

Abstract
Genome-wide nucleotide variation in non-African populations of Drosophila melanogaster is a subset of variation found in East sub-Saharan African populations, suggesting a bottleneck in the history of the former. We implement an approximate Bayesian approach to infer the timing, duration, and severity of this putative bottleneck and ask whether this inferred model is sufficient to account for patterns of variability observed at 115 loci scattered across the X chromosome. We estimate a recent bottleneck 0.006Ne generations ago, somewhat further in the past than suggested by biogeographical evidence. Using various proposed statistical tests, we find that this bottleneck model is able to predict the majority of observed features of diversity and linkage disequilibrium in the data. Thus, while precise estimates of bottleneck parameters (like inferences of selection) are sensitive to model assumptions, our results imply that it may be unnecessary to invoke frequent selective sweeps associated with the dispersal of D. melanogaster from Africa to explain patterns of variability in non-African populations.
THERE is considerable interest in using population genetic approaches to identify regions of the genome that underlie population- or species-specific adaptations. More generally, such scans of the genome can also answer basic evolutionary questions such as the relative importance of selection and nonadaptive factors (such as changes in the size and structure of populations over time) in shaping patterns of genome variability. The rapid increase in our ability to survey population-level nucleotide variability for larger and larger portions of the genome yields increasing statistical power to distinguish among alternative population genetic models.
The most common approach to hypothesis testing has been model based, comparing statistics summarizing features of the observed data, such as the distribution of polymorphism frequencies, to the expectations of a null model (e.g., Tajima 1989; Fu and Li 1993; Fay and Wu 2000). The most commonly used null model assumes a large, panmictic, population with neutral mutations occurring according to the infinite-sites model, hereafter referred to as the standard neutral model (SNM) (see Hudson 1983). Significant departures from the SNM are usually interpreted as evidence for the recent action of natural selection (e.g., Harr et al. 2002; Glinka et al. 2003). However, natural selection is not a unique alternative hypothesis to the SNM. Deviations from the demographic assumptions of the SNM, such as population bottlenecks, can result in the observation of significant departures in the same direction as that expected under selective sweep models (e.g. Andolfatto 2001; Przeworski 2002; Lazzaro and Clark 2003; Haddrill et al. 2005b).
In Drosophila melanogaster, the observation that variation is substantially reduced outside of Africa (Begun and Aquadro 1993) has focused much attention on understanding the cause of this difference between populations. The species is believed to have an African origin and to have colonized other continents only recently (David and Capy 1988; Lachaise et al. 1988). While this scenario is consistent with a population bottleneck being the cause of the reduction in diversity outside of Africa, details of the demographic history of the species are lacking. The timing of emigration from Africa is based on biogeographic rather than direct evidence, although estimates based on genetic data have been argued to be consistent with inference made from biogeography (e.g., Baudry et al. 2004).
In addition to the reduction in diversity, several lines of evidence suggest that demographic effects have had a profound influence on variability in these populations. The observation of shared haplotypes among non-African populations suggests that derived populations share a common history (Baudry et al. 2004). In both D. melanogaster and its sibling species, D. simulans, there is too much linkage disequilibrium (LD) in natural populations relative to expectations based on genetic maps (Andolfatto and Przeworski 2000). This excess of LD is not as pronounced in a Zimbabwe sample of D. melanogaster (Andolfatto and Wall 2003; Haddrill et al. 2005b), a putative ancestral population of the species. A recent survey of consistently sampled noncoding loci from the D. melanogaster X chromosome reveals a significant excess of LD relative to diversity in all population samples relative to Eastern Africa (Haddrill et al. 2005b), suggesting a common population process in all derived populations that leads to an increase in LD. While recent bottlenecks are able to explain the increase in LD (scaled by diversity) in derived populations, simulations suggest that selective sweeps at random loci have little to no effect on LD scaled to levels of diversity (Wall et al. 2002; Haddrill et al. 2005b).
To date, formal inference on bottleneck parameters has been lacking. There are two major reasons for this. First, a lack of consistent sampling of multilocus data sets from both African and non-African samples has been an impediment, with such data sets only recently having been reported for relatively few loci in a large number of populations (Baudry et al. 2004; Haddrill et al. 2005b) or a large number of loci in fewer populations (Glinka et al. 2003). Second, inference of the model parameters is computationally demanding for these multiparameter models, and methods relying on summary statistics to estimate the likelihood of the data require long run times (e.g., Akey et al. 2004; Tenaillon et al. 2004). A recent attempt to estimate bottleneck parameters from a multilocus data set was unable to simultaneously account for both the excess of rare alleles, as measured by Tajima's (1989) D, and the large number of invariant loci in a Netherlands population (Glinka et al., 2003), leading the authors to conclude that recent directional selection has likely occurred outside of Africa. In contrast, Haddrill et al. (2005b) pointed out that allowing for a broader range of bottleneck parameters may lead to a different conclusion. However, the bottleneck parameters explored by both studies were not obtained by an inference procedure, but rather were found by looking for parameter combinations with the same expected level of polymorphism as that seen in the Netherlands sample. Thus, it is still not clear what the most likely parameters of such a model are that are compatible with the data.
In this study, we describe an approximate Bayesian approach to inferring bottleneck parameters from data that are consistently sampled both from a derived and from an ancestral population. This approach allows joint inference of the timing, duration, and severity of a bottleneck, and the uncertainty of the estimates can be quantified. The method is applied to a 115-locus data set sampled from D. melanogaster populations from Zimbabwe, Africa, and the Netherlands (Glinka et al. 2003; Haddrill et al. 2005b). As might be expected on the basis of biogeographical data (Lachaise et al. 1988), we find evidence for a recent, severe bottleneck in the Netherlands. Several lines of evidence suggest that this bottleneck is an appropriate demographic null model for the Netherlands population. First, the bottleneck model is used to obtain “demographically corrected” P-values for five commonly used summaries of the data. The distribution of these P-values is approximately uniform, in sharp contrast to when the SNM is used as the demographic null. Second, the bottleneck model also has high predictive power for multilocus summaries of the data. These results suggest that a recent severe reduction in effective population size can account for most features of polymorphism data from non-African samples of D. melanogaster.
METHODS
Data:
The data analyzed consist of 115 loci sampled from both a Zimbabwe, Africa, and a Leiden, Netherlands, population. A total of 105 of these loci were sampled in an average of 12 individuals from each population and consist of intergenic and intron sequences (Glinka et al. 2003). The remaining 9 intronic and one 5′ upstream region had a mean sample size of 20 per population (Haddrill et al. 2005b). The length of each region sequenced in both studies was ∼500 bp, and a single line of D. simulans was sequenced as an outgroup. All loci are sampled from the high-recombining portion of the X chromosome, where there is little evidence for significant variation in recombination rate on the basis of comparisons of genetic and physical maps (Charlesworth 1996).
Polymorphisms inferred from alignments of all 115 loci were systematically filtered for consistency as described in Haddrill et al. (2005b). This procedure was applied to all five populations from Haddrill et al. (2005b) and to the two population samples from Glinka et al. (2003) before generating separate polymorphism tables for each population.
Approximate Bayesian inference:
We took an approximate Bayesian approach (Pritchard et al. 1999; Beaumont et al. 2002; Marjoram et al. 2003; Przeworski 2003) to obtain k independent samples from the joint posterior distribution of model parameters by rejection sampling. Briefly, a set of summary statistics, 𝒮, is calculated from the observed data and from data simulated with parameters drawn from a prior distribution, and the parameter values are accepted if 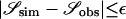 , where ɛ is a fixed set of tolerances.
, where ɛ is a fixed set of tolerances.
Estimating mutation and recombination parameters:
The joint posterior distribution of θ, the population mutation rate, and ρ, the population recombination rate, were estimated for the 115 loci from the Zimbabwe population using the rejection sampling algorithm described in Haddrill et al. (2005b). Briefly, this is an approximate Bayesian method that samples from uniform priors on θ and ρ and uses rejection sampling based on summary statistics to simulate from the approximate joint posterior distribution of parameters. The prior distributions used for the Zimbabwe sample were θ ∼ Uniform(0.005, 0.02) and ρ ∼ Uniform(0.03, 0.15), with ɛ = 0.1 for both parameters. For the Netherlands sample, θ ∼ Uniform(0, 0.01), ρ ∼ Uniform(0, 0.015), and ɛ = 0.35.
Bottleneck model:
The bottleneck model considered here is illustrated in Figure 1. The model is constrained such that the effective population size recovers to the prebottleneck size (N0), and changes in effective size occur instantaneously. The model then has six parameters, the sample size (n) and five unknowns: the coalescent mutation rate (θ = 4N0μ, where N0 is the prebottleneck effective population size and μ is the neutral mutation rate per generation), the coalescent recombination rate (ρ = 4N0r), the time of recovery from the bottleneck (tr), the duration of the bottleneck (d) and the severity of the bottleneck ( ), and the ratio of the effective size during the bottleneck (Nb) to N0. All times in the simulation are measured in units of 4N0 generations. We scale N0 to 1, which constrains 0 < f < 1 and ensures that θ is on the scale of the ancestral population size (i.e., N0 before the bottleneck, going forward in time).
), and the ratio of the effective size during the bottleneck (Nb) to N0. All times in the simulation are measured in units of 4N0 generations. We scale N0 to 1, which constrains 0 < f < 1 and ensures that θ is on the scale of the ancestral population size (i.e., N0 before the bottleneck, going forward in time).
Figure 1.

A simple stepwise bottleneck model.
Inference of bottleneck parameters:
We implemented an approximate Bayesian method, based on summary statistics of the data, to make inferences under the simple stepwise bottleneck model (Figure 1). The method is approximate in two senses. First, we used rejection sampling based on summary statistics in place of explicit estimation of likelihoods. Second, as described below, the method is not fully Bayesian as we substitute point estimates for the true values of two nuisance parameters, mutation and recombination rates, instead of integrating over them (due to constraints on computational time).
In our implementation, we chose to summarize the data according to the variance of nucleotide diversity (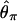 ) per locus, the number of haplotypes (K), and H, a standardized summary of the site-frequency spectrum related to that proposed by Fay and Wu (2000) (J. Shapiro and C.-I Wu, personal communication). The numerator of H depends on
) per locus, the number of haplotypes (K), and H, a standardized summary of the site-frequency spectrum related to that proposed by Fay and Wu (2000) (J. Shapiro and C.-I Wu, personal communication). The numerator of H depends on 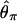 and
and
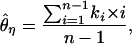 |
(1) |
where ki is the number of occurrences in the data of a derived mutation of size i in the sample. The expression for H is
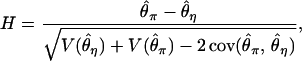 |
(2) |
where
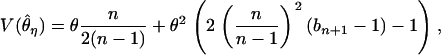 |
(3) |
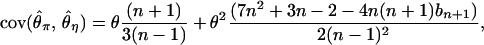 |
(4) |
and
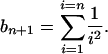 |
(5) |
The expression for 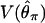 in (2) is Ewens' (2004, p. 357) Equation 11.12. As the true value of θ is an unknown, we substitute the moment estimator
in (2) is Ewens' (2004, p. 357) Equation 11.12. As the true value of θ is an unknown, we substitute the moment estimator 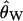 (Watterson 1975) in the above equations. In our implementation, we consider the value of H to be undefined for data sets with zero segregating sites. We therefore exclude such loci from the calculation of the variance of H across loci, meaning that the summary of the data used for inference should be written as V(H|S > 0). We note that the calculation of H depends on accurate inference of the ancestral state of a mutation. Specifically, violations of the infinite-sites model result in more negative values of this statistic (e.g., Fay and Wu 2000), and accounting for this deviation can improve the fit of the statistic to the model (see Haddrill et al. 2005b, for a discussion of this in the context of Fay and Wu's Hfw).
(Watterson 1975) in the above equations. In our implementation, we consider the value of H to be undefined for data sets with zero segregating sites. We therefore exclude such loci from the calculation of the variance of H across loci, meaning that the summary of the data used for inference should be written as V(H|S > 0). We note that the calculation of H depends on accurate inference of the ancestral state of a mutation. Specifically, violations of the infinite-sites model result in more negative values of this statistic (e.g., Fay and Wu 2000), and accounting for this deviation can improve the fit of the statistic to the model (see Haddrill et al. 2005b, for a discussion of this in the context of Fay and Wu's Hfw).
The approximate Bayesian approach is extremely computationally intensive, so we make the simplifying assumption that the Zimbabwe population is at demographic equilibrium. In practice, this means that we assume that  and
and 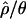 are the true values of θ and
are the true values of θ and 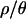 , respectively. We also assume that the ratio
, respectively. We also assume that the ratio  is uniform across loci. This assumption reduces the problem to inferring three unknowns (tr, d, and f) and allows us to simulate only the derived population (i.e., the Netherlands), at the cost of not accounting for uncertainty about the true values of θ and ρ.
is uniform across loci. This assumption reduces the problem to inferring three unknowns (tr, d, and f) and allows us to simulate only the derived population (i.e., the Netherlands), at the cost of not accounting for uncertainty about the true values of θ and ρ.
Our estimates of θ for each locus were obtained using the HKA framework (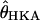 per site) (Hudson et al. 1987), which uses the available divergence data to take into account variation in the neutral mutation rate. Values of
per site) (Hudson et al. 1987), which uses the available divergence data to take into account variation in the neutral mutation rate. Values of  were based on estimates from the Zimbabwe sample, using an approximate Bayesian procedure (Haddrill et al. 2005b, and see above), for which the maximum a posteriori estimate was
were based on estimates from the Zimbabwe sample, using an approximate Bayesian procedure (Haddrill et al. 2005b, and see above), for which the maximum a posteriori estimate was 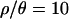 . In addition, we also used
. In addition, we also used 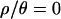 , 1, and 7 to explore the effect of assuming no, very little, or intermediate levels of recombination, on the inference of bottleneck parameters for these data. We used uniform prior distributions on tr, d, and f. Under the model (Figure 1), only f is bounded, so in practice we need to put limits on the priors on tr and d so that the priors are proper probability distributions.
, 1, and 7 to explore the effect of assuming no, very little, or intermediate levels of recombination, on the inference of bottleneck parameters for these data. We used uniform prior distributions on tr, d, and f. Under the model (Figure 1), only f is bounded, so in practice we need to put limits on the priors on tr and d so that the priors are proper probability distributions.
In the current application, we simulate 115 loci with recombination for each replicate, and the computational time required is substantial. There is also a trade-off between computational time and accuracy (the value of ɛ used) (Beaumont et al. 2002). To maximize the computational efficiency, we first obtained 103 samples from the joint posterior for each of the four values of 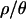 , using a relatively liberal ɛ. Then, for
, using a relatively liberal ɛ. Then, for 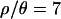 and 10, we obtained a further 104 samples using a smaller ɛ and new priors based on the 1st and 99th percentiles calculated (in R) (R Development Core Team 2004) from the three marginal posterior distributions of the initial 103 samples. The ranges of the priors, and the values of ɛ used, are listed in Table 1. As Table 2 shows, the posterior densities for
and 10, we obtained a further 104 samples using a smaller ɛ and new priors based on the 1st and 99th percentiles calculated (in R) (R Development Core Team 2004) from the three marginal posterior distributions of the initial 103 samples. The ranges of the priors, and the values of ɛ used, are listed in Table 1. As Table 2 shows, the posterior densities for 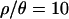 and 7 are contained within the limits of the uniform priors (Table 1). For
and 7 are contained within the limits of the uniform priors (Table 1). For 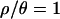 and 0, the sizes of the priors were increased, and the inference procedure rerun, until the posteriors were contained within the bounds of the priors.
and 0, the sizes of the priors were increased, and the inference procedure rerun, until the posteriors were contained within the bounds of the priors.
TABLE 1.
Prior distributions and values of ε used for bottleneck inference
| ρ/θ | tr | d | f | ε |
|---|---|---|---|---|
| 0 | 0–0.20 | 0–2 | 0–0.75 | 1, 1, 1 |
| 1 | 0–0.20 | 0–1 | 0–0.75 | 1, 1, 1 |
| 7 | 0–0.03 | 0.003–0.15 | 0.001–0.15 | 0.5, 0.5, 0.5 |
| 0.0002–0.017 | 0.0048–0.091 | 0.008–0.15 | 0.25, 0.25, 0.5 | |
| 10 | 0–0.02 | 0.003–0.10 | 0.001–0.15 | 0.5, 0.5, 0.5 |
| 0.0001–0.0123 | 0.0038–0.072 | 0.007–0.128 | 0.25, 0.25, 0.5 |
For each value of ρ/θ, the first line corresponds to parameter values used to obtain an initial 103 samples from the joint posterior distribution of bottleneck parameters. Where present, the second line corresponds to parameter values used to obtain a further 104 samples. The three values listed under ε refer to the tolerance used for the summaries of diversity ( ), the number of haplotypes (K), and the site frequency spectrum (H), respectively. See text for details about model parameters and summary statistics.
), the number of haplotypes (K), and the site frequency spectrum (H), respectively. See text for details about model parameters and summary statistics.
TABLE 2.
Summaries of marginal posterior distributions of bottleneck parameters
| ρ/θ
|
|||||
|---|---|---|---|---|---|
| 0 | 1 | 7 | 10 | ||
| tr | MAP | 1.2 × 10−2 | 8.3 × 10−3 | 4.8 × 10−3 | 4.2 × 10−3 |
| 2.5% | 7.5 × 10−4 | 5.8 × 10−4 | 6.3 × 10−4 | 4.0 × 10−4 | |
| 97.5% | 0.11 | 5.4 × 10−2 | 1.4 × 10−2 | 0.01 | |
| d | MAP | 0.26 | 0.12 | 2.1 × 10−2 | 1.5 × 10−2 |
| 2.5% | 5.6 × 10−2 | 2.6 × 10−2 | 7.1 × 10−3 | 5.5 × 10−3 | |
| 97.5% | 1.92 | 0.73 | 7.5 × 10−2 | 5.7 × 10−2 | |
| tb | MAP | 0.32 | 0.13 | 3.2 × 10−2 | 2.2 × 10−2 |
| 2.5% | 0.10 | 2.6 × 10−2 | 7.1 × 10−3 | 1.2 × 10−2 | |
| 97.5% | 1.95 | 0.73 | 7.5 × 10−3 | 5.9 × 10−2 | |
| f | MAP | 0.44 | 0.20 | 4.7 × 10−2 | 2.9 × 10−2 |
| 2.5% | 8.6 × 10−2 | 0.04 | 1.4 × 10−2 | 1.0 × 10−2 | |
| 97.5% | 0.63 | 0.48 | 0.13 | 0.11 | |
All times are in units of 4Ne generations. Marginal distributions are summarized by maximum a posteriori (MAP) estimates and by 2.5th and 97.5th percentiles.
The rejection sampling algorithm is as follows:
Draw random values of tr, d, and f from the prior distributions for each parameter.
Simulate 115 independent loci under the bottleneck model specified in step one. For each locus, the value of θ per site is taken to be
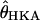 from the Zimbabwe sample for the locus. Recombination rates are determined by the value
from the Zimbabwe sample for the locus. Recombination rates are determined by the value 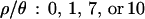 . The sample size and number of sites for each locus are taken from the data.
. The sample size and number of sites for each locus are taken from the data.Summarize the 115 simulated loci by the variance across loci of
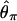 , K, and H.
, K, and H.If the summaries are within ɛ of the observed summaries of the data, accept tr, d, and f; otherwise reject.
Return to step 1 until k acceptances are recorded.
Our program to perform the above algorithm was written in C++. The coalescent simulations are performed by system calls to a slightly modified version of Hudson's (2002) program, and simulation output was processed using libsequence (Thornton 2003).
The initial 103 samples were obtained on a single 2.4-Ghz Pentium 4 processor. For 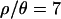 and 10, it took 1 week each to obtain the samples, and the acceptance rates were 1.9 × 10−3 and 1.8 × 10−3, respectively. Approximately 1.5–2 weeks each were required for
and 10, it took 1 week each to obtain the samples, and the acceptance rates were 1.9 × 10−3 and 1.8 × 10−3, respectively. Approximately 1.5–2 weeks each were required for 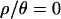 and 1, with acceptance rates of 3.1 × 10−4 and 6.0 × 10−4, respectively. Obtaining the subsequent 104 samples, with the lower ɛ, took ∼10 and 14 days on a cluster of 94 2.0-Ghz Apple G5 processors for
and 1, with acceptance rates of 3.1 × 10−4 and 6.0 × 10−4, respectively. Obtaining the subsequent 104 samples, with the lower ɛ, took ∼10 and 14 days on a cluster of 94 2.0-Ghz Apple G5 processors for 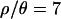 and 10, respectively. The acceptance rates were 5 × 10−4 and 3.9 × 10−4 for
and 10, respectively. The acceptance rates were 5 × 10−4 and 3.9 × 10−4 for 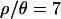 and 10, respectively. These acceptance rates correspond to 2.3 × 109 (
and 10, respectively. These acceptance rates correspond to 2.3 × 109 (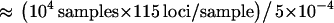 ) coalescent simulations for
) coalescent simulations for 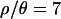 and to 2.9 billion for
and to 2.9 billion for 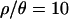 . We found that it was not possible in practice to obtain 104 samples for
. We found that it was not possible in practice to obtain 104 samples for 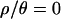 and 1 with smaller values of ɛ, as the acceptance rate dropped to a handful of acceptances per week per processor.
and 1 with smaller values of ɛ, as the acceptance rate dropped to a handful of acceptances per week per processor.
Posterior predictive simulations:
Multilocus summaries:
To assess the fit of the simulated posterior distribution of bottleneck parameters to the data, we performed predictive simulations by sampling from the joint posterior distribution (e.g., Gelman et al. 2003, p. 159). Predictive simulations have two uses. First, they allow us to study the fit of a model to the data and therefore play a role in model checking. Second, performing coalescent simulations with bottleneck parameters chosen according to their posterior probability allows “posterior predictive” P-values to be estimated for each locus. These P-values can be used to identify any individual loci that may be outliers under the bottleneck model.
For these simulations, the data set was summarized by the median and the variance of several summary statistics, and coalescent simulations were performed under the models used for inference. The summary statistics explored were S, the number of segregating sites in the sample, 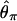 per site, Tajima's (1989) D, H (Equation 2), Fay and Wu's (2000) Hfw, K, the number of haplotypes in the sample, and Kd, the haplotype diversity of the sample. For each statistic, we calculated two probabilities: the probability that the observed median is too low and the probability that the observed variance is too large, given the model. We also examined the number of invariant loci (the F0-statistic of Glinka et al. 2003) and calculated P(F0 ≥ F0,obs). To estimate these P-values, 2.5 × 105 simulations of a 115-locus data set were performed. The observed values of these multilocus summaries are in supplemental Table A at http://www.genetics.org/supplemental/.
per site, Tajima's (1989) D, H (Equation 2), Fay and Wu's (2000) Hfw, K, the number of haplotypes in the sample, and Kd, the haplotype diversity of the sample. For each statistic, we calculated two probabilities: the probability that the observed median is too low and the probability that the observed variance is too large, given the model. We also examined the number of invariant loci (the F0-statistic of Glinka et al. 2003) and calculated P(F0 ≥ F0,obs). To estimate these P-values, 2.5 × 105 simulations of a 115-locus data set were performed. The observed values of these multilocus summaries are in supplemental Table A at http://www.genetics.org/supplemental/.
Single-locus tests:
We also obtained P-values for several summary statistics of each of the 115 loci . For each locus, we ran 2.5 × 105 coalescent simulations to estimate P-values. To compare with the results of standard hypothesis testing, we also obtained P-values using the SNM as the demographic null model. Again, 2.5 × 105 replicates were run per locus. To be consistent with the simulations from the bottleneck model, these simulations were run using the 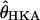 per site, for each locus, and the values of
per site, for each locus, and the values of  used for parameter inference.
used for parameter inference.
The summary statistics examined were the same as those described above. All P-values were two-tailed, estimated from a simulated null distribution to within 0.001. This was done by adjusting the size of the rejection region from zero to one in steps of 0.001 and calculating the corresponding percentiles for the two-sided rejection region using the numeric routines in the GNU Scientific Library (http://sources.redhat.com/gsl). It is perhaps more common to calculate the one-sided probability P(X ≤ x), because the lower tail is of particular interest when the alternative hypothesis is assumed to be the occurrence of a recent selective sweep at nearby linked sites. However, there is a two-sided signal in the data, and correction for multiple tests using the false discovery rate (FDR) (Storey 2002) requires that P-values be calculated correctly, namely that they are two-sided when there is a two-sided signal. To apply the FDR method, we used the qvalue routines (http://faculty.washington.edu/∼jstorey/qvalue/).
RESULTS
Levels of diversity and linkage disequilibrium:
Both the power of neutrality tests and the posterior distributions of bottleneck parameters (as shown below) depend on ρ, the population recombination rate. We therefore estimated ρ, θ, and 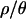 for the 115 loci sampled in the putatively ancestral Zimbabwe population, using an approximate Bayesian approach (as implemented by Haddrill et al. 2005b; K. Thornton, unpublished data). The posterior distributions are shown in Figure 2, and point estimates and percentiles of the posterior distributions of θ, ρ, and
for the 115 loci sampled in the putatively ancestral Zimbabwe population, using an approximate Bayesian approach (as implemented by Haddrill et al. 2005b; K. Thornton, unpublished data). The posterior distributions are shown in Figure 2, and point estimates and percentiles of the posterior distributions of θ, ρ, and 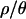 are shown in Table 3. The maximum a posteriori estimate of
are shown in Table 3. The maximum a posteriori estimate of 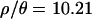 , with a 95% credibility interval of 8.45–12.37. Compared with Zimbabwe, the Netherlands sample shows significantly reduced diversity and significantly increased linkage disequilibrium, as measured both by
, with a 95% credibility interval of 8.45–12.37. Compared with Zimbabwe, the Netherlands sample shows significantly reduced diversity and significantly increased linkage disequilibrium, as measured both by 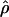 and by
and by 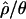 .
.
Figure 2.

Marginal posterior distributions of θ and ρ per site in Zimbabwe (solid line) and the Netherlands (dashed line). The parameters θ and ρ were jointly estimated from the 115-locus data set in both populations, using a rejection sampling scheme (see methods). These marginal distributions are summarized in Table 3.
TABLE 3.
Estimates of mutation and recombination rates in 115 X-linked loci in Zimbabwe and the Netherlands
| Population | Parameter | MAP | Mean | 0.025 | 0.975 |
|---|---|---|---|---|---|
| Zimbabwe | θ | 0.0096 | 0.0096 | 0.0091 | 0.0103 |
| ρ | 0.098 | 0.0994 | 0.0832 | 0.1170 | |
| ρ/θ | 10.205 | 10.322 | 8.450 | 12.369 | |
| The Netherlands | θ | 0.0033 | 0.0033 | 0.0029 | 0.0037 |
| ρ | 0.0032 | 0.0039 | 0.0019 | 0.0070 | |
| ρ/θ | 0.955 | 1.188 | 0.0532 | 2.222 |
Estimates were obtained using an approximate Bayesian method (Haddrill et al. 2005b). The quantity ρ/θ is a ratio, and the ratio of means and the mean of the ratios were very similar. Values of θ and ρ are per site.
The relative excess of LD in non-African samples described is consistent with results obtained by a variety of related methods (Andolfatto and Przeworski 2000; Wall et al. 2002; Andolfatto and Wall 2003; Haddrill et al. 2005b). These results suggest that non-African samples depart significantly from the assumptions of the standard neutral model. Simulations of bottleneck and selective sweep models, where both models are parameterized to reduce 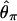 by the same amount, have revealed that population bottlenecks reduce
by the same amount, have revealed that population bottlenecks reduce 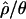 relative to its true value. In contrast, steady-state sweep models have little to no effect on
relative to its true value. In contrast, steady-state sweep models have little to no effect on 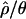 at randomly chosen loci, despite reducing both
at randomly chosen loci, despite reducing both 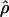 and
and 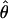 relative to the true parameter values (Wall et al. 2002; Haddrill et al. 2005b).
relative to the true parameter values (Wall et al. 2002; Haddrill et al. 2005b).
Inference on bottleneck parameters:
Joint posterior distributions of bottleneck parameters were obtained by a rejection sampling algorithm for four different values of 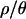 (0, 1, 7, and 10). The value
(0, 1, 7, and 10). The value 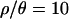 is based on the maximum a posteriori (MAP) estimate for the Zimbabwe data (Figure 2 and Table 3). The remaining values of
is based on the maximum a posteriori (MAP) estimate for the Zimbabwe data (Figure 2 and Table 3). The remaining values of 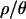 (0, 1, and 7) were used to explore the effects of assuming either no or intermediate levels of recombination on the inference procedure.
(0, 1, and 7) were used to explore the effects of assuming either no or intermediate levels of recombination on the inference procedure.
The marginal posterior distributions for tr, the time at which Ne recovered to N0, d, the duration of the bottleneck, tb, the timing of the bottleneck (which is given by the distribution of the sum tr + d), and f, the bottleneck severity, are shown in Figure 3 and summarized in Table 2. One important feature of Table 2 and Figure 3 is that both point estimates of parameters and percentiles of the marginal posterior distributions depend on the assumed value of 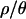 . As
. As 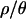 increases, the inferred bottleneck parameters show more evidence for a relatively recent, short, and severe bottleneck compared to
increases, the inferred bottleneck parameters show more evidence for a relatively recent, short, and severe bottleneck compared to 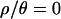 . Although the marginal posterior distributions in Figure 3 are based on different numbers of samples and different-sized priors for
. Although the marginal posterior distributions in Figure 3 are based on different numbers of samples and different-sized priors for 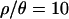 and 7 compared to
and 7 compared to 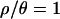 and 0 (see Table 1), it is unlikely that these differences account for the difference in results between values of
and 0 (see Table 1), it is unlikely that these differences account for the difference in results between values of 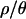 .
.
Figure 3.

Marginal posterior distributions of bottleneck parameters with four values of 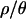 . The parameters are tr, the time at which the population recovered from the bottleneck, d, the duration of the bottleneck, tb, the time of the crash in population size, and f, the bottleneck severity. Time is measured in units of 4Ne generations, and smaller values of f correspond to more severe bottlenecks (see Figure 1). These marginal distributions are summarized in Table 2.
. The parameters are tr, the time at which the population recovered from the bottleneck, d, the duration of the bottleneck, tb, the time of the crash in population size, and f, the bottleneck severity. Time is measured in units of 4Ne generations, and smaller values of f correspond to more severe bottlenecks (see Figure 1). These marginal distributions are summarized in Table 2.
The timescale of the bottleneck parameters is in units of 4N0 generations that can be converted into absolute time if N0 and the generation time are known. One parameter of particular interest is the time in the past at which the effective size crashed (labeled tb on the scale of 4N0 generations in Figure 1), because it represents our best estimate of the timing of dispersal from Africa, if this dispersal was associated with a bottleneck. The time in years of the crash is then 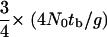 , where g is the number of generations per year. The factor of
, where g is the number of generations per year. The factor of 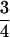 assumes equal effective sizes for males and females for the X-linked loci analyzed here. For these data, the mean
assumes equal effective sizes for males and females for the X-linked loci analyzed here. For these data, the mean 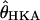 is 0.011 per site in Zimbabwe. Assuming a neutral mutation rate of 1.5 × 10−8/year (Li 1997, p. 191), and g = 10, we obtain N0 = 2.4 million. Assuming this N0, the MAP estimate of the time in years (and 95% credibility intervals) of the population crash is 16,000 (9000–43,000), 23,000 (12,000–57,000), 98,000 (43,100–540,000), and 236,000 (75,000–1,400,000), for
is 0.011 per site in Zimbabwe. Assuming a neutral mutation rate of 1.5 × 10−8/year (Li 1997, p. 191), and g = 10, we obtain N0 = 2.4 million. Assuming this N0, the MAP estimate of the time in years (and 95% credibility intervals) of the population crash is 16,000 (9000–43,000), 23,000 (12,000–57,000), 98,000 (43,100–540,000), and 236,000 (75,000–1,400,000), for 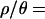 10, 7, 1, and 0, respectively (Figure 4). Recent studies have suggested moderate sexual selection (Charlesworth 2001) and a lower nucleotide substitution rate for introns than for synonymous sites (Haddrill et al. 2005a) in D. melanogaster. Incorporating these details would increase our estimate of N0 and thus push our estimates of tb further into the past. The reliability of these estimates given uncertainties in the model (i.e., misspecification) are discussed below.
10, 7, 1, and 0, respectively (Figure 4). Recent studies have suggested moderate sexual selection (Charlesworth 2001) and a lower nucleotide substitution rate for introns than for synonymous sites (Haddrill et al. 2005a) in D. melanogaster. Incorporating these details would increase our estimate of N0 and thus push our estimates of tb further into the past. The reliability of these estimates given uncertainties in the model (i.e., misspecification) are discussed below.
Figure 4.

Timing of bottleneck in the Netherlands. Times are converted from units of 4Ne generations into years, assuming Ne = 2.4 million and 10 generations per year.
Posterior predictive simulations:
We made several assumptions about demography in Africa and about variation in mutation and recombination rates across loci to simplify the inference procedure. It is therefore reasonable to ask how well the simulated posterior distributions predict the data. Two questions are of interest here. First, How good is the overall fit? Second, Are there individual loci that are outliers under the bottleneck models described by the posterior distributions? The latter question is also relevant to the so-called “hitchhiking mapping” approach to identifying genomic regions that have undergone recent directional selection (Harr et al. 2002; Kauer et al. 2002; Glinka et al. 2003; Kauer et al. 2003; Orengo and Aguade 2004). In the hitchhiking mapping framework, individual loci that reject the bottleneck model may be considered candidate loci that have undergone recent selective sweeps, noting that rejecting this simple demographic model is not a formal test for positive selection. Additionally, we actually have four different bottleneck models obtained by the approximate Bayesian method (one for each value of 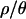 ) and it is of interest to ask whether or not the fit of the model to the data is affected by assumptions about how much recombination is occurring.
) and it is of interest to ask whether or not the fit of the model to the data is affected by assumptions about how much recombination is occurring.
Multilocus summaries:
To address the overall fit of the bottleneck model to the observed data, we summarized the data by the median and by the variance of several summaries of the data. For a given summary statistic, a bottleneck will tend to change the median value, and increase the variance, of the statistic relative to the standard neutral model. Using coalescent simulations, we therefore estimated P(Median(X) ≤ Median(X)obs) and P(V(X) ≥ V(X)obs) under each of the bottleneck models. The estimated P-values for each bottleneck model are shown in Table 4. As 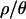 moves from 0 to 10, fewer and fewer of these multilocus summaries are significant at the 5% level. With
moves from 0 to 10, fewer and fewer of these multilocus summaries are significant at the 5% level. With 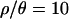 , the only outliers are a median Tajima's D, which is too low, and an overly large variance of H. It is worth noting that a correction for multiple tests is appropriate in interpreting Table 4, as none of the test statistics are independent. A Bonferroni correction would result in an adjusted rejection region of
, the only outliers are a median Tajima's D, which is too low, and an overly large variance of H. It is worth noting that a correction for multiple tests is appropriate in interpreting Table 4, as none of the test statistics are independent. A Bonferroni correction would result in an adjusted rejection region of 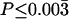 , suggesting no significant departures from the bottleneck model assuming
, suggesting no significant departures from the bottleneck model assuming 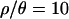 (the point estimate from the Zimbabwe sample). Additionally, the MAP estimates obtained with
(the point estimate from the Zimbabwe sample). Additionally, the MAP estimates obtained with 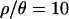 do an excellent job of predicting the median observed values of all summaries of the data except for Tajima's D (supplemental Table A at http://www.genetics.org/supplemental/).
do an excellent job of predicting the median observed values of all summaries of the data except for Tajima's D (supplemental Table A at http://www.genetics.org/supplemental/).
TABLE 4.
P-values for multilocus summaries of the data, evaluated under four different bottleneck models and accounting for uncertainty in estimates of bottleneck parameters

|
||||
|---|---|---|---|---|
| Statistic | 0 | 1 | 7 | 10 |
| M(S) | 0.29 | 0.53 | 0.67 | 0.65 |
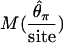 |
0.21 | 0.39 | 0.35 | 0.30 |
| M(K) | 0.32 | 0.61 | 0.96 | 0.96 |
| M(Kd) | 0.03 | 0.09 | 0.31 | 0.32 |
| M(Hfw) | 0 | 6.2 × 10−4 | 0.28 | 0.46 |
| M(H) | 0 | 4 × 10−5 | 0.16 | 0.32 |
| M(D) | 0.44 | 0.32 | 0.02 | 0.01 |
| V(S) | 0.77 | 0.68 | 0.18 | 0.12 |
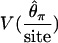 |
0.73 | 0.74 | 0.49 | 0.43 |
| V(K) | 0.24 | 0.30 | 0.19 | 0.20 |
| V(Kd) | 8 × 10−6 | 4.3 × 10−4 | 0.04 | 0.05 |
| V(Hfw) | 0.29 | 0.20 | 0.07 | 0.06 |
| V(H) | 0 | 8.8 × 10−5 | 3 × 10−3 | 5 × 10−3 |
| V(D) | 3.8 × 10−3 | 0.02 | 0.13 | 0.13 |
| F0 | 0.01 | 0.04 | 0.31 | 0.35 |
M refers to the median value and V to the variance, of the summary statistic, calculated across loci. F0 refers to the probability of observing greater ≥14 of 115 loci with zero segregating sites. The P-values are not corrected for multiple tests, and the tests are not independent. A Bonferroni correction would result in a significance level of  .
.
Individual loci:
The distribution of P-values among loci is also informative about the fit of the null model to the data (e.g., Przeworski et al. 2001; Orengo and Aguade 2004; Thornton and Long 2005). When data consist of a large number of independent samples from the null model, the distribution of P-values for a continuous summary statistic should be Uniform(0, 1). Figure 5 compares the distribution of P-values evaluated under the SNM to those obtained under the bottleneck model with 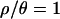 and 10. When two-sided P-values are obtained under the standard neutral model for the 115 loci, the modes of the distributions of P-values are shifted to the left, visually indicating a poor fit to the null model. In contrast, the distributions are much more uniform when evaluated by posterior predictive simulations from the bottleneck model.
and 10. When two-sided P-values are obtained under the standard neutral model for the 115 loci, the modes of the distributions of P-values are shifted to the left, visually indicating a poor fit to the null model. In contrast, the distributions are much more uniform when evaluated by posterior predictive simulations from the bottleneck model.
Figure 5.

Distributions of P-values for the observed data under equilibrium and bottleneck models. Shown are the distributions of two-sided P-values for four summary statistics. The P-values were obtained for both the standard neutral model (solid bars) and a bottleneck model described by a posterior distribution conditional on the data (shaded bars). The value 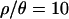 is the maximum a posteriori estimate based on the Zimbabwe data (Table 3).
is the maximum a posteriori estimate based on the Zimbabwe data (Table 3).
The distributions of P-values in Figure 5 provide a visual means to evaluate the fit of a model to data. The fit can be quantified by fitting a Beta(a, b) density to the observed distribution of P-values using the fitdistr routines in the MASS (Venables and Ripley 1999) library for R. If the data are consistent with the null model the corresponding distribution of P-values should be best fit by a uniform distribution, i.e., Beta(1, 1). However, if there are too many outliers, the distribution of two-sided P-values will be significantly left skewed, corresponding to a Beta(a < 1, b > 1) density. The estimates of the parameters of the Beta density are in supplemental Table B at http://www.genetics.org/supplemental/.
When the P-values are evaluated under the inferred bottleneck model, they are much closer to being uniformly distributed (Figure 5 and supplemental Table B at http://www.genetics.org/supplemental/). If we assume that the point estimates of the parameters of the Beta density are normally distributed, then the standard errors in supplemental Table B can be used to obtain confidence intervals. Point estimates significantly different from Beta(1, 1) are indicated in supplemental Table B. Even if the assumption about the distribution of the estimator does not hold, it is clear from supplemental Table B that the distribution of P-values under a bottleneck model is closer to Beta(1, 1) than that under the SNM for a majority of the summary statistics (Figure 5). We found no outliers when  , the most likely estimate of the recombination rate.
, the most likely estimate of the recombination rate.
The distribution of two-sided P-values was also used to identify outlier loci, i.e., loci incompatible with the bottleneck model. The FDR (Storey 2002) was used to correct for multiple tests wherever possible; otherwise a Bonferroni correction was applied. The number of loci that are significant outliers after correcting for multiple tests is shown in Table 5. The pertinent feature of Table 5 is that, for all values of 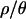 examined, the number of outlier loci is drastically reduced when P-values are obtained using the bottleneck as the demographic null model, compared to using the SNM as the null. In fact, we found no outliers when using our inferred bottleneck parameters (with
examined, the number of outlier loci is drastically reduced when P-values are obtained using the bottleneck as the demographic null model, compared to using the SNM as the null. In fact, we found no outliers when using our inferred bottleneck parameters (with 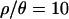 ) as the null model.
) as the null model.
TABLE 5.
Number of outlier loci under standard neutral and bottleneck models
|
D
|
H
|
Hfw
|
K
|
Kd
|
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNM
|
BN
|
SNM
|
BN
|
SNM
|
BN
|
SNM
|
BN
|
SNM
|
BN
|
|||||||||||
 |
B | S | B | S | B | S | B | S | B | S | B | S | B | S | B | S | B | S | B | S |
| 10 | 81 | 78 | 0 | 0 | 28 | 28 | 0 | 0 | 0 | 0 | 0 | 0 | 110 | 106 | 0 | 0 | 27* | 27* | 0 | 0 |
| 7 | 80 | 78 | 12 | 0 | 27 | 25 | 0 | 1 | 0 | 0 | 0 | 0 | 106 | 101 | 0 | 0 | 24* | 24* | 0 | 0 |
| 1 | 55 | 28 | 0 | 0 | 13 | 11 | 0 | 0 | 0 | 0 | 0 | 0 | 55 | 55 | 0 | 0 | 14* | 115 | 0 | 0 |
| 0 | 1 | 1 | 0 | 0 | 7 | 1 | 2 | 2 | 0 | 0 | 0 | 0 | 39 | 30 | 0 | 0 | 57 | 57 | 0 | 0 |
Outlier loci were determined by the calculation of two-sided P-values and then by controlling the false discovery rate (FDR) at 0.05. The FDR method implemented in qvalue (Storey 2002) can use either a bootstrapping or a smoothing method while estimating parameters. The results of both methods are presented here (B and S, respectively). SNM, standard neutral model; BN, bottleneck. The bottlenecks are simulated from the joint posterior distributions obtained with the values of 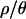 indicated. In some cases, the distribution of P-values is too skewed, resulting in a negative point estimate of the fraction of P-values that come from the null model. In this case, a Bonferroni correction is applied instead, reporting the number of loci with a P-value of ≤0.05/115. Such values are marked with an asterisk.
indicated. In some cases, the distribution of P-values is too skewed, resulting in a negative point estimate of the fraction of P-values that come from the null model. In this case, a Bonferroni correction is applied instead, reporting the number of loci with a P-value of ≤0.05/115. Such values are marked with an asterisk.
DISCUSSION
Timing of a bottleneck in Europe:
D. melanogaster is believed to have an African origin, having colonized the rest of the world only recently. The evidence for this comes primarily from geological evidence that favorable conditions for the migration of the species from Africa into Europe existed 6000–10,000 years ago (reviewed in Lachaise et al. 1988). However, our estimates of the time of a putative population crash in the history of the Netherlands population (∼16,000 years ago) have credibility intervals that barely overlap with this range (Figure 4). Estimating this time relies on an estimate of the effective size (Ne), which in turn relies on several other unknown parameters (notably, the number of generations per year, the ratio of males to females in the population, and the mutation rate). On the basis of best guesses of these parameters (Andolfatto and Przeworski 2000; Charlesworth 2001), the effective population size—and thus the time estimate—is unlikely to be much smaller but could easily be severalfold larger. Interestingly, we show that estimates of the time of the bottleneck obtained assuming little or no recombination suggest radically older bottlenecks, with a lower 2.5% credibility limit >10,000 years ago, highlighting the importance of accounting for recombination in our inference procedure.
Baudry et al. (2004) recently proposed an estimate of a bottleneck for non-African samples occurring ∼6400 years ago. Their estimate does not assume an explicit number for Ne, but does assume a star-like genealogy, which is approximately correct for a very severe bottleneck, if considering the number of derived mutations specific to non-African samples. Their estimate of the timing of the bottleneck is much lower than any of those obtained here (Figure 4). However, their estimate likely corresponds to tr, the time at which Ne has recovered, rather than to tb, the time of the bottleneck (Figure 1). Moving backward in time, the rate of coalescence increases by 1/f at time tr (and quickly approaches 1/f when modeling exponential recovery from the bottleneck). Therefore, when the bottleneck is severe, lineages leading to private mutations in derived population samples are more likely to have reached common ancestors at times closer to tr than to tb. Thus, our estimate of tr (∼3000 years ago, with 95% credibility interval 290–7400 years ago) and td (∼16,000 years ago) is consistent with the estimate of Baudry et al. (2004) .
The fit of a bottleneck to multilocus summaries of the data:
Given the evidence for a recent and severe bottleneck in the history of non-African populations of D. melanogaster, we can address the question of how well our inferred bottleneck model predicts the data overall. In this study, we chose to explore the predictive power of the posterior distributions to explain several statistical summaries (medians and variances) of the 115-locus data set. Similar approaches were taken by Glinka et al. (2003) and Haddrill et al. (2005b); however, in those cases bottleneck parameters were not estimated from the data but were illustrative explorations of some possible parameter values. Our approach is more similar to that of Ometto et al. (2005), with the difference that our estimates of P-values take into account our uncertainty in parameter estimates rather than relying on point estimates.
The multilocus posterior predictive simulations suggest that a population bottleneck can account for both the median and the variance of most summaries of the 115-locus data set (Table 4). The degree to which this holds depends on the amount of recombination assumed in the simulations. When inference was made assuming no recombination (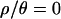 ), 6 of 15 summaries of the multilocus data set had observed values incompatible with the bottleneck model at the 5% significance level. In particular, when
), 6 of 15 summaries of the multilocus data set had observed values incompatible with the bottleneck model at the 5% significance level. In particular, when 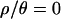 , the observed number of invariant loci (F0) is too large, yet the observed median Tajima's D is compatible. However, under the bottleneck model we inferred using our best estimate of the recombination rate (i.e., using the posterior distribution of bottleneck parameters obtained with
, the observed number of invariant loci (F0) is too large, yet the observed median Tajima's D is compatible. However, under the bottleneck model we inferred using our best estimate of the recombination rate (i.e., using the posterior distribution of bottleneck parameters obtained with 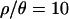 ), we find that of the 16 summaries of the data we investigated, the median D is too low (P = 0.01) and the variance of H too high (P = 0.005, see Table 4).
), we find that of the 16 summaries of the data we investigated, the median D is too low (P = 0.01) and the variance of H too high (P = 0.005, see Table 4).
However, there are at least three reasons that the somewhat poor fit of some aspects of the frequency spectrum of polymorphisms to our bottleneck model does not lead us to necessarily invoke recent positive selection. First, since Tajima's D and the variance of Hfw are just 2 of 16 correlated summaries investigated, it is appropriate to apply a correction for multiple tests. In fact, neither statistic is unusual after applying a Bonferroni correction (Table 4 legend) and we must conclude that we cannot reject a simple bottleneck model.
Second, we have assumed in our model that the ancestral population was at equilibrium (among other assumptions, see below). A recent bottleneck is expected to make the mean and median of Tajima's D more positive, relative to the standard neutral model. However, if the ancestral population was not at equilibrium, the Tajima's D of the bottlenecked population would become more positive relative to the expectation of D in the ancestral population. An example of this is shown in Figure 6, which plots the distribution of D for two cases. In Figure 6a, we show the distribution of D-values for a derived population that is recently bottlenecked from an ancestral population at equilibrium, corresponding to our modeling assumptions. In Figure 6b, we show the distribution of D-values for a similarly bottlenecked population derived from an exponentially growing ancestral population. As expected, the mean, the median, and the variance of the summary statistic depend on the demography of the ancestral population. When the bottlenecked population is derived from a population with a negative D (due in this case to population growth), the expectation of D is closer to zero than when the ancestral population is at demographic equilibrium. While these results are intended to be illustrative, they imply that the observed median D-value in the Netherlands would be less unusual if the ancestral population had a negative average Tajima's D, as is indeed observed in the Zimbabwe population (see Glinka et al. 2003; Haddrill et al. 2005b).
Figure 6.


The effect of a bottleneck on Tajima's D in a derived population when the ancestral population is not at demographic equilibrium. Distributions of Tajima's D were obtained from 105 coalescent simulations with n = 12 in both populations, θ = 0.01 per site,  , and 1000 bp. The bottleneck parameters are based on the MAP estimates for each of the marginal distributions inferred when
, and 1000 bp. The bottleneck parameters are based on the MAP estimates for each of the marginal distributions inferred when 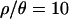 (Figure 2). (a) The derived population is bottlenecked from an ancestor at demographic equilibrium. (b) The growth parameter in the ancestral population was set to give a mean Tajima's D close to the −0.56 observed in the Zimbabwe sample. Vertical lines indicate the median D for each population.
(Figure 2). (a) The derived population is bottlenecked from an ancestor at demographic equilibrium. (b) The growth parameter in the ancestral population was set to give a mean Tajima's D close to the −0.56 observed in the Zimbabwe sample. Vertical lines indicate the median D for each population.
Finally, while it is possible that exponential growth in Africa or recent positive selection in the Netherlands are behind the poor fit of Tajima's D to the bottleneck model, several other possible misspecifications of the model include past bottlenecks or population subdivision in Africa (see Haddrill et al. 2005b), weakly deleterious alleles segregating in both African and non-African populations, frequent positive selection in Africa, and the assumption of infinite sites. Thus positive selection is not a unique alternative to our null model.
The fit of a bottleneck to individual loci:
A second way to view the explanatory power of a bottleneck model more directly bears on the question of how much power we have to detect individual loci that have undergone a recent episode of positive directional selection. A widely used approach to this problem has been to construct a test of the null hypothesis that the data are drawn from a population evolving according to the standard neutral model and to evaluate this hypothesis by coalescent simulation (see Hudson 1993 for a description of one of the most widely applied methods). A genomics-era approach to the problem of detecting loci under selection has been termed hitchhiking mapping (Harr et al. 2002; Schlotterer 2003). This approach relies on the collection of large multilocus data sets and considers loci that fall in the tails of the distribution (as measured by some summary of the data) to be “candidate genes,” by which it is meant that they are the loci most likely to have experienced a recent selective sweep. Rejection of the null hypothesis at outlier loci is commonly cited as evidence for positive selection (e.g., Harr et al. 2002).
A limitation of the hitchhiking mapping approach is that what one considers to be unusual depends strongly on what is assumed to be the norm. A reliance on the tails of the distribution to detect loci subject to recent selection may be problematic if the standard neutral model is violated genome-wide, which appears to be the case in Drosophila species, particularly for non-African populations (e.g., Andolfatto and Przeworski 2000; Wall et al. 2002; Andolfatto and Wall 2003; Haddrill et al. 2005b). In terms of identifying loci that are truly under selection, the false positive rate may be higher than nominal if a genome-wide departure from the assumptions of the SNM is due to a model with a larger variance in possible outcomes than to the standard neutral model. This is precisely the case for a population bottleneck under a wide range of parameters. While some versions of hitchhiking mapping have been claimed to correct for demographic effects by relying on the comparison of multiple populations, simulations suggest that statistics like lnRV (and lnRH) are indeed sensitive to demographic departures from the SNM (see Table 6 in Schlotterer 2002).
To detect outlier loci, we performed simulations from joint posterior distributions of bottleneck parameters to obtain posterior predictive P-values for the observed data from the Netherlands sample. These are analogous to demographically corrected probabilities (e.g., Akey et al. 2004), but take into account uncertainty in the estimates of the demographic parameters. Regardless of the value of 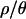 used to estimate bottleneck parameters, the significance of the canonical signals of recent selective sweeps effectively disappears (Table 5), including the predicted excess of low-frequency alleles (Braverman et al. 1995), the excess of high-frequency derived alleles (Fay and Wu 2000), and the reduction both in the number of haplotypes and in haplotype diversity (Strobeck 1987; Fu 1996; Depaulis and Veuille, 1998).
used to estimate bottleneck parameters, the significance of the canonical signals of recent selective sweeps effectively disappears (Table 5), including the predicted excess of low-frequency alleles (Braverman et al. 1995), the excess of high-frequency derived alleles (Fay and Wu 2000), and the reduction both in the number of haplotypes and in haplotype diversity (Strobeck 1987; Fu 1996; Depaulis and Veuille, 1998).
Note that a lack of outlier loci is observed for all values of 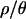 . However, the reasons why there are few outlier loci for both
. However, the reasons why there are few outlier loci for both 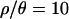 and 0 may be quite different. In the first case, the recombination rates used in the simulations come from estimates in Zimbabwe, and we expect that taking recombination into account should increase the power to detect outlier loci when performing the posterior predictive simulations. Given that the multilocus summaries of the data are well predicted by the bottleneck model for
and 0 may be quite different. In the first case, the recombination rates used in the simulations come from estimates in Zimbabwe, and we expect that taking recombination into account should increase the power to detect outlier loci when performing the posterior predictive simulations. Given that the multilocus summaries of the data are well predicted by the bottleneck model for 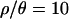 , the lack of outlier loci suggests that this bottleneck model is an adequate null model for the data. On the other hand, while multilocus summaries are poorly predicted by the bottleneck model for
, the lack of outlier loci suggests that this bottleneck model is an adequate null model for the data. On the other hand, while multilocus summaries are poorly predicted by the bottleneck model for 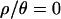 , there are also few outlier loci. This is likely an issue of power; assuming no recombination when data are sampled from recombining regions leads to overly conservative tests (Przeworski et al. 2001), which can result in a lack of power to detect departures from a model that is a relatively poor predictor of the data.
, there are also few outlier loci. This is likely an issue of power; assuming no recombination when data are sampled from recombining regions leads to overly conservative tests (Przeworski et al. 2001), which can result in a lack of power to detect departures from a model that is a relatively poor predictor of the data.
We note that estimating posterior predictive P-values should not be interpreted as a test for positive selection, as this procedure does not explicitly compare the fit of a model of demography vs. a model of demography-plus-selection to the data. Even though we fail to detect a single outlier locus under our bottleneck model, the existence of such outliers would not necessarily implicate positive selection. Indeed, just as for the interpretation of the fit of multilocus summaries of the data above, positive selection is just one of many possible ways that the null model could be misspecified. A formal test for positive selection would have the form of inference under a mixture model where all loci are subject to nonequilibrium demography, and some fraction of the loci (estimated from the data) are also subject to sweeps. A simplified version of this approach was recently applied to maize data (Wright et al. 2005), where sweeps were modeled as a second bottleneck occurring at some loci.
Accuracy of the estimation procedure:
The issue of accuracy of our bottleneck estimates is difficult to address as it depends both on the accuracy of the inference procedure when the null model is true and on the sensitivity of the procedure to misspecification of the null. A thorough exploration of the performance of our method is not computationally feasible and beyond the scope of this study. However, our preliminary simulations suggest that our procedure performs reasonably well when assuming a bottleneck scenario similar to the one we have estimated (see Testing the inference procedure in the supplemental information at http://www.genetics.org/supplemental/). Of more concern is how robust our inference procedure is to assumptions of the model and we have made several simplifying assumptions that are potential misspecifications. Like inferences of selection, inferences of bottleneck parameters may be sensitive to misspecification of the model used.
In particular, the demographic and selective history of the ancestral population may be relevant to parameter inference in derived populations (as discussed above). However, fully accounting for ancestral population history is currently computationally impracticable. Several previous rejections of simple bottleneck models in favor of selection (in various study systems) have assumed an equilibrium ancestral population despite evidence for nonequilibrium (putatively) ancestral populations (Glinka et al. 2003; Akey et al. 2004; Tenaillon et al. 2004; Schmid et al. 2005; Ometto et al. 2005; Stajich and Hahn 2005; Wright et al. 2005). As suggested by our ancestral population growth example (see above), accounting for demographic history of the ancestral population may considerably improve the fit of demographic models to the data, potentially making inferences of selection unnecessary in some of these cases. Despite making the simplifying assumption of an equilibrium ancestor, we find no overwhelming statistical evidence ruling out a recent bottleneck as a plausible and sufficient explanation for variability patterns documented in the Netherlands D. melanogaster population. It is unclear that accounting for ancestral demography (and uncertainty in this demography) will strengthen inferences in favor of selection.
Acknowledgments
The authors thank Doris Bachtrog, Deborah Charlesworth, and Jeff Jensen for critical reading of the manuscript. We thank Josh Shapiro and Chung-I Wu for making available the formula for the H-statistic. K.T. is supported by an Alfred P. Sloan Foundation postdoctoral fellowship in computational molecular biology. P.A. is supported by an Alfred P. Sloan foundation fellowship in computational molecular biology.
References
- Akey, J. M., M. A. Eberle, M. J. Rieder, C. S. Carlson, M. D. Shriver et al., 2004. Population history and natural selection shape patterns of genetic variation in 132 genes. PloS Biol. 2: 1591–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andolfatto, P., 2001. Adaptive hitchhiking effects on genome variability. Curr. Opin. Genet. Dev. 11: 635–641. [DOI] [PubMed] [Google Scholar]
- Andolfatto, P., and M. Przeworski, 2000. A genome-wide departure from the standard neutral model in natural populations of Drosophila. Genetics 156: 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andolfatto, P., and J. D. Wall, 2003. Linkage disequilibrium patterns across a recombination gradient in African Drosophila melanogaster. Genetics 165: 1289–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudry, E., B. Viginier and M. Veuille, 2004. Non-African populations of Drosophila melanogaster have a unique origin. Mol. Biol. Evol. 21: 1482–1491. [DOI] [PubMed] [Google Scholar]
- Beaumont, M. A., W. Zhang and D. J. Balding, 2002. Approximate Bayesian computation in population genetics. Genetics 162: 2025–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begun, D. J., and C. F. Aquadro, 1993. African and North-American populations of Drosophila melanogaster are very different at the DNA level. Nature 365: 548–550. [DOI] [PubMed] [Google Scholar]
- Braverman, J. M., R. R. Hudson, N. L. Kaplan, C. H. Langley and W. Stephan, 1995. The hitchhiking effect on the site frequency-spectrum of DNA polymorphisms. Genetics 140: 783–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth, B., 1996. Background selection and patterns of genetic diversity in Drosophila melanogaster. Genet. Res. 68: 131–149. [DOI] [PubMed] [Google Scholar]
- Charlesworth, B., 2001. The effect of life-history and mode of inheritance on neutral genetic variability. Genet. Res. 77: 153–166. [DOI] [PubMed] [Google Scholar]
- David, J. R., and P. Capy, 1988. Genetic variation of Drosophila melanogaster natural populations. Trends Genet. 4: 106–111. [DOI] [PubMed] [Google Scholar]
- Depaulis, F., and M. Veuille, 1998. Neutrality tests based on the distribution of haplotypes under an infinite-site model. Mol. Biol. Evol. 15(12): 1788–1790. [DOI] [PubMed] [Google Scholar]
- Ewens, W., 2004. Mathematical Population Genetics I. Theoretical Introduction, Ed. 2. Springer-Verlag, Berlin/Heidelberg, Germany/New York.
- Fay, J., and C.-I Wu, 2000. Hitchhiking under positive Darwinian selection. Genetics 155: 1405–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y.-X., 1996. New statistical tests of neutrality for DNA samples from a population. Genetics 146: 557–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y. X., and W. H. Li, 1993. Statistical tests of neutrality of mutations. Genetics 133: 693–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman, A., J. B. Carlin, H. S. Stern and D. B. Rubin, 2003. Bayesian Data Analysis, Ed. 2. Chapman & Hall/CRC Press, London/New York/Cleveland/Boca Raton, FL.
- Glinka, S., L. Ometto, S. Mousset, W. Stephan and D. D. Lorenzo, 2003. Demography and natural selection have shaped genetic variation in Drosophila melanogaster: a multilocus approach. Genetics 165: 1269–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddrill, P., B. Charlesworth, D. Halligan and P. Andolfatto, 2005. a Patterns of intron sequence evolution in Drosophila are dependent upon length and GC content. Genome Biol. 6: R67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddrill, P., K. Thornton, P. Andolfatto and B. Charlesworth, 2005. b Multilocus patterns of nucleotide variability and the demographic and selection history of Drosophila melanogaster populations. Genome Res. 15: 790–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harr, B., M. Kauer and C. Schlotterer, 2002. Hitchhiking mapping: a population-based fine-mapping strategy for adaptive mutations in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 99(20): 12949–12954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., 1983. Properties of a neutral allele model with intragenic recombination. Theor. Popul. Biol. 23: 183–201. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., 1993. The how and why of generating gene genealogies, in Mechanisms of Molecular Evolution, edited by A. G. Clark and N. Takahata. Sinauer Press, Sunderland, MA.
- Hudson, R. R., 2002. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18: 337–338. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., M. Kreitman and M. Aguade, 1987. A test of neutral molecular evolution based on nucleotide data. Genetics 116: 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauer, M. O., B. Zangerl, D. Dieringer and C. Schlotterer, 2002. Chromosomal patterns of microsatellite variability contrast sharply in African and non-African populations of Drosophila melanogaster. Genetics 160: 247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachaise, D., M.-L. Cairou, J. R. David, F. Lemeunier, L. Tsacas et al., 1988. Historical biogeography of the Drosophila melanogaster species subgroup. Evol. Biol. 22: 159–226. [Google Scholar]
- Lazzaro, B. P., and A. G. Clark, 2003. Molecular population genetics of inducible antibacterial peptide genes in Drosophila melanogaster. Mol. Biol. Evol. 20: 914–923. [DOI] [PubMed] [Google Scholar]
- Li, W. H., 1997. Molecular Evolution. Sinauer Associates, Sunderland, MA.
- Marjoram, P., J. Molitor, V. Plagnol and S. Tavare, 2003. Markov chain Monte Carlo without likelihoods. Proc. Natl. Acad. Sci. USA 100: 15324–15328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ometto, L., S. Glinka, D. DeLorenzo and W. Stephan, 2005. Inferring the effects of demography and selection on Drosophila melanogaster populations from a chromosome-wide scan of DNA variation. Mol. Biol. Evol. 22: 2119–2130.15987874 [Google Scholar]
- Orengo, D., and M. Aguade, 2004. Detecting the footprint of positive selection in a European population of Drosophila melanogaster: multilocus pattem of variation and distance to coding regions. Genetics 167: 1759–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard, J. K., M. T. Seielstad, A. Perez-Lezaun and M. W. Feldman, 1999. Population growth of human Y chromosomes: a study of Y chromosome microsatellites. Mol. Biol. Evol. 16: 1791–1798. [DOI] [PubMed] [Google Scholar]
- Przeworski, M., 2002. The signature of positive selection at randomly chosen loci. Genetics 160: 1179–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przeworski, M., 2003. Estimating the time since the fixation of a beneficial allele. Genetics 164: 1667–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przeworski, M., J. D. Wall and P. Andolfatto, 2001. Recombination and the frequency spectrum in Drosophila melanogaster and Drosophila simulans. Mol. Biol. Evol. 18: 291–298. [DOI] [PubMed] [Google Scholar]
- R Development Core Team, 2004. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. (http://www.R-project.org).
- Schlotterer, C., 2002. A microsatellite-based multilocus screen for the identification of local selective sweeps. Genetics 160: 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlotterer, C., 2003. Hitchhiking mapping–functional genomics from the population genetics perspective. Trends Genet. –38.19:32. [DOI] [PubMed] [Google Scholar]
- Schmid, K. J., S. Ramos-Osnins, H. Ringys-Beckstein, B. Weisshaar and T. Mitchell-Olds, 2005. A multilocus sequence survey in Arabidopsis thaliana reveals a genome-wide departure from a neutral model of DNA sequence polymorphism. Genetics 169: 1601–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stajich, J. E., and M. W. Hahn, 2005. Disentangling the effects of demography and selection in human history. Mol. Biol. Evol. 22: 63–73. [DOI] [PubMed] [Google Scholar]
- Storey, J. D., 2002. A direct approach to false discovery rates. J. R. Stat. Soc. Ser. B 64: 479–498. [Google Scholar]
- Strobeck, C., 1987. Average number of nucleotide differences in a sample from a single subpopulation: a test for population subdivision. Genetics 117: 149–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenaillon, M. I., J. U'Ren, O. Tenaillon and B. S. Gaut, 2004. Selection versus demography: A multilocus investigation of the domestication process in maize. Mol. Biol. Evol. 21(7): 1214–1225. [DOI] [PubMed] [Google Scholar]
- Thornton, K., 2003. libsequence: a C++ class library for evolutionary genetic analysis. Bioinformatics 19: 2325–2327. [DOI] [PubMed] [Google Scholar]
- Thornton, K., and M. Long, 2005. Excess of amino acid substitutions relative to polymorphism between X–linked duplications in Drosophila melanogaster. Mol. Biol. Evol. 22: 273–284. [DOI] [PubMed] [Google Scholar]
- Venables, W. N., and B. D. Ripley, 1999. Modern Applied Statistics with S-Plus, Ed. 3. Springer-Verlag, Berlin/Heidelberg, Germany/New York.
- Wall, J. D., P. Andolfatto and M. Przeworski, 2002. Testing models of selection and demography in Drosophila simulans. Genetics 162: 203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson, G. A., 1975. Number of segregating sites in genetic models without recombination. Theor. Popul. Biol. 7: 256–276. [DOI] [PubMed] [Google Scholar]
- Wright, S. I., I. V. Bi, S. G. Schroeder, M. Yamasaki, J. F. Doebley et al., 2005. The effects of artificial selection on the maize genome. Science 308: 1310–1314. [DOI] [PubMed] [Google Scholar]