Marchini et al. 2004; Hirschhorn and Daly 2005). Moreover, the existence of population genetic structure or demographic processes, such as range expansions or retreats, might produce signatures on the allele frequency spectrum similar to those produced by the action of natural selection and mislead the interpretation of standard neutrality tests, such as Tajima's D. We used 21 highly polymorphic (average of 15 alleles per locus) nuclear microsatellites (nuSSRs), covering most P. taeda linkage groups, to test for population structure or demographic processes. The nuSSR data were kindly provided by C. Dana Nelson (Southern Institute of Forest Genetics, U.S. Department of Agriculture) and included 94 trees sampled from roughly the same range as the sequence data presented here (see supplemental Table S3 at http://www.genetics.org/supplemental/).
To test for population structure, we first used a model-based clustering algorithm (Structure software; Pritchard et al. 2000; Rosenberg et al. 2002), which constructs groups of populations without any prior geographical information. Models with a putative number of clusters (K parameter) from one to four, noncorrelated allele frequencies, and both burn-in, to minimize the effect of the starting configuration, and run-length periods of 106 were run. Second, we computed genetic differentiation estimates (F-statistics, based on a nested ANOVA following Weir and Cockerham 1984) among the three geographical regions included in the sample (Gulf Coast, Northeast, and Southeast). Both a permutation test (10,000 permutations) and a jackknifed estimator over loci were used to test for significance of population genetic structure among regions.
To test for genomewide departures from neutrality, such as those produced by demographic processes, the Ewens–Watterson test of neutrality (Watterson 1978, 1986), with probabilities calculated on the basis of both homozygosity and Fisher's exact tests (Ewens–Watterson–Slatkin's exact test; Slatkin 1994, 1996), was performed using the program Arlequin v. 2000 (Schneider et al. 2000). The Ewens–Watterson test enables the detection of deviations from the neutral model as either a deficit or an excess of homozygosity relative to the neutral equilibrium expectation, given the number of alleles found at a locus. It should be noted that homozygosity excess is a typical genomewide signature of population expansion (Payseur et al. 2002; Luikart et al. 2003). Once the test was computed for each of the 21 nuSSR loci, a Mann–Whitney U-test was used to detect whether expected and observed homozygosity values were drawn from the same distribution. The Bonferroni correction for multiple testing was applied when necessary.
Nucleotide variation and neutrality tests:
Analyses of sequence data were performed using DnaSP v. 4.0 (Rozas et al. 2003). Nucleotide diversity was estimated by Watterson's θw (Watterson 1975) and π, the average number of pairwise nucleotide differences among sequences in a sample (Nei and Li 1979). Heterogeneity of sequence variation across loci was assessed using coalescence simulations without recombination. A number of statistical analyses were conducted to identify genes or amino acid sites departing from the standard neutral model of evolution. Tajima's (1989) D-statistic was computed for each locus for both the full sequence and a sliding window (window length and step size of 100 and 25 sites, respectively). Tajima's D-statistic reflects the difference between π and θw. At mutation–drift equilibrium, the expected value of D is close to zero. The Fs-test statistic for neutrality (Fu 1997), based on the haplotype (gene) frequency distribution conditional on the value of θ (estimated by π), was also calculated. Both Tajima's D- and Fu's Fs-test statistics can also reflect demographic changes (Fu 1997; Sano and Tachida 2005). To compute tests that required data from an outgroup, putative orthologs of 14 genes were obtained from P. pinaster, a European species that might have diverged from P. taeda ∼120 million years ago (Krupkin et al. 1996). For 8 genes, we used sequences from GenBank (accession nos.: AL751338, lp3-1; BX255067, dhn-1; BX677401, lp5-like; BX252032, sod-chl; BX681838, sams-2; AY641535, pal-1; CR393126, ccoaomt-1; and AJ309112, ppap12) and, for the other six, we used sequences obtained directly from P. pinaster megagametophyte DNA using the same primer pairs for sequencing as in P. taeda (genes dhn-2, rd21A-like, pp2c, Aqua-MIP, erd-3, and ug-2_498; A. Soto and M. T. Cervera, unpublished data). Then, we computed: (1) Fay and Wu's H-test (Fay and Wu 2000), on the basis of the relative excess of high-frequency-derived alleles expected immediately after a selective sweep; (2) the Hudson–Kreitman–Aguadé (HKA) test (Hudson et al. 1987), which tests for decoupling between polymorphism and divergence in a particular region; and (3) the McDonald–Kreitman (MK) test (McDonald and Kreitman 1991), on the basis of the comparison of synonymous and nonsynonymous substitutions within and between species. HKA tests were done comparing each gene against every other one. Finally, to detect positive selection at single amino acid sites, we estimated the rates of nonsynonymous and synonymous changes at each site in a sequence alignment using likelihood-based methods as implemented in the on-line DataMonkey package (Kosakovsky-Pond and Frost 2005a,b). For these analyses, we used both a conservative single-likelihood ancestor-counting (SLAC) method, related to that of Suzuki–Gojobori (Suzuki and Gojobori 1999), and a fixed-effects likelihood (FEL) method, which directly estimates nonsynonymous and synonymous substitution rates at each site and is more adequate for data sets with a moderate number of sequences (n = 20–40; Kosakovsky-Pond and Frost 2005a).
LD, haplotype diversity, and selection of htSNPs for association mapping:
The LD descriptive statistic r2 (Hill and Robertson 1968) was calculated, only on the basis of informative sites (frequency of 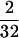 = 0.063), using Tassel software (http://www.maizegenetics.net/index.php?page=bioinformatics/tassel/index.html). The r2 statistic summarizes both recombination and mutation history and it is less sensitive to sample size than other common LD statistics such as D′ (Flint-García et al. 2003). Statistical significance of r2 was computed with a one-tailed Fisher's exact test and applying Bonferroni corrections for multiple testing. The decay of linkage disequilibrium with physical distance was estimated using nonlinear regression of LD between polymorphic sites, as estimated by r2, and the distance, in base pairs, between sites (Remington et al. 2001; Ingvarsson 2005). To adjust the nonlinear function, we used the r2 expectation provided by Hill and Weir (1988) for drift–recombination equilibrium with a low level of mutation and an adjustment for sample size n,
= 0.063), using Tassel software (http://www.maizegenetics.net/index.php?page=bioinformatics/tassel/index.html). The r2 statistic summarizes both recombination and mutation history and it is less sensitive to sample size than other common LD statistics such as D′ (Flint-García et al. 2003). Statistical significance of r2 was computed with a one-tailed Fisher's exact test and applying Bonferroni corrections for multiple testing. The decay of linkage disequilibrium with physical distance was estimated using nonlinear regression of LD between polymorphic sites, as estimated by r2, and the distance, in base pairs, between sites (Remington et al. 2001; Ingvarsson 2005). To adjust the nonlinear function, we used the r2 expectation provided by Hill and Weir (1988) for drift–recombination equilibrium with a low level of mutation and an adjustment for sample size n,
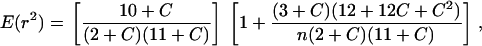 |
(1) |
where C is the population recombination parameter. Equation 1 was fitted using the Gauss–Newton algorithm implemented in the proc nlin of SAS v. 8.0 statistical package (SAS Institute, Cary, NC). Haplotypic diversity (He) was computed following Nei (1987). We identified htSNPs, i.e., those representing common allelic variants, on line using HaploblockFinder software (Zhang and Jin 2003; http://cgi.uc.edu/cgi-bin/kzhang/haploBlockFinder.cgi/) and a threshold of r2 = 0.2 to define LD blocks. Power in association studies (for a fixed sample size) is significantly reduced with low frequency of alleles (Wang et al. 2005). Then, htSNPs were selected considering minor allele frequencies (MAFs) corresponding only to common (MAF > 5%) and frequent (MAF > 15%) SNPs. Given the low level of LD found in pine, which resulted in short LD blocks, other approaches to identify htSNPs, such as the identification of LD subgroups within LD blocks (see Takeuchi et al. 2005 for details), did not perform well and are not shown.
RESULTS
Thirty-two gametes were sequenced for each of 18 candidate gene loci, resulting in ∼324 kb (32 × 10,116 bp) of DNA sequence data (Table 1). Approximately 60% of the sequence data were obtained from coding regions. We found insertion/deletions (indels) in 13 genes, ranging from 1 to 67 bp (average of ∼8 bp). Five genes (dhn-1, dhn-2, lp5-like, rd21A-like, and pp2c) had indels within the coding region, including a 30-bp indel in dhn-1. The lengths of indels within coding regions were multiples of 3 bp, so they did not result in a shift of reading frame. Finally, highly variable TA microsatellite regions were observed in lp3-1 and ug-2_498 DNA sequences. Indels and microsatellite regions were excluded in further analyses.
TABLE 1.
Candidate genes for drought tolerance in P. taeda
| Candidate gene
|
Putative gene function
|
Linkage group
|
bp screened
|
Indelsb (bp)
|
|||||
|---|---|---|---|---|---|---|---|---|---|
| Sourcea | Total | 5′ UTR | Exon | Intron | 3′ UTR | ||||
| lp3-1 | Water-stress-inducible protein | 3 | 2 | 365 | 136 | 229 | 2 (13) | ||
| lp3-3 | Water-stress-inducible protein | 4 | 2 | 468 | 305 | 163 | 0 | ||
| dhn-1 | Dehydrin | 1 | 8 | 673 | 560 | 113 | 3 (38) | ||
| dhn-2 | Dehydrin | 3 | NS | 531 | 439 | 92 | 1 (3) | ||
| lp5-like | Putative cell wall protein, similar to lp5 in Pinus taeda | 3 | NS | 496 | 62 | 434 | 3 (24) | ||
| mt-like | Similar to metallothionein | 1 | 6 | 403 | 90 | 79 | 234 | 0 | |
| sod-chl | Cu/Zn superoxide dismutase, nuclear gene for chloroplast product | 3 | Unlinked | 692 | 168 | 524 | 5 (6) | ||
| ferritin | Ferritin | 3 | NS | 605 | 157 | 263 | 185 | 2 (9) | |
| rd21A-like | Cysteine protease (Pseudotzain), similar to rd21A in Arabidopsis | 2 | 7 | 1,000 | 159 | 579 | 262 | 5 (73) | |
| sams-2 | S-adenosylmethionine synthetase 2 | 3 | 8 | 541 | 347 | 194 | 0 | ||
| pal-1 | Phenylalanine ammonia-lyase 1 | 3 | 6 | 394 | 246 | 148 | 0 | ||
| ccoaomt-1 | Caffeoyl-CoA-O-methyltransferase 1 | 3 | 6 | 499 | 259 | 240 | 1 (16) | ||
| cpk3 | Calcium-dependent protein kinase | 1 | 1 | 630 | 377 | 187 | 66 | 1 (1) | |
| ppap12 | Uncertain, possible wall-associated protein kinase | 4 | 8 | 378 | 378 | 0 | |||
| pp2c | Protein phosphatase 2C, similar to ABI1 in Arabidopsis | 1 | 10 | 638 | 461 | 177 | 1 (3) | ||
| Aqua-MIP | Aquaporin, membrane intrinsic protein | 1 | 2 | 611 | 264 | 347 | 1 (8) | ||
| erd3 | Early response to drought 3 | 2 | NS | 882 | 622 | 204 | 56 | 1 (4) | |
| ug-2_498 | Unknown | 3 | NS | 310 | — | — | — | — | 1 (23) |
| Total | 10,116 | 221 | 5,822 | 2,651 | 1,112 | 27 (221) | |||
Notation of linkage groups follows the reference genetic map of Brown et al. (2001); NS, locus not segregating in the reference mapping populations; UTR, untranslated region.
Candidate gene source: 1, public databases (DDBJ/EMBL/GenBank); 2, North Carolina State University unigene set; 3, “electronic” Northerns using root EST libraries with different drought-stress treatments; 4, differential expression under drought as shown by conifer literature. See further details in the text.
Number of indels (total indel length).
Population structure and demographic processes:
No population structure or apparent demographic processes, such as range expansion, were found using 21 nuclear microsatellites. The model-based clustering analyses showed a typical pattern of unstructured populations (Pritchard and Wen 2004): plateaus in the estimate of the log-likelihood of the data were not reached, the proportion of the sample assigned to each population was roughly symmetric (for K = 3, for example, 30.3, 41.7, and 28.0% of samples were assigned to each group), and most individuals were given as admixed (see supplemental Figure S1 at http://www.genetics.org/supplemental/). Additional evidence of lack of population structure within the sampling range was provided by genetic differentiation estimates among the three geographical regions sampled in this study (Gulf Coast, Northeast, and Southeast). Indeed, genetic differentiation was extremely low (Fst = 0.0019) and nonsignificant as shown by both a jackknifed estimator over loci and a permutation test. The Ewens–Watterson test, after correcting for multiple testing using Bonferroni, was unable to detect any departure from neutrality, estimates of observed minus expected homozygosity being distributed around zero (i.e., about equal numbers of loci showing excess or deficit of homozygosity). The Mann–Whitney U-test could not reject the hypothesis of expected and observed homozygosity sets of values being samples drawn from the same distribution (P = 0.4311), also supporting the lack of genomewide departures from neutrality.
Nucleotide variation and neutrality tests:
In total, we found 196 segregating sites, corresponding to 1 SNP per 50 bp (Table 2 and supplemental Table S4 at http://www.genetics.org/supplemental/). Two genes (rd21A-like and ccoaomt-1) had triallelic variants and the least frequent allele was recoded as missing data for further analyses. Of the 196 segregating sites, 37 (∼20%) were nonsynonymous substitutions. Average nucleotide diversity at silent sites, πsil, was 0.00853, fivefold the diversity found at nonsynonymous sites (πa = 0.00166). Nucleotide variation was slightly higher at synonymous sites than in noncoding regions (πsyn = 0.00909 and πnoncoding = 0.00631; see supplemental Table S4), but these differences were not statistically significant. Average frequency of the less common nucleotide variant was similar at silent and nonsynonymous sites (17.16 and 13.58%, respectively) and frequency distributions for silent and nonsynonymous sites were not significantly different (P = 0.7145, Kolmogorov–Smirnov test). Coalescence simulations (implemented in DnaSP v. 4.0) showed lower values of πtot than the average for lp3-3, ferritin, pp2c, and erd3 (Table 2). Nucleotide variation, all sites considered, was higher than the average for only one gene, ccoaomt-1 (0.01179).
TABLE 2.
Nucleotide variation and haplotypic diversity in 18 candidate gene loci for drought tolerance
| Nucleotide diversity
|
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total
|
Nonsynonymous sites
|
Silent sites
|
Haplotype diversity
|
|||||||||||
| Candidate gene | L | S | θw | πa | L | S | θw | πa | L | S | θw | πa | Nh (singl.) | He (SD) |
| lp3-1 | 351 | 18 | 12.70 | 8.77 | 108 | 1 | 2.29 | 0.58 | 243 | 17 | 17.40 | 12.47 | 14 (7) | 0.91 (0.03) |
| lp3-3 | 466 | 3 | 1.59 | 0.97* | 230 | 2 | 2.10 | 1.42 | 236 | 1 | 1.08 | 0.53** | 4 (1) | 0.42 (0.10) |
| dhn-1 | 627 | 13 | 5.08 | 4.15 | 407 | 3 | 1.83 | 1.72 | 220 | 10 | 11.30 | 8.81 | 10 (6) | 0.77 (0.06) |
| dhn-2 | 521 | 14 | 6.58 | 7.61 | 328 | 4 | 3.03 | 2.82 | 193 | 10 | 12.88 | 16.04 | 9 (2) | 0.88 (0.04) |
| lp5-like | 472 | 22 | 11.58 | 10.60 | 293 | 6 | 5.09 | 4.51* | 179 | 16 | 22.15 | 20.52* | 11 (4) | 0.90 (0.03) |
| mt-like | 403 | 9 | 5.55 | 5.10 | 73 | 2 | 6.77 | 2.50 | 330 | 7 | 5.27 | 5.68 | 7 (1) | 0.80 (0.04) |
| sod-chl | 686 | 19 | 6.88 | 7.80 | 129 | 2 | 3.86 | 4.11 | 557 | 17 | 7.57 | 8.65 | 9 (4) | 0.77 (0.06) |
| ferritin | 595 | 7 | 2.92 | 1.28* | 120 | 1 | 2.07 | 0.52 | 475 | 6 | 3.14 | 1.48** | 6 (3) | 0.52 (0.10) |
| rd21A-like | 924 | 26 | 6.96 | 7.69 | 441 | 5 | 2.82 | 3.63 | 483 | 21 | 10.80 | 11.45 | 12 (5) | 0.88 (0.04) |
| sams-2 | 539 | 6 | 2.75 | 3.60 | 263 | 0 | 0.00 | 0.00* | 276 | 6 | 5.40 | 7.06 | 6 (2) | 0.74 (0.05) |
| pal-1 | 394 | 6 | 3.78 | 2.74 | 185 | 1 | 1.34 | 0.34 | 209 | 5 | 5.95 | 4.88 | 8 (4) | 0.70 (0.07) |
| ccoaomt-1 | 480 | 13 | 6.68 | 11.79* | 182 | 0 | 0.00 | 0.00* | 298 | 13 | 10.83 | 19.11* | 4 (1) | 0.68 (0.04) |
| cpk3 | 627 | 8 | 3.16 | 3.55 | 297 | 1 | 0.84 | 0.59 | 330 | 7 | 5.26 | 6.22 | 7 (2) | 0.78 (0.05) |
| ppap12 | 375 | 10 | 6.57 | 8.08 | 292 | 7 | 5.94 | 5.15* | 83 | 3 | 9.02 | 18.72* | 7 (4) | 0.70 (0.06) |
| pp2c | 635 | 1 | 0.39 | 0.10*** | 343 | 0 | 0.00 | 0.00* | 292 | 1 | 0.86 | 0.22*** | 2 (1) | 0.06 (0.06) |
| aqua-MIP | 600 | 5 | 2.06 | 1.74 | 190 | 0 | 0.00 | 0.00* | 410 | 5 | 3.03 | 2.55* | 7 (3) | 0.74 (0.05) |
| erd3 | 877 | 6 | 1.70 | 0.43*** | 477 | 2 | 1.04 | 0.26 | 400 | 4 | 2.48 | 0.62*** | 5 (4) | 0.24 (0.10) |
| ug-2_498 | 287 | 10 | 8.65 | 5.26 | — | — | — | — | — | — | — | — | 7 (3) | 0.75 (0.05) |
| Average (SD) | 548 | 11 (7) | 5.31 (3.40) | 5.07 (3.59) | 256 | 2 (2) | 2.30 (2.10) | 1.66 (1.78) | 307 | 9 (6) | 7.91 (5.83) | 8.53 (6.85) | 7.50 (3.00) | 0.68 (0.23) |
L, length in base pairs; S, number of segregating sites; Nh (singl.), number of haplotypes (number of singletons); He (SD), Nei's haplotypic diversity (standard deviation). Indels are excluded from the estimates. Nucleotide diversity estimates (θw and π) are ×103.
Values that are significantly smaller or larger than the average are indicated: *P < 0.05; **P < 0.01; ***P < 0.001.
A number of neutrality tests were applied to find evidence of positive selection in our candidate gene set (Table 3) but only a few genes gave any significant result and no positive selection acting at particular amino acid sites was found (as shown by rates of nonsynonymous and synonymous changes at each site from sequence alignments). Both Tajima's D- and Fu's Fs-test statistics were negative and significantly different from zero for erd3, revealing an excess of rare variants and a greater number of haplotypes than expected, respectively. This pattern of polymorphism is commonly associated with genetic hitchhiking or a recent increase in population size. The HKA test rejected neutrality only in two pairwise comparisons (with lp3-1, P < 0.010; and sod-chl, P < 0.098) involving this gene, and MK and Fay and Wu's H-tests were not significant. The latter results are relevant because tests based on comparison between nucleotide classes (synonymous vs. nonsynonymous), such as the MK test, or the excess of derived variants at high frequency (Fay and Wu's H-test) are robust to deviations from the standard neutral model due to demographic processes. Tajima's D- and Fu's Fs-test statistics at ccoaomt-1 indicated an excess of variants at intermediate frequencies and fewer haplotypes than expected, respectively. Indeed, all haplotypes at ccoaomt-1 belong to two clearly differentiated lineages separated by 11 mutational steps with the majority of the variation existing between, not within, lineages (Figure 1a). This skew in the site frequency spectra is consistent with the maintenance of a balanced polymorphism. However, none of the neutrality tests conducted using outgroup sequences was significant, and evidence of natural selection acting on this gene was unclear.
TABLE 3.
Neutrality tests and detection of positive (+) or negative (−) selection at single amino acid sites
| Selection at single amino acid sites
|
|||||||
|---|---|---|---|---|---|---|---|
| Neutrality testsa
|
SLAC
|
FEL
|
|||||
| Candidate gene | Tajima's D | Fu's Fs | Fay and Wu's H | (+) | (−) | (+) | (−) |
| lp3-1 | −1.051 | −4.873 | −2.625 | 0 | 0 | 0 | 1 |
| lp3-3 | −0.897 | −1.371 | — | 0 | 0 | 0 | 0 |
| dhn-1 | −0.599 | −1.801 | 0.440 | 0 | 0 | 0 | 1 |
| dhn-2 | 0.513 | 0.506 | 0.746 | 0 | 0 | 0 | 2 |
| lp5-like | −0.292 | −0.066 | −2.891 | 0 | 0 | 0 | 4 |
| mt-like | −0.247 | −0.257 | — | 0 | 0 | 0 | 0 |
| sod-chl | 0.458 | 1.629 | 0.552 | 0 | 0 | 0 | 1 |
| ferritin | −1.631 | −2.398 | — | 0 | 0 | 0 | 0 |
| rd21A-like | 0.369 | 0.662 | 0.931 | 0 | 0 | 0 | 2 |
| sams-2 | 0.863 | 0.413 | 0.798 | 0 | 0 | 0 | 1 |
| pal-1 | −0.772 | −3.495 | 0.226 | 0 | 0 | 0 | 1 |
| ccoaomt-1 | 2.489* | 8.553** | −1.210 | 0 | 0 | 0 | 3 |
| cpk3 | 0.370 | 0.007 | — | 0 | 0 | 0 | 2 |
| ppap12 | 0.716 | 1.057 | −0.137 | 0 | 0 | 0 | 3 |
| pp2c | −1.142 | −1.265 | 0.060 | 0 | 0 | 0 | 1 |
| aqua-MIP | −0.422 | −2.471 | 0.480 | 0 | 0 | 0 | 1 |
| erd3 | −2.102* | −3.272* | 0.363 | 0 | 0 | 0 | 0 |
| ug-2_498 | −1.224 | −1.235 | −1.044 | — | — | — | — |
Fay and Wu's H-test was computed using as an outgroup putative ortholog sequences from maritime pine (Pinus pinaster), a European pine species. SLAC, single-likelihood ancestor-counting method; FEL, fixed-effects-likelihood method.
Significance levels for neutrality tests are also given: *P < 0.05; **P < 0.01; ***P < 0.001.
Figure 1.

Single-nucleotide polymorphisms (SNPs) and haplotype structure for ccoaomt-1. (a) Polymorphic sites and haplotype network. The size of the circle is proportional to the frequency of the haplotype in the sample. (b) Geographical distribution of haplotype lineages, A and B, including nine additional sequences from the northern and western Mississippi Valley range of P. taeda. Only first-generation selections from undisturbed natural forest stands are shown. Numbers next to symbols indicate sample size.
Finally, the sliding-window analyses revealed statistically significant values of Tajima's D in a few regions within the ug-2_498 (Tajima's D = −2.0084 at 126–248 bp) and ppap12 (Tajima's D = 2.159–2.712 at 226–378 bp) genes.
LD, haplotype diversity, and selection of htSNPs for association mapping:
Linkage disequilibrium within the sequenced gene regions varied, depending on the candidate gene locus, from very low (e.g., lp3-3, aqua-MIP, ferritin) to high (e.g., ppap-12, ccoaomt-1). We did not find any evidence of tight LD among sites from different genes, not even for those that putatively reside on the same chromosome (see, for instance, Figure 2, linkage group 8; similar results in other linkage groups are not shown). Decay of LD within genes was rapid (Figure 3). A nonlinear fitting of the squared correlation of allele frequencies (r2) as a function of distance between sites showed expected values of ∼0.20 at 800 bp. In a sample of 32 sequences, we found from 2 (pp2c; He = 0.06) to 14 (lp3-1; He = 0.91) haplotypes per candidate gene locus, with an average of 7.5 (He = 0.68). Selection of htSNPs based on construction of LD blocks was relatively successful, considering the low level of LD found within most genes (10 of 16 genes had average pairwise r2 ≤ 0.20; Table 4). We found from 1 (ccoaomt-1) to 14 (rd21A-like) and 0 (lp3-3, aqua-MIP, ferritin, and pal-1) to 8 (rd21A-like) LD blocks for MAFs of 0.05 and 0.15, respectively. For common SNPs (MAF > 5%), we identified 94 htSNPs (of 139 available), resulting in a reduction in genotyping effort of 32.27%. The reduction of genotyping effort was increased to 39.74% (47 htSNPs of 78 available) when only frequent (MAF > 15%) SNPs were considered.
Figure 2.

Linkage disequilibrium (as estimated by r2) plots for five drought-response candidate genes, including three (dhn-1, sams-2, and ppap-12) that map in the same linkage group (LG 8; see Brown et al. 2001). The significance of linkage disequilibrium was estimated using Fisher's exact test (P) and applying Bonferroni corrections. Only sites with minor allele frequency >0.15 are shown.
Figure 3.

Scatter plot of the squared correlation of allele frequencies (r2) as a function of distance between sites for 18 candidate genes in P. taeda. A nonlinear fitting was done following Remington et al. (2001) (see details in the text). Lower and upper 95% confidence intervals are represented with thin lines. For comparison, the LD-decay curve from Brown et al. (2004) is also shown (dashed line).
TABLE 4.
Average pairwise LD (estimated by r2) and selection of haplotype-tagging SNPs (htSNPs) for common and frequent SNPs found in 18 candidate gene loci for drought tolerance
| Candidate gene
|
Sites (bp)
|
Average pairwise r2
|
Common SNPs (MAF > 5%)
|
Frequent SNPs (MAF > 15%)
|
||||
|---|---|---|---|---|---|---|---|---|
| SNPs | LD blocks | htSNPs | SNPs | LD blocks | htSNPs | |||
| lp3-1 | 365 | 0.19 | 13 | 8 | 8 | 3 | 3 | 3 |
| lp3-3 | 468 | 0.01 | 2 | 2 | 2 | 1 | 0 | 1 |
| dhn-1 | 673 | 0.39 | 9 | 5 | 5 | 4 | 2 | 3 |
| dhn-2 | 531 | 0.20 | 13 | 10 | 11 | 7 | 5 | 6 |
| lp5-like | 496 | 0.44 | 18 | 7 | 9 | 5 | 5 | 4 |
| mt-like | 403 | 0.16 | 6 | 5 | 6 | 4 | 3 | 4 |
| sod-chl | 692 | 0.17 | 15 | 8 | 11 | 12 | 3 | 5 |
| ferritin | 605 | 0.02 | 2 | 2 | 2 | 0 | 0 | 0 |
| rd21A-like | 1,000 | 0.24 | 20 | 14 | 16 | 14 | 8 | 10 |
| sams-2 | 541 | 0.28 | 5 | 3 | 3 | 3 | 1 | 1 |
| pal-1 | 394 | 0.13 | 4 | 3 | 3 | 1 | 0 | 1 |
| ccoaomt-1 | 499 | 0.90 | 12 | 1 | 2 | 12 | 1 | 2 |
| cpk3 | 630 | 0.18 | 7 | 6 | 7 | 4 | 2 | 3 |
| ppap12 | 378 | 0.66 | 6 | 2 | 2 | 5 | 1 | 1 |
| pp2c | 638 | — | — | — | — | — | — | — |
| Aqua-MIP | 611 | 0.04 | 4 | 4 | 4 | 1 | 0 | 1 |
| erd3 | 882 | — | — | — | — | — | — | — |
| ug-2_498 | 310 | 0.18 | 3 | 2 | 3 | 2 | 1 | 2 |
| Total | 10,116 | 0.30 | 139 | 82 | 94 | 78 | 35 | 47 |
MAF, minor allele frequency.
DISCUSSION
This study reports nucleotide diversity and LD estimates for 18 drought-tolerance candidate genes in P. taeda. Several neutrality tests, using or not using outgroup sequences, were performed to identify candidate genes that might be under natural selection. Our study provides insights on optimal SNP genotyping strategies for our ongoing association mapping studies in pines, including SNP selection and potential biases due to population structure. Indeed, using putatively neutral markers (21 nuSSRs) evenly distributed along most P. taeda linkage groups, we did not find any evidence of population structure, which confirms previous reports showing absence of population genetic structure within the eastern Mississippi Valley range of P. taeda (see, for instance, Al-Rabab'ah and Williams 2002). Despite the moderate level of LD and its rapid decay within genes, the use of htSNPs would reduce SNP genotyping effort by ∼30–40%, 50–100 SNPs being enough to represent common allelic variants in the sequenced candidate gene loci.
The average level of variation (πsil = 0.00853) found in candidate genes for drought-stress response in P. taeda was similar to the one in wood- and disease-related candidates in this species (see review in Neale and Savolainen 2004). Levels of silent variation in pal-1 for P. taeda (this study) and P. sylvestris L. (Dvornyk et al. 2002) were also similar (πsil ≈ 0.00490) and at the lower range of those of the genes studied here. Most standing variation in forest trees is normally found within populations (see, for instance, Hamrick et al. 1992). The extensive sampling of Florida, which is considered a putative Pleistocene glacial refugium of the species (Schmidtling et al. 1999; Al-Rabab'ah and Williams 2002), resulted in only slightly higher nucleotide variation estimates than those in previous studies of the species [average of 0.00604 vs. 0.00580, based on five gene fragments from our study, ccoaomt-1, pal-1, sams-2, ug_2-498, and lp3-1, that we also sequenced in Brown et al.'s (2004) set of samples], the difference not being significant (P = 0.281) as shown by a pairwise signed rank test (n = 5). Bottlenecks, as those that might have occurred in forest trees during Pleistocene range shifts, can generate substantial LD due to a reduction in population size with accompanying genetic drift (Flint-García et al. 2003; Rafalski and Morgante 2004). Levels of LD in this study were lower than those found in Brown et al. (2004) (see Figure 3), which might reflect more stable population dynamics in the putative glacial refugium of Florida. Compared also with Brown et al. (2004), we found a larger range in nucleotide diversity in our study, where maximum per gene silent diversity (0.02052; lp5-like) was 100-fold the minimum estimate (0.00022; pp2c). The nucleotide diversity found in pine, compared with that in other plants, was moderate (see supplemental Table S5 at http://www.genetics.org/supplemental/), which, as first noted by Dvornyk et al. (2002), does not meet predictions based on their life history or other studies based on molecular markers, such as allozymes or RAPDs (Hamrick et al. 1992; Nybom and Bartish 2000). Indeed, pines are highly outcrossing organisms showing generally large effective population sizes and higher heterozygosity than other plants (expected heterozygosity of 0.163–0.193 in P. taeda based on 18 allozymes; Schmidtling et al. 1999). It is striking, then, that average nucleotide variation in P. taeda (and other pines; see, for instance, Pot et al. 2005) was consistently lower than that in Arabidopsis thaliana, the model selfing species. Estimates based on divergence time from related species showed mutation rates in pines (∼0.5–1.5 × 10−10/year; Dvornyk et al. 2002; Brown et al. 2004) two orders of magnitude lower than those in angiosperms, including Arabidopsis (Dvornyk et al. 2002 and references therein). A lower overall rate of sequence evolution might explain the increasing evidence of low to moderate nucleotide diversity in pines.
A number of neutrality tests were conducted to identify genes or sites departing from standard neutral patterns. A selective sweep might have occurred at the early-response-to-drought-3 (erd3) gene, which had reduced nucleotide variation, as shown by pairwise HKA tests, and an excess of less frequent variants. This polymorphism pattern can result from genetic hitchhiking (Braverman et al. 1995; see Olsen et al. 2002 for an example in plants). However, Fay and Wu's H-test did not find any excess of derived variants at high frequency for this gene (Fay and Wu's H = 0.363, P = 0.4140), which is a unique pattern produced by genetic hitchhiking (Fay and Wu 2000). The observed site frequency spectrum might also have resulted from population expansion. Despite the lack of evidence of population expansion shown by our nuSSR survey, a relatively recent population expansion for the southern pines (note that pollen morphology among species of southern pines, including P. taeda, is indistinguishable) within the study range is supported by palynological data showing a steady increase of pine presence beginning 7000 years before present (Watts and Hansen 1994). Because the survival of P. taeda seedlings is strongly limited by the average annual minimum temperature (Schmidtling 2001), range expansions and retreats in response to changing climatic conditions are expected in this species. Further evidence of population expansion in P. taeda from the southeastern United States comes from the skewed Tajima's D distribution (∼70% of genes giving negative estimates of D) of the ∼50 genes currently sequenced in our laboratory (our unpublished data). A skew of the Tajima's D distribution toward negative values is a typical genomewide signature of population growth (Sano and Tachida 2005 and references therein).
One other gene, Caffeoyl-CoA-O-methyltransferase (ccoaomt-1), a methylating enzyme involved in lignification, had an excess of intermediate variants (significant positive Tajima's D), fewer haplotypes than expected (significant positive Fu's Fs), and high within-gene LD (average pairwise r2 of 0.90), resulting in a polymorphism pattern characterized by the existence of two distinctly major haplotype lineages at similar frequencies (named dimorphism; see Figure 1a). This gene also showed higher variation than the average in silent sites but lower variation in nonsynonymous sites (πsil of 0.01911 and null πa vs. averages of 0.00853 and 0.00166, respectively). The two haplotype lineages did not show any geographical pattern, both lineages being present in all the major biogeographical zones of the P. taeda range (see Figure 1b). All 13 polymorphic sites found in the sequenced fragment (see Table 1) were silent mutations and, consequently, we were not able to compute the MK test for this gene or identify a replacement polymorphism causing the singular haplotype structure found in ccoaomt-1. Pairwise HKA tests, which consider variable mutation rates across the genome, did not show an excess of polymorphism relative to the other loci, used here as reference. In a scenario of no population structure and population expansion, demography and population factors do not provide any satisfactory explanation for dimorphism in this gene. Dimorphism has often been considered as the outcome of the long-term action of balancing selection in different genes and species [PgiC in Leavenworthia species (Filatov and Charlesworth 1999); RPS5 and Rpm1 resistance genes in Arabidopsis (Stahl et al. 1999; Tian et al. 2002)]. However, this pattern is also compatible with a constant-size neutral model with no recombination (see Aguadé 2001 for FAH1 and F3H in Arabidopsis) and evidence of natural selection acting in ccoaomt-1 remains inconclusive. Full-length sequencing of this gene, including the promoter region, is advisable. Olsen et al. (2002) found two promoter haplogroups, weakly associated with flower developmental traits, in the TFL1 gene of Arabidopsis that appear to be maintained by selection. Further evidence of natural selection for this gene might also come from our ongoing association studies where ∼900 P. taeda clones will be used to test ccoaomt-1 haplotype differences in performance for adaptive traits related to growth, drought-stress response, and resistance to fungal disease.
In conifers, a candidate-gene-based strategy for association mapping is favored. Genomewide scans are implausible for conifers because of the number of SNPs needed to cover the large genome and because of the general lack of intergenic LD (Neale and Savolainen 2004). Our results are relevant to define SNP genotyping strategies for our ongoing association mapping of drought-stress tolerance candidate genes in pines. Genes or portions of genes showing departure from the standard neutral model will be given priority, in particular ccoaomt-1, where a balanced polymorphism might have caused dimorphism at nearby linked regions. In total, we identified 196 polymorphisms, including 139 common SNPs (i.e., SNPs with minor allele frequency >5%) suitable for association mapping, in 18 candidate gene loci for drought-stress response in P. taeda. Pine genes might be structured in short blocks within which common variants are in strong LD but among which recombination has left little LD. Then, genotyping strategies based on htSNPs would produce only moderate reductions in genotyping effort. Depending on the minor allele frequency chosen, we found that genotyping of 50–100 SNPs would suffice to represent common allelic variants, resulting in reductions of genotyping effort of ∼30–40% in P. taeda association studies.
Acknowledgments
We thank K. Krutovsky, M. Heuertz, and P. G. Goicoechea for valuable comments and discussions. G. P. Gill, R. J. Kuntz, J. Beal, and J. Manares provided technical assistance in the lab. We thank A. Soto and M. T. Cervera, and P. Garnier-Géré, who provided unpublished sequence data and unpublished nucleotide diversity estimates, respectively, for P. pinaster. C. Dana Nelson [Southern Institute of Forest Genetics, U.S. Department of Agriculture (USDA)] produced the nuclear microsatellite data. The work of S. C. González-Martínez was supported by a Fulbright/MECD scholarship at University of California (Davis) and by the “Ramón y Cajal” fellowship (RC02-2941). This research was supported by the Allele Discovery for Genes Controlling Economic Traits in Loblolly Pine project funded in the framework of the Initiative for Future Agriculture and Food Systems (USDA).
References
- Aguadé, M., 2001. Nucleotide sequence variation at two genes of the phenylpropanoid pathway, the FAH1 and F3H genes, in Arabidopsis. Mol. Biol. Evol. 18: 1–9. [DOI] [PubMed] [Google Scholar]
- Al-Rabab'ah, M., and C. G. Williams, 2002. Population dynamics of Pinus taeda L. based on nuclear microsatellites. For. Ecol. Manage. 163: 263–271. [Google Scholar]
- Braverman, J. M., R. R. Hudson, N. L. Kaplan, C. H. Langley and W. Stephan, 1995. The hitchhiking effect on the site frequency spectrum of DNA polymorphisms. Genetics 140: 783–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, G. R., E. E. Kadel, III, D. L. Bassoni, K. L. Kiehne, B. Temesgen et al., 2001. Anchored reference loci in loblolly pine (Pinus taeda L.) for integrating pine genomics. Genetics 159: 799–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, G. R., D. L. Bassoni, G. P. Gill, J. R. Fontana, N. C. Wheeler et al., 2003. Identification of quantitative trait loci influencing wood property traits in loblolly pine (Pinus taeda L.). III. QTL verification and candidate gene mapping. Genetics 164: 1537–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, G. R., G. P. Gill, R. J. Kuntz, C. H. Langley and D. B. Neale, 2004. Nucleotide variation and linkage disequilibrium in loblolly pine. Proc. Natl. Acad. Sci. USA 101: 15255–15260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler, IV, E. S., and J. M. Thornsberry, 2002. Plant molecular diversity and applications to genomics. Curr. Opin. Plant Biol. 5: 107–111. [DOI] [PubMed] [Google Scholar]
- Burns, R. M., and B. H. Honkala, 1990. Silvics of North America: 1. Conifers. 2. Hardwoods. Agriculture Handbook 654. U.S. Department of Agriculture, Forest Service, Washington, DC (http://www.na.fs.fed.us/spfo/pubs/silvics_manual/table_of_contents.htm).
- Chang, S., J. D. Puryear, M. A. D. L. Dias, E. A. Funkhouser, R. J. Newton et al., 1996. Gene expression under water deficit in loblolly pine (Pinus taeda): isolation and characterization of cDNA clones. Physiol. Plant. 97: 139–148. [Google Scholar]
- Dubos, C., and C. Plomion, 2001. Drought differentially affects expression of a PR-10 protein in needles of maritime pine (Pinus pinaster Ait.) seedlings. J. Exp. Bot. 358: 1143–1144. [DOI] [PubMed] [Google Scholar]
- Dubos, C., and C. Plomion, 2003. Identification of water-deficit responsive genes in maritime pine (Pinus pinaster Ait.) roots. Plant Mol. Biol. 51: 249–262. [DOI] [PubMed] [Google Scholar]
- Dubos, C., G. Le-Provost, D. Pot, F. Salin, C. Lalane et al., 2003. Identification and characterization of water-stress-responsive genes in hydroponically grown maritime pine (Pinus pinaster) seedlings. Tree Physiol. 23: 169–179. [DOI] [PubMed] [Google Scholar]
- Dvornyk, V., A. Sirviö, M. Mikkonen and O. Savolainen, 2002. Low nucleotide diversity at the pal1 locus in the widely distributed Pinus sylvestris. Mol. Biol. Evol. 19: 179–188. [DOI] [PubMed] [Google Scholar]
- Ewing, B., L. Hillier, M. Wendl and P. Green, 1998. Basecalling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8: 175–185. [DOI] [PubMed] [Google Scholar]
- Fay, J. C., and C.-I Wu, 2000. Hitchhiking under positive Darwinian selection. Genetics 155: 1405–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filatov, D. A., and D. Charlesworth, 1999. DNA polymorphism, haplotype structure and balancing selection in the Leavenworthia PgiC locus. Genetics 153: 1423–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint-García, S. A., J. M. Thornsberry and E. S. Buckler, IV, 2003. Structure of linkage disequilibrium in plants. Annu. Rev. Plant Biol. 54: 357–374. [DOI] [PubMed] [Google Scholar]
- Ford, M. J., 2002. Applications of selective neutrality tests to molecular ecology. Mol. Ecol. 11: 1245–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y. X., 1997. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147: 915–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Gil, M. R., M. Mikkonen and O. Savolainen, 2003. Nucleotide diversity at two phytochrome loci along a latitudinal cline in Pinus sylvestris. Mol. Ecol. 12: 1195–1206. [DOI] [PubMed] [Google Scholar]
- Gordon, D., C. Abajian and P. Green, 1998. Consed: a graphical tool for sequence finishing. Genome Res. 8: 195–202. [DOI] [PubMed] [Google Scholar]
- Hamrick, J. L., M. J. Godt and S. L. Sherman-Broyles, 1992. Factors influencing levels of genetic diversity in woody plant species. New For. 6: 95–124. [Google Scholar]
- Hill, W. G., and A. Robertson, 1968. Linkage disequilibrium in finite populations. Theor. Appl. Genet. 38: 226–231. [DOI] [PubMed] [Google Scholar]
- Hill, W. G., and B. S. Weir, 1988. Variances and covariances of squared linkage disequilibria in finite populations. Theor. Popul. Biol. 33: 54–78. [DOI] [PubMed] [Google Scholar]
- Hirschhorn, J. N., and M. J. Daly, 2005. Genome-wide association studies for common diseases and complex traits. Nat. Rev. Genet. 6: 95–108. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., M. Kreitman and M. Aguadé, 1987. A test of neutral molecular evolution based on nucleotide data. Genetics 116: 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram, J., and D. Bartels, 1996. The molecular basis of dehydratation tolerance in plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 47: 377–403. [DOI] [PubMed] [Google Scholar]
- Ingvarsson, P. K., 2005. Nucleotide polymorphism and linkage disequilibrium within and among natural populations of European aspen (Populus tremula L., Salicaceae). Genetics 169: 945–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis, S. B., M. A. Taylor, M. R. MacLeod and H. V. Davies, 1996. Cloning and characterisation of the cDNA clones of three genes that are differentially expressed during dormancy-breakage in the seeds of Douglas fir (Pseudotsuga menziesii). J. Plant Physiol. 147: 559–566. [Google Scholar]
- Kado, T., H. Yoshimaru, Y. Tsumura and H. Tachida, 2003. DNA variation in a conifer, Cryptomeria japonica (Cupressaceae sensu lato). Genetics 164: 1547–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpinska, B., M. Karlsson, H. Schinkel, S. Streller, K. H. Süss et al., 2001. A novel superoxide-dismutase with a high isoelectric point in higher plants. Expression, regulation, and protein localization. Plant Physiol. 126: 1668–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosakovsky-Pond, S. L., and S. D. W. Frost, 2005. a Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 22: 1208–1222. [DOI] [PubMed] [Google Scholar]
- Kosakovsky-Pond, S. L., and S. D. W. Frost, 2005. b Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21: 2531–2533. [DOI] [PubMed] [Google Scholar]
- Kreitman, M., 2000. Methods to detect selection in populations with applications to the human. Annu. Rev. Genomics Hum. Genet. 1: 539–559. [DOI] [PubMed] [Google Scholar]
- Krupkin, A. B., A. Liston and S. H. Strauss, 1996. Phylogenetic analysis of the hard pines (Pinus subgenus Pinus, Pinaceae) from chloroplast restriction site analysis. Am. J. Bot. 83: 489–498. [Google Scholar]
- Krutovsky, K. V., and D. B. Neale, 2005. Nucleotide diversity and linkage disequilibrium in cold hardiness and wood quality related candidate genes in Douglas fir. Genetics 171: 2029–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L., X. H. Zhang, C. P. Joshi and V. L. Chiang, 1998. Compression stress responsive expression of ferritin (accession no AF028072) and peroxidase genes (accession no AF028073) in developing xylem of loblolly pine (Pinus taeda). Plant Physiol. 116: 1604. [Google Scholar]
- Long, A. D., and C. H. Langley, 1999. The power of association studies to detect the contribution of candidate genetic loci to variation in complex traits. Genome Res. 9: 720–731. [PMC free article] [PubMed] [Google Scholar]
- Luikart, G., P. R. England, D. Tallmon, S. Jordan and P. Taberlet, 2003. The power and promise of population genomics: from genotyping to genome typing. Nat. Rev. Genet. 4: 981–994. [DOI] [PubMed] [Google Scholar]
- Marchini, J., L. R. Cardon, M. S. Phillips and P. Donnelly, 2004. The effects of human population structure on large genetic association studies. Nat. Genet. 36: 512–517. [DOI] [PubMed] [Google Scholar]
- McDonald, J. H., and M. Kreitman, 1991. Adaptive protein evolution at the Adh locus in Drosophila. Nature 351: 652–654. [DOI] [PubMed] [Google Scholar]
- Neale, D. B., and O. Savolainen, 2004. Association genetics of complex traits in conifers. Trends Plant Sci. 9: 325–330. [DOI] [PubMed] [Google Scholar]
- Nei, M., 1987. Molecular Evolutionary Genetics. Columbia University Press, New York.
- Nei, M., and W. H. Li, 1979. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA 76: 5269–5273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton, R. J., E. A. Funkhouser, F. Fong and C. G. Tauer, 1991. Molecular and physiological genetics of drought tolerance in forest species. For. Ecol. Manage. 43: 225–250. [Google Scholar]
- Nybom, H., and I. V. Bartish, 2000. Effects of life history traits and sampling strategies on genetic diversity estimates obtained with RAPD markers in plants. Perspect. Plant Ecol. Evol. Syst. 3: 93–114. [Google Scholar]
- Olsen, K. M., A. Womack, A. R. Garrett, J. I. Suddith and M. D. Purugganan, 2002. Contrasting evolutionary forces in the Arabidopsis thaliana floral developmental pathway. Genetics 160: 1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan, V., M. A. D. L. Dias and R. J. Newton, 1997. Expression analysis of a gene family in loblolly pine (Pinus taeda L.) induced by water-deficit stress. Plant Mol. Biol. 35: 801–807. [DOI] [PubMed] [Google Scholar]
- Payseur, B. A., A. D. Cutter and M. W. Nachman, 2002. Searching for evidence of positive selection in the human genome using patterns of microsatellite variability. Mol. Biol. Evol. 7: 1143–1153. [DOI] [PubMed] [Google Scholar]
- Pot, D., L. McMillan, C. Echt, G. Le-Provost, P. Garnier-Géré et al., 2005. Nucleotide variation in genes involved in wood formation in two pine species. New Phytol. 167: 101–112. [DOI] [PubMed] [Google Scholar]
- Pritchard, J. K., and W. Wen, 2004. Documentation for Structure Software Version 2. Department of Human Genetics, University of Chicago, Chicago (http://pritch.bsd.uchicago.edu).
- Pritchard, J. K., M. Stephens and P. Donnelly, 2000. Inference of population structure using multilocus genotype data. Genetics 155: 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafalski, A., and M. Morgante, 2004. Corn and humans: recombination and linkage disequilibrium in two genomes of similar size. Trends Genet. 20: 103–111. [DOI] [PubMed] [Google Scholar]
- Remington, D. L., J. M. Thornsberry, Y. Matsouka, L. M. Wilson, S. R. Whitt et al., 2001. Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc. Natl. Acad. Sci. USA 98: 11479–11484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard, S., M. J. Morency, C. Drevet, L. Jouanin and A. Séguin, 2000. Isolation and characterization of a dehydrin gene from white spruce induced upon wounding, drought and cold stresses. Plant Mol. Biol. 43: 1–10. [DOI] [PubMed] [Google Scholar]
- Rosenberg, N. A., and M. Nordborg, 2002. Genealogical trees, coalescent theory and the analysis of genetic polymorphisms. Nat. Rev. Genet. 3: 380–390. [DOI] [PubMed] [Google Scholar]
- Rosenberg, N. A., J. K. Pritchard, J. L. Weber, H. M. Cann, K. K. Kidd et al., 2002. Genetic structure of human populations. Science 298: 2381–2385. [DOI] [PubMed] [Google Scholar]
- Rozas, J., J. C. Sánchez-del-Barrio, X. Messeguer and R. Rozas, 2003. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19: 2496–2497. [DOI] [PubMed] [Google Scholar]
- Sano, A., and H. Tachida, 2005. Gene genealogy of test statistics of neutrality under population growth. Genetics 169: 1687–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidtling, R. C., 2001. Southern Pine Seed Sources. USDA, GTR SRS-44, Asheville, NC.
- Schmidtling, R. C., E. Carroll and T. LaFarge, 1999. Allozyme diversity of selected and natural loblolly pine populations. Silvae Genet. 48: 35–45. [Google Scholar]
- Schneider, S., D. Roessli and L. Excoffier, 2000. Arlequin Ver. 2000: A Software for Population Genetics Data Analysis. Genetics and Biometry Laboratory, University of Geneva, Geneva.
- Seki, M., A. Kamei, K. Yamaguchi-Shinozaki and K. Shinozaki, 2003. Molecular responses to drought, salinity and frost: common and different paths for plant protection. Curr. Opin. Biotechnol. 14: 194–199. [DOI] [PubMed] [Google Scholar]
- Slatkin, M., 1994. An exact test for neutrality based on the Ewens sampling distribution. Genet. Res. 64: 71–74. [DOI] [PubMed] [Google Scholar]
- Slatkin, M., 1996. A correction to the exact test based on the Ewens sampling distribution. Genet. Res. 68: 259–260. [DOI] [PubMed] [Google Scholar]
- Stahl, M. G., G. Dwyer, R. Mauricio, M. Kreitman and J. Bergelson, 1999. Dynamics of disease resistance polymorphism at the Rpm1 locus of Arabidopsis. Nature 400: 667–671. [DOI] [PubMed] [Google Scholar]
- Suzuki, Y., and T. Gojobori, 1999. A method for detecting positive selection at single amino acid sites. Mol. Biol. Evol. 16: 1315–1328. [DOI] [PubMed] [Google Scholar]
- Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi, F., K. Yanai, T. Morii, Y. Ishinaga, K. Taniguchi-Yanai et al., 2005. Linkage disequilibrium grouping of single nucleotide polymorphisms (SNPs) reflecting haplotype phylogeny for efficient selection of tag SNPs. Genetics 170: 291–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temesgen, B., G. R. Brown, D. E. Harry, C. S. Kinlaw, M. M. Sewell et al., 2001. Genetic mapping of expressed sequence tag polymorphism (ESTP) markers in loblolly pine (Pinus taeda L.). Theor. Appl. Genet. 102: 664–675. [Google Scholar]
- Tian, D., H. Araki, E. Stahl, J. Bergelson and M. Kreitman, 2002. Signature of balancing selection in Arabidopsis. Proc. Natl. Acad. Sci. USA 99: 11525–11530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tranbarger, T. J., and S. Misra, 1996. Structure and expression of a developmentally regulated cDNA encoding a cysteine protease (pseudotzain) from Douglas-fir. Gene 172: 221–226. [DOI] [PubMed] [Google Scholar]
- Wang, W. Y. S., B. J. Barratt, D. G. Clayton and J. A. Todd, 2005. Genome-wide association studies: theoretical and practical concerns. Nat. Rev. Genet. 6: 109–118. [DOI] [PubMed] [Google Scholar]
- Watkinson, J. I., A. A. Sioson, C. Vasquez-Robinet, M. Shukla, D. Kumar et al., 2003. Photosynthetic acclimation is reflected in specific patterns of gene expression in drought-stressed loblolly pine. Plant Physiol. 133: 1702–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson, G. A., 1975. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7: 256–276. [DOI] [PubMed] [Google Scholar]
- Watterson, G. A., 1978. The homozygosity test of neutrality. Genetics 88: 405–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson, G. A., 1986. The homozygosity test after a change in population size. Genetics 112: 899–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts, W. A., and B. C. S. Hansen, 1994. Pre-Holocene and Holocene pollen records of vegetation history from the Florida peninsula and their climatic implications. Paleogeogr. Paleoclimatol. Paleoecol. 109: 163–176. [Google Scholar]
- Weir, B. S., and C. C. Cockerham, 1984. Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370. [DOI] [PubMed] [Google Scholar]
- Zhang, K., and L. Jin, 2003. HaploBlockFinder: haplotype block analyses. Bioinformatics 19: 1300–1301. [DOI] [PubMed] [Google Scholar]