

Abstract
Clostridium perfringens is an important human and animal pathogen that causes a number of diseases that vary in their etiology and severity. Differences between strains regarding toxin gene composition and toxin production partly explain why some strains cause radically different diseases than others. However, they do not provide a complete explanation. The purpose of this study was to determine if there is a phylogenetic component that explains the variance in C. perfringens strain virulence by assessing patterns of genetic polymorphism in genes (colA gyrA, plc, pfoS, and rplL) that form part of the core genome in 248 type A strains. We found that purifying selection plays a central role in shaping the patterns of nucleotide substitution and polymorphism in both housekeeping and virulence genes. In contrast, recombination was found to be a significant factor only for the virulence genes plc and colA and the housekeeping gene gyrA. Finally, we found that the strains grouped into five distinct evolutionary lineages that show evidence of host adaptation and the early stages of speciation. The discovery of these previously unknown lineages and their association with distinct disease presentations carries important implications for human and veterinary clostridial disease epidemiology and provides important insights into the pathways through which virulence has evolved in C. perfringens.
CLOSTRIDIUM perfringens is a spore-forming, low G + C gram-positive anaerobe that is found in soil and in the gastrointestinal tract of vertebrates (Johnson and Gerding 1997; McClane 1997). Pathogenic strains of C. perfringens cause a variety of human and animal diseases including food poisoning, antibiotic-associated diarrhea, gas gangrene, necrotic enteritis, necrotizing colitis, sudden death syndrome, and enterotoxemia (Lindsay 1996; Songer 1996; Gibert et al. 1997; Meer et al. 1997; Rood 1998; Brynestad and Granum 2002; Bos et al. 2005). C. perfringens causes this wide array of diseases as a result of being able to produce at least 15 toxins (Rood 1998; Petit et al. 1999; Smedley et al. 2004). Many of these toxins lie on plasmids, whereas others are found in the C. perfringens chromosome. The presence/absence of these toxins forms the basis of the C. perfringens typing scheme (reviewed in Petit et al. 1999).
The illnesses caused by C. perfringens that are familiar to most persons are food poisoning and gas gangrene. Clostridial food poisoning causes abdominal discomfort and diarrhea within hours of consumption (Johnson and Gerding 1997; McClane 1997; Meer et al. 1997; Brynestad and Granum 2002). Most persons suffering from this type of food poisoning recover within 24–48 hr. However, if left untreated, severe dehydration or death may occur in infants, the elderly, the debilitated, or the immune compromised. Gas gangrene (also known as clostridial myonecrosis) is an extremely serious disease, requiring both antibiotic and surgical treatment (Present et al. 1990; Bryant and Stevens 1997; Bryant 2003). It is usually acquired when wounds become exposed to soil or other environmental material. If left untreated, the disease results in extensive tissue necrosis requiring surgical intervention that often involves limb amputation; unsuccessful treatment results in systemic shock and death (Present et al. 1990; Bryant and Stevens 1997; Bryant 2003).
It is not well understood why C. perfringens produces illnesses that vary so greatly in severity. Clearly, the presence of certain toxins is one explanation, but it falls short in many instances. For example, strains that cause food poisoning may differ from those that cause gas gangrene only by the presence of an enterotoxin gene in the former, yet food poisoning strains have never been found to cause gas gangrene. One reason may be that the strains differ in their genetic composition and modes of gene expression. For example, the presence of the enterotoxin gene is associated with food poisoning in addition to non-food-borne antibiotic-associated diarrhea and sporadic diarrhea (AAD–SD) (Sarker et al. 1999, 2000; Fisher et al. 2005). The genetic background of food poisoning strains differs from that of the AAD–SD strains, as revealed by the existence of the enterotoxin gene on the chromosome of food poisoning strains vs. the plasmid in the case of AAD–SD strains (Collie and McClane 1998; Sarker et al. 1999, 2000; Fisher et al. 2005). The fact that a strain's genetic background is associated with a specific type of disease suggests that evolutionary divergence has driven the phenotypic (especially the disease-causing) differences among C. perfringens strains. The purpose of this study was to investigate this question through the analysis of genetic variability patterns across several genes that make up the C. perfringens “core genome.”
The core genome consists of the set of genes that are present in all members of a species, implying that they are required for essential cellular functions (Dykhuizen and Green 1991; Lan and Reeves 2001; Wertz et al. 2003). Because these genes are expected to be highly conserved, the evolution of the core genome is expected to be homogeneous across all of its component genes. In this study, we assessed patterns of genetic polymorphism across both housekeeping and virulence loci. This approach allowed us to investigate whether or not the core genome evolves homogeneously or if the evolutionary processes affecting some core genes, such as housekeeping genes, are substantially different from those affecting other core genes, such as virulence genes. While virulence genes are not usually considered to be part of the core genome, there are exceptions. In this study, we examined two such exceptions: the plc gene, which encodes the α-toxin, a phospholipase C that is also a primary determinant of C. perfringens virulence (Long and Truscott 1976; Baba et al. 1992; Awad et al. 1995; Rood 1998); and the colA gene, which encodes the κ-toxin, a collagenase that facilitates tissue necrosis (Bryant and Stevens 1997; Rood 1998; Petit et al. 1999).
MATERIALS AND METHODS
We obtained 247 C. perfringens strains isolated from various human clinical (151 strains), veterinary (46 strains), and retail food (50 strains) sources. Human clinical strains were acquired from the Loyola University Medical Center, and veterinary strains were acquired from the University of Illinois Veterinary Diagnostic Laboratory. Retail food strains were isolated at the U.S. Department of Agriculture–Agricultural Research Service (USDA–ARS), National Center for Agricultural Utilization Research from various meats (beef, chicken, pork, and venison) using the method of Rhodehamel and Harmon (2001). This method entails selective anaerobic isolation of C. perfringens colonies on tryptose–sulfite–cycloserine (TSC) agar containing egg yolk emulsion (50%) at 37°, in which colonies are presumed to be C. perfringens if they form a black pigment. All strains (human, veterinary, and food) were verified as being C. perfringens through the use of a multiplexed polymerase chain reaction (PCR) amplification of C. perfringens-specific toxin genes (Meer and Songer 1997; Bueschel et al. 2003). This test was also used to classify strains into their various toxinotypes (A, B, C, D, or E) (Petit et al. 1999), which is the most common clinical system for subtyping C. perfringens pathogenic strains. All strains were found to be type A and have been deposited into the USDA–ARS Culture Collection (http://nrrl.ncaur.usda.gov), where they are available free of charge.
From each strain, we obtained the nucleotide sequences of the complete plc coding region [1194 aligned base pairs (bp)] and the plc 3′ noncoding flanking region (FR) (197 aligned bp), as well as the partial coding sequence of the central portion of the colA gene (aligned 924 bp). In addition, we obtained nucleotide sequence data from several housekeeping genes located in different parts of the C. perfringens genome. The housekeeping genes included: the gyrase subunit A gene (gyrA) (918 aligned bp), the regulatory protein gene pfoS (519 aligned bp), the pfoS 5′ FR (246 aligned bp), the 50S ribosomal protein gene rplL (363 aligned bp), and the rplL 5′ FR (318 aligned bp). In total, for each strain, we analyzed 4679 aligned bp, of which 2315 bases represented virulence genes (plc and colA) and 2364 bases represented housekeeping genes and noncoding regions. In addition, we included homologous nucleotide sequence data from the complete genome of C. perfringens strain 13 (Shimizu et al. 2002). With these completed genome data, the total number of strains analyzed was 248.
PCR amplifications were performed with Platinum Taq DNA Polymerase High Fidelity (Invitrogen Life Technologies, Carlsbad, CA) under standard reaction conditions. The oligonucleotide primer sequences and PCR annealing temperatures are listed in Table 1. Amplification products were purified using Montage PCR Cleanup filter plates (Millipore, Billerica, MA). Sequencing reactions were conducted using the ABI BigDye version 3.0 sequencing kit (Applied Biosystems, Foster City, CA) following the manufacturer's suggested protocol but at one-fourth the recommended volume. Reaction products were purified via ethanol precipitation and run on an ABI3730 genetic analyzer (Applied Biosystems). DNA sequences were edited using Sequencher version 4.1.2 (Gene Codes, Ann Arbor, MI) and added to an alignment containing the genes extracted from the published genome (strain 13). The sequences have been deposited in GenBank under accession nos. DQ183189–DQ184423. In addition, during the course of our studies we found that the strains formed distinct groups. The taxonomic identity of these unique groups was investigated by amplifying and sequencing a fragment of the 16S ribosomal RNA (rRNA) gene using the primers and PCR protocol in Rooney et al. (2005). The sequencing reactions were performed as described above.
TABLE 1.
Primer sequences and annealing temperatures for genes and flanking regions
| Locus | Primers (5′–3′)a | Annealing temperature |
|---|---|---|
| colA | TAG GAA CAA AGG CGC AAG AT | 55° |
| TTC TCC TTG TCC CCA CAT TC | ||
| gyrA | GCT TGT TGA CGG ACA TGG TA | 54° |
| AGA TTG CAG CAG CTT GCT TT | ||
| plc | ATT TCA GTG CAA GTG TTA ATC | 58° |
| CAT CAA TTC CAA CTT CTC C | ||
| rplL | GCC ACG TCT TTG ACT TTT GC | 55° |
| CGA TTT CGC TAA GGA AAA CAA | ||
| pfoS | CGG GTA TAG GCA TAC AAA AGG A | 52° |
| GTG CAG TTG CAA CCA CTG TT |
For each locus, the forward primer is listed above the reverse primer.
Analyses of nucleotide sequence polymorphism and recombination were conducted using two computer programs. The program DnaSP 4.0 (Rozas et al. 2003) was used to calculate estimates of nucleotide diversity (π) and the number of segregating (polymorphic) sites (S). This program was also used to conduct Tajima's test for neutrality (Tajima 1989). The significance of Tajima's test was assessed through coalescent simulation (10,000 replications) using estimated mutation and recombination rates. In addition, the degree of codon usage bias for each gene was assessed through computation of the scaled χ2-statistic (66), using DnaSP version 4.0. Analyses of recombination were performed using the methods of Hudson and Kaplan (1985) and McVean et al. (2002). The Hudson and Kaplan method entails the estimation of the minimum number of recombination events per sequence (RM), while the McVean et al. method employs a likelihood permutation test (LPT) to assess the probability of recombination. DnaSP 4.0 was used to estimate RM and to infer nucleotides involved in recombination events. The program LDhat (McVean et al. 2002) was used to conduct the LPT and to compute estimates of Watterson's θ (Watterson 1975) and the recombination parameter ρ (McVean et al. 2002).
Analyses of positive Darwinian selection at individual codon sites were performed using two methods. The first was the single-likelihood ancestor counting (SLAC) method (Kosakovsky Pond and Frost 2005), which uses a likelihood method to infer ancestral codons and then parsimony to infer subsequent changes. The second method used was the full-likelihood method for detecting position selection at single amino acid sites (Nielsen and Yang 1998; Yang and Nielsen 2002; Yang et al. 2005). Under this method, a likelihood-ratio test (LRT) is first used to assess the probability of positive selection. If the test is significant, the Bayes empirical Bayes (BEB) procedure is then used to calculate the posterior probability that a given codon is under positive Darwinian selection (Yang et al. 2005). Under this method, two models (M2a vs. M1a and/or M7a vs. M8a) are compared using a likelihood-ratio test to assess the probability of positive selection. If positive selection is confirmed, a BEB analysis is conducted to determine which sites are significantly influenced by positive Darwinian selection.
Phylogenetic analyses were conducted using the computer program MEGA3 (Kumar et al. 2004). Kimura's (1980) two-parameter distances were computed and used to generate phylogenetic trees with the neighbor-joining method (Saitou and Nei 1987). The statistical reliability of internal branches was assessed from 1000 bootstrap pseudoreplicates.
RESULTS
Analysis of genetic variability and recombination:
The results from polymorphism analyses are summarized in Tables 2 and 3. Most protein-coding loci (excluding noncoding flanking sequence) displayed moderate levels of polymorphism, with average π-values (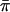 ) for the entire gene or FR ranging from 0.007 to 0.053 (Table 2). When
) for the entire gene or FR ranging from 0.007 to 0.053 (Table 2). When 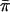 was examined at nonsynonymous (
was examined at nonsynonymous (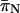 ) and synonymous (
) and synonymous (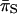 ) sites, we found that
) sites, we found that 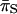 was greater than
was greater than 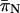 at all loci (Table 3) and that the former ranged from 0.019 to 0.047 while the latter ranged from 0.001 to 0.005 (Table 3). In addition, we found that FRs possessed higher levels of variability than the respective coding regions that they flanked (Table 2). In contrast, the rplL gene and FR present an interesting exception to these patterns. In this case, estimates of
at all loci (Table 3) and that the former ranged from 0.019 to 0.047 while the latter ranged from 0.001 to 0.005 (Table 3). In addition, we found that FRs possessed higher levels of variability than the respective coding regions that they flanked (Table 2). In contrast, the rplL gene and FR present an interesting exception to these patterns. In this case, estimates of 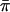 (= 0.007) were identical for the gene and the FR. When the estimates of
(= 0.007) were identical for the gene and the FR. When the estimates of 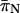 and
and 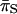 for the coding region were compared to the estimate of
for the coding region were compared to the estimate of 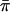 for the FR, we found that
for the FR, we found that 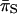 was about two times higher. A higher level of
was about two times higher. A higher level of  vs. FR or intergenic
vs. FR or intergenic 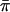 could result due to purifying selection acting upon regulatory elements within the FR (Hughes and Friedman 2004). In fact, regulatory elements are found within the rplL FR in Proteobacteria (e.g., Escherichia coli; Morgan et al. 1984), Spirochaetes (e.g., Borrelia burgdorferi; Alekshun et al. 1997), and Firmicutes [e.g., Bacillus subtilis (Boor et al. 1995) and Staphylococcus aureus (Aboshkiwa et al. 1995)]. While we do not have experimental evidence for the presence of regulatory elements in this region of the C. perfringens genome, it is likely because the operon in which rplL is found is under intense purifying selection and is one of the most highly conserved operons in bacteria (Dabbs 1984). In fact, relative to what was observed at other loci, we found a low number of alleles and segregating sites at both the rplL coding region and FR (Table 2), indicating that this region of the C. perfringens genome is highly constrained. A selective sweep could have potentially produced these patterns as well. However, given that this region is so highly conserved across long evolutionary time periods, the most logical explanation is strong purifying selection.
could result due to purifying selection acting upon regulatory elements within the FR (Hughes and Friedman 2004). In fact, regulatory elements are found within the rplL FR in Proteobacteria (e.g., Escherichia coli; Morgan et al. 1984), Spirochaetes (e.g., Borrelia burgdorferi; Alekshun et al. 1997), and Firmicutes [e.g., Bacillus subtilis (Boor et al. 1995) and Staphylococcus aureus (Aboshkiwa et al. 1995)]. While we do not have experimental evidence for the presence of regulatory elements in this region of the C. perfringens genome, it is likely because the operon in which rplL is found is under intense purifying selection and is one of the most highly conserved operons in bacteria (Dabbs 1984). In fact, relative to what was observed at other loci, we found a low number of alleles and segregating sites at both the rplL coding region and FR (Table 2), indicating that this region of the C. perfringens genome is highly constrained. A selective sweep could have potentially produced these patterns as well. However, given that this region is so highly conserved across long evolutionary time periods, the most logical explanation is strong purifying selection.
TABLE 2.
Polymorphism and recombination parameters
| Locus | l | h | 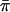 |
S | D | RM | θ | ρ | ρ/θ | PLPT |
|---|---|---|---|---|---|---|---|---|---|---|
| pfoS | 519 | 41 | 0.014 | 40 | −0.797 | 7 | 9.349 | 42.126 | 4.506 | 0.345 |
| pfoS FR | 246 | 46 | 0.053 | 52 | −0.614 | 9 | 11.832 | 2.01 | 0.17 | 0.892 |
| rplL | 363 | 9 | 0.007 | 12 | −1.876 | 0 | 4.415 | 0 | 0 | 0.937 |
| rplL FR | 318 | 12 | 0.007 | 11 | −1.478 | 0 | 3.643 | 15.578 | 4.276 | 0.042 |
| plc | 1194 | 104 | 0.014 | 131 | −1.808 | 21 | 25.111 | 111.558 | 4.443 | 0 |
| plc FR | 197 | 39 | 0.002 | 24 | −1.453 | 2 | 7.096 | 10.302 | 1.452 | 0.009 |
| colA | 924 | 94 | 0.011 | 103 | −1.714 | 15 | 20.136 | 65.65 | 3.26 | 0 |
| gyrA | 918 | 49 | 0.007 | 44 | −1.08 | 7 | 9.868 | 37 | 3.75 | 0.012 |
| All (concatenated) | 4679 | 180 | 0.009 | 417 | −1.434 | 67 | 72.303 | 230.461 | 3.187 | 0 |
| No toxin locia | 2364 | 118 | 0.008 | 159 | −1.278 | 27 | 29.615 | 83.236 | 2.181 | 0 |
| Toxin loci | 2315 | 164 | 0.011 | 258 | −1.474 | 40 | 45.47 | 146.794 | 3.228 | 0 |
l, number of nucleotides; h, number of haplotypes; D, Tajima's D calculated from the total number of segregating sites; PLPT, P-value for the LPT. All other abbreviations are defined in materials and methods.
All loci are concatenated except for toxin loci (plc, plc FR, and colA).
TABLE 3.
Estimates of protein-coding region variability and codon usage bias
| Locus | 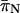 |
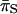 |
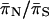 |
GC3a | Scaled χ2 | Scaled χ2-corrected |
|---|---|---|---|---|---|---|
| pfoS | 0.004 | 0.042 | 0.095 | 8.2 | 1.139 | 0.324 |
| rplL | 0.004 | 0.019 | 0.21 | 14.3 | 1.216 | 0.4 |
| plc | 0.005 | 0.047 | 0.106 | 21.2 | 0.674 | 0.156 |
| colA | 0.004 | 0.036 | 0.111 | 16.6 | 0.886 | 0.222 |
| gyrA | 0.001 | 0.029 | 0.035 | 15.5 | 1.067 | 0.316 |
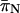 and
and 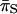 were calculated using the Nei and Gojobori (1986) method.
were calculated using the Nei and Gojobori (1986) method.
G + C content at third codon positions.
Estimates of recombination parameters are shown in Table 2. In analyses of concatenated sequence data, results of both tests of recombination were significant, as well as when only housekeeping loci or only toxin loci were analyzed (Table 2). However, when individual protein-coding regions and noncoding regions were analyzed separately, the LPT was significant for plc, plc FR, colA, gyrA, and rplL FR (Table 2). In the preceding paragraph, we described higher levels of nucleotide variability at rplL synonymous sites in contrast to the rplL FR sites and indicated that this result is evidence for recombination in the absence of natural selection. However, the estimate of RM was 0 for both the rplL gene and FR. Although RM is an estimate of the minimum number of recombination events and not an estimate of the total number, it does suggest that the results of the LPT may be too liberal here. Given that there are only 9 and 12 alleles at the coding region and FR, respectively, sample size may be confounding the LPT for the rplL FR.
The availability of the crystal structure for the C. perfringens α-toxin (Naylor et al. 1998) and the results from a number of structure–function studies (Alape-Girón et al. 2000; Jepson and Titball 2000; Walker et al. 2000; Nagahama et al. 2002; Sakurai et al. 2004) allow for thorough analysis of recombination involving the plc gene. The α-toxin possesses two domains (Titball et al. 1991; Naylor et al. 1998). The N-terminal domain contains the active site and the C-terminal domain contains substrate-binding sites. We identified several nucleotide sites involved in recombination events (herein referred to as RM sites), using the Hudson and Kaplan method (Table 4). Most of these sites occur at third codon positions (Figure 1). It is well known that polymorphism in coding regions is higher at third codon positions (Nei 1987; Graur and Li 2000). What was unexpected is that there were no RM site pairs that spanned both domains, indicating that plc recombination is domain specific.
TABLE 4.
Recombination and positive Darwinian selection in the plc gene
| Amino acid
|
RM codon sites
|
Positive selection
|
Functional region
|
|||
|---|---|---|---|---|---|---|
| First | Second | Third | SLAC | BEB | ||
| S-9a | X | X | ||||
| S-12 | X | |||||
| S-13 | X | X | ||||
| S-27 | X | |||||
| 6 | X | |||||
| 19 | X | X | ||||
| 26 | ||||||
| 44 | X | |||||
| 47 | X | |||||
| 60 | X | Signal loop | ||||
| 68 | X | Active site; signal loop | ||||
| 92 | X | |||||
| 152 | X | Active site | ||||
| 156 | X | |||||
| 160 | X | |||||
| 167 | X | X | X | |||
| 174 | X | |||||
| 194 | X | |||||
| 200 | X | |||||
| 212 | X | |||||
| 218 | X | |||||
| 222 | X | |||||
| 225 | X | |||||
| 233 | X | |||||
| 247 | X | Linker | ||||
| 267 | X | |||||
| 298 | X | Substrate binding | ||||
| 305 | X | Substrate binding | ||||
| 310 | X | |||||
| 320 | X | |||||
| 342 | X | |||||
| 345 | X | |||||
| 358 | X | |||||
Residues that serve as active sites or substrate-binding sites are shown in italics.
Amino acid residues marked with “S-” are found in the 28-residue signal peptide leader sequence, which is cleaved off the mature molecule.
Figure 1.

Frequency of recombinant sites identified at each codon position among all protein-coding loci analyzed in this study.
Analysis of codon usage bias and Hill–Robertson effects:
C. perfringens possesses a highly A + T-biased genome, displaying an average G + C content of 28.4% (Shimizu et al. 2002). This large degree of bias is reflected in the G + C content at third codon positions (GC3) and the scaled χ2-values listed in Table 2. In studies of eukaryotic taxa, a negative correlation has been found between the degree of codon usage bias and the strength of recombination (Kliman and Hey 1993; Comeron et al. 1999; McVean and Charlesworth 2000). In the case of bacteria, similar results have been found (Sharp et al. 2005), although there are certain notable exceptions (Feil et al. 2001). Because a high amount of codon usage bias in bacteria is believed to be the result of selection for translational efficiency (Sharp and Li 1987; Bulmer 1991), the negative correlation between recombination and codon usage bias may be attributed to the Hill–Robertson effect in which the efficacy of natural selection is reduced in regions of low recombination (Hill and Robertson 1966; Felsenstein 1974). In our study, we found a significantly negative correlation between ρ and both the scaled χ2-statistic (r = −0.97; P = 0.007) and the corrected scaled χ2-statistic (r = −0.98; P = 0.003).
Analysis of positive Darwinian selection:
To determine if any of the amino acid residues in the protein-coding regions were under positive Darwinian selection, we conducted an analysis using the SLAC and likelihood methods. There were no codons under positive Darwinian selection at the P < 0.05 level (SLAC method) for the gyrA, colA, and rplL genes. Similarly, the LRT was not significant for these genes under models M1a and M2a or under models M7a and M8a. However, the pfoS and plc genes were found to be under positive selection with the SLAC and likelihood methods. In pfoS, codon 11 (Table 4) was found to be under positive selection at the P < 0.05 level using both SLAC and BEB methods. In addition, this site was found to be subject to recombination using the Hudson and Kaplan method. Patterns similar to what was observed at pfoS were also found for the plc gene. In this case, three sites (codons 19, 167, and 345) were inferred to be under selection at the P < 0.05 level (Table 3) using the SLAC method. Under the BEB method, only two sites (codons S-13 and 167) were found to be under positive selection at the P < 0.05 level. Except for codon 345, these codons were also found to be involved in recombination events.
Phylogenetic analysis:
We conducted phylogenetic analyses on each gene and corresponding FR separately as well as combined. We observed that strain relationships differed between loci, which is not surprising in light of the moderate to high levels of recombination detected (Table 2). Because recombination will confound attempts to reconstruct the phylogenetic relationships among strains (Schierup and Hein 2000; Worobey 2001), a better way to assess clustering patterns is to compare trees reconstructed from loci that display statistically significant evidence of recombination (Figure 2) vs. trees reconstructed from loci that do not (Figure 3). Indeed, we observed that the tree in Figure 2 showed 173 unique haplotypes while the tree in Figure 3 had only 72 unique haplotypes. This disparity is consistent with recombination causing a higher level of variability rather than clonality, although it also should be kept in mind that the tree in Figure 2 was based on 3551 bp whereas the tree in Figure 3 was based on 1128 bp. More importantly, although the two trees were different, there was a considerable amount of congruence between loci concerning the formation of five distinct strain groups (Figures 2 and 3). Because of this consistency and the fact that nonrecombining loci should better reflect the true phylogenetic relationships between strains than loci subject to recombination, we herein focus on the tree reconstructed from the former (Figure 3).
Figure 2.

Phylogeny of C. perfringens strains reconstructed from loci subject to statistically significant levels of recombination (Table 2). Only nonidentical sequences are shown. Numbers along branches represent bootstrap values. Evolutionary lineages are represented by different colors. Strains are identified by their USDA–ARS (NRRL) accession numbers except for the Shimizu et al. (2002) genome-sequencing strain, which is denoted as “Genome.”
Figure 3.

Phylogeny of C. perfringens strains reconstructed from loci that evolve clonally (Table 2). Only nonidentical sequences are shown. Numbers along branches represent bootstrap values. Evolutionary lineages are represented by different colors. Strains are identified by their USDA–ARS (NRRL) accession numbers except for the Shimizu et al. (2002) genome-sequencing strain, which is denoted as “Genome.”
Interestingly, the five major groups shown in Figure 3 are also separable on the basis of their disease pathology/ecology and, therefore, represent ecologically diversified lineages. Lineage I is the largest, with >93% of all strains in our sample, and is the most diverse, possessing strains that are found in all sources sampled. Both pathogenic and nonpathogenic strains fall within this lineage. The former include various gangrene strains of human and animal origin, human nosocomial strains, human antibiotic-associated diarrhea strains, and bovine hemorrhagic enteritis strains that also possess the β2-toxin. Lineage II consists of strains isolated from peritoneal abscesses or septicemic blood of humans. Lineage III consists of strains isolated from cases of bovine and equine hemorrhagic enteritis. These strains differ from the lineage I isolates that cause bovine hemorrhagic enteritis in that they do not possess the β2-toxin. Lineage IV consists of a lamb enterotoxaemia isolate. And lineage V is composed of human food-borne gastrointestinal (GI) disease isolates. When we examined the level of 16S rRNA gene sequence divergence between representatives of each lineage, we found that they all displayed >99% sequence similarity between each other, suggesting that the lineages have not diverged enough to be considered unique species. However, because they show evidence of ecological specialization, they appear to be in the early stages of speciation.
DISCUSSION
Evolution of the core genome:
Over the last several years a number of studies on genetic variability of bacterial pathogens have been conducted using an approach known as “multilocus sequence typing” (MLST) (Maiden et al. 1998), in which short gene fragments of ∼300–500 bp are amplified and sequenced from seven loci spread across the genome (Maiden et al. 1998; Urwin and Maiden 2003; Feil and Enright 2004). In addition to their public health importance, these studies have offered a wealth of opportunities to investigate the mechanisms that influence bacterial evolution. In fact, because MLST studies focus on housekeeping genes, they provide an assessment of which mechanisms have the most impact on the evolution of the core genome, which consists of the set of genes that are present in all members of a species and, therefore, is presumably required for its long-term evolutionary survival (Dykhuizen and Green 1991; Lan and Reeves 2000, 2001; Wertz et al. 2003). Without doubt, MLST studies have reinforced the importance of recombination in the evolution of most bacterial genomes. However, is it reasonable to expect that all genes in the core genome will evolve in the same manner? Or put another way, will the effects of the evolutionary processes influencing the core genome be homogeneous across all of its components? To answer this question, we compared patterns of genetic variability between two different toxin genes and representative housekeeping genes.
The plc and colA toxin genes were found to undergo similar modes of evolution in which purifying selection and recombination were identified as the processes leaving the strongest imprint on genetic variability. While there was some difference in the amount of recombination between plc and colA, as shown by a greater ρ/θ ratio in the latter (4.443) than in the former (3.26) (Table 2), this difference is minor considering the difference in the ability between the α-toxin and κ-toxin to cause infection. On this alone, we expected substantially different patterns of variability between plc and colA. This result (or lack thereof) was unexpected. If one subscribes to the homogeneous core genome hypothesis, a possible explanation is that plc and colA are influenced by similar patterns of evolution because both are members of the core genome. Yet, if this were true, it would stand in direct conflict with the fact that pfoS and rplL, which are both housekeeping genes and members of the core genome, are not significantly influenced by recombination (Table 2). As such, our study indicates that the evolution of the core genome is not homogeneous, which raises another issue. Why do some core genes undergo high levels of recombination whereas others do not?
Unfortunately, there is no easy answer for this question, although there are two possibilities. First, recombination may be constrained to parts of the genome that contain the proper signals and binding sites (among other features) for the process to occur. Thus, the reason why some genes undergo higher levels of recombination is that they are located in a region of the genome amenable for recombination, whereas other genes may not be. A second explanation for why some genes undergo higher levels of recombination is because it is selectively advantageous (Felsenstein 1974). In this study, the majority of the recombinant sites that we detected in genes occurred at third codon positions (Table 4; Figure 1). Since changes at third codon positions are predominantly silent, the large number of recombinant sites at these positions suggests that any selective advantage of recombination is probably slight, at best. If recombination involved nonsynonymous sites, the argument for its selective advantage would be stronger. However, detecting selection in the presence of recombination poses considerable methodological problems (Anisimova et al. 2003; Shriner et al. 2003). For example, in the case of plc, certain amino acids were inferred to be under positive Darwinian selection, but these codons were also involved in recombination events (Table 4). There was one possible exception in the plc gene (Table 4), but the amino acid site is not functionally important and was shown to be under positive selection only using the SLAC method, but not the BEB method. In summary, the above results cast doubt on the idea of a strong selective advantage of recombination at the level of the individual gene. However, it should be pointed out that because polymorphism is much higher at third positions than at second positions, it is reasonable to expect that recombination would be easier to detect at the former than at the latter, particularly since the latter is on the low side. Therefore, we cannot be completely rule out that there is not a selective advantage to recombination at the level of the individual gene.
Nonetheless, recombination can provide the genome with a means with which to buffer against the accumulation of slightly deleterious mutations (i.e., Muller's ratchet) (Muller 1964; Felsenstein 1974) and to bolster the efficacy of natural selection (i.e., the Hill–Robertson effect) (Hill and Robertson 1966; Felsenstein 1974). When looking at each gene individually, the importance of slightly deleterious mutations may seem trivial, particularly over short-term evolutionary time. However, if examined on a genomewide scale over long-term evolutionary time, recombination has clear selective advantages. For example, if the frequency of slightly deleterious mutations per gene in our data set is calculated following Tajima (1989), the average is 0.207 per gene, which is quite low. Yet, when considering that the C. perfringens genome contains 2660 predicted genes (Shimizu et al. 2002), a more substantial number of slightly deleterious mutations (551) across the entire genome may be extrapolated. Consequently, recombination clearly imparts a selective advantage at the genome level, which explains why it is so prevalent in the genomes of many bacterial species.
Phylogenetic history, lineage formation, and disease presentation:
It has been suggested that recombination serves as a cohesive force that prevents strain divergence, thereby maintaining the “integrity” of a species (Cohan 2002a,b). In this study, we found that C. perfringens is composed of five strain groups that are associated with certain disease pathologies. The existence of these evolutionary lineages suggests that recombination has become less frequent between them; otherwise such clear delineations would not be evident. Interestingly, the lineages are associated with certain disease pathologies (e.g., lineage V and food-borne GI illness), suggesting an ecological separation. Certainly, if these lineages exist in distinct ecological niches, they would find fewer opportunities to recombine, which may also indicate that the lineages are in the early stages of speciation. For example, lineages IV and V both contain GI disease isolates associated with food. Lineage IV is associated with lamb enterotoxemia (a disease in which GI illness results from overeating during periods of high temperatures), while lineage V is associated with human food poisoning. These two lineages are closely related (both cluster with 99% bootstrap support; Figure 3), but have apparently diverged as a result of adaptation to different hosts and, hence, to ecological niches. The same is true for the other lineages, with the exception of lineage I. This lineage is highly diverse and is composed of nonpathogenic strains isolated from food as well as various gangrenous strains, human antibiotic-associated diarrhea strains, and human nosocomial strains. Considering that this lineage is the most “ecologically” diverse, it is probably the ancestral lineage from which the others are derived.
The fact that C. perfringens possesses five distinct evolutionary lineages is interesting not only from the perspective of speciation studies but also from a practical standpoint. Characterizing disease isolates according to their lineage would enhance public health efforts aimed at monitoring, tracking, and controlling sources of infection and outbreak in C. perfringens as well as in other bacteria, such as Listeria monocytogenes (Ward et al. 2004). Determining the frequency with which each lineage is associated with disease outbreaks would identify lineages prone to epidemic spread vs. those that are not. Thus, the accuracy and precision of public health monitoring, tracking, and control efforts would be greatly improved through the identification of the specific lineages of C. perfringens. Moreover, characterizing the evolutionary lineages that comprise bacterial species in terms of their phylogeny and virulence will reveal how virulence evolves, including the pathways by which strains acquire pathogenicity to humans or other organisms.
For example, C. perfringens-induced GI disease in humans can be either food borne or non-food borne, in which the latter is sporadic or antibiotic associated in nature. The food-borne illness form generally lasts 24–48 hr and is caused by strains carrying a chromosomal enterotoxin gene, whereas the non-food-borne illness form is caused by strains carrying an episomal (plasmid-borne) enterotoxin gene and lasts 1–2 weeks with more pronounced disease symptoms (abdominal cramping and diarrhea) (McClane 1996, 1997; Carman 1997; Johnson and Gerding 1997). The chromosomal and episomal enterotoxin genes are not separable on the basis of their nucleotide or protein sequences, suggesting that the genes in the chromosome and plasmid either (1) frequently interchange or, if not, (2) have only recently diverged. One wonders how this duality between chromosomal vs. episomal enterotoxin genes has evolved to result in different pathologies. In our study, we examined three strains involved in AAD–SD: all were found in lineage I and all contained episomal enterotoxin genes. In contrast, all human food poisoning strains were chromosomal enterotoxin bearing and part of lineage V, which is limited to the food environment (specifically meat derived from farm animals). Our data suggest that the reason for the duality between the food-borne and AAD–SD GI illnesses caused by C. perfringens is because the strains causing those respective diseases represent distinct evolutionary lineages. This indicates that there is a link between the type of disease produced by a strain and its particular lineage or genetic background.
A recent study (Fisher et al. 2005) found that the episomal enterotoxin gene and the β2-toxin gene occur on the same plasmid in cases of C. perfringens-induced AAD–SD and that the β2-toxin is an accessory toxin in these cases. In animals, the β2-toxin causes a severe, often fatal form of hemorrhagic enteritis (Gibert et al. 1997; Manteca et al. 2002). In our study, we found that hemorrhagic enteritis in cattle can be caused by both lineage III strains and certain lineage I strains, but the β2-toxin occurs only among lineage I strains. We suggest that the plasmid containing the enterotoxin and the β2-toxin gene is derived from an animal-adapted lineage. Most likely, it is lineage IV, which we found to harbor both chromosomal and episomal enterotoxin genes. We predict that the plasmid containing the enterotoxin gene evolved first in this lineage and was subsequently transferred to lineage I, where it transformed nonpathogenic strains into pathogenic ones via the presence of this plasmid. The reason why AAD–SD disease results when these strains are introduced into the human GI tract is because the original genetic background of the plasmid may have been an animal-adapted lineage (probably lineage IV). However, due to unknown human-specific factors, the toxin genes on the plasmid induce a much more severe form of disease in ruminants than in humans. Likewise, lineage V may be an animal-adapted lineage that induces disease symptoms (food poisoning) when introduced into the human GI tract, but not in animals. The presence of the chromosomal gene and the sister-group relationship to lineage V (Figure 3) suggest that lineage IV also gave rise to this lineage.
Our study's finding of a link between a strain's lineage and the type of disease caused by it provides an important insight into the etiology and pathogenesis of the different illnesses caused by C. perfringens. In fact, our finding of a lineage-specific plasmid distribution is further evidence of a barrier to genetic exchange between lineages. As such, our study has important ramifications for C. perfringens disease source tracking, surveillance, and risk assessment. For example, AAD–SD is presumed to be a non-food-borne GI illness. But do cattle that harbor lineage I strains with an episomal enterotoxin gene sometimes get sent to slaughter and become the source of meats that are eventually consumed by the public? The answer to that question might explain the erratic incidence of human AAD–SD. More importantly, the results of our study call for the development of potential intervention strategies and therapies based on targeting the individual lineage responsible for a specific disease, as opposed to the development of a “shotgun” approach aimed at the entire suite of C. perfringens diversity.
Acknowledgments
We thank H. N. Kline [U.S. Department of Agriculture-Agricultural Research Service (USDA–ARS), Peoria, IL] for technical assistance and G. McVean for assistance with the computer program LDHat. We are grateful to J. A. Lindsay (USDA–ARS national program staff), T. J. Ward (USDA–ARS, Peoria, IL), P. J. Oefner (Stanford University), and two anonymous reviewers for helpful comments. We thank J. S. Novak (USDA–ARS, Wyndmoor, PA) for providing certain enterotoxigenic strains. The mention of manufacturer names or trade products does not imply that they are endorsed or recommended by the USDA over other manufacturers or similar products not mentioned.
References
- Aboshkiwa, M. A., G. C. Rowland and G. Coleman, 1995. Nucleotide and deduced amino acid sequence of the gene for a novel protein with a possible regulatory function encoded in the beta operon of Staphylococcus aureus. FEMS Microbiol. Lett. 181: 305–309. [DOI] [PubMed] [Google Scholar]
- Alape-Girón, A., M. Flores-Díaz, I. Guillouard, C. E. Naylor, R. W. Titball et al., 2000. Identification of residues critical for toxicity in Clostridium perfringens phospholipase C, the key toxin in gas gangrene. Eur. J. Biochem. 267: 5191–5197. [DOI] [PubMed] [Google Scholar]
- Alekshun, M., M. Kashlev and I. Schwartz, 1997. Molecular cloning and characterization of Borrelia burgdorferi rpoB. Gene 186: 227–235. [DOI] [PubMed] [Google Scholar]
- Anisimova, M., R. Nielsen and Z. Yang, 2003. Effect of recombination on the accuracy of the likelihood method for detecting positive selection at amino acid sites. Genetics 164: 1229–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awad, M. M., A. E. Bryant, D. L. Stevens and J. I. Rood, 1995. Virulence studies on chromosomal alpha-toxin and theta-toxin mutants constructed by allelic exchange provide genetics evidence for the essential role of alpha-toxin in Clostridium perfringens-mediated gas gangrene. Mol. Microbiol. 15: 191–202. [DOI] [PubMed] [Google Scholar]
- Baba, E., A. L. Fuller, J. M. Gilbert, S. G. Thayer and L. R. Mcdougald, 1992. Effects of Eimeria brunetti infection and dietary zinc on experimental induction of necrotic enteritis in broiler chickens. Avian Dis. 36: 59–62. [PubMed] [Google Scholar]
- Boor, K. J., M. L. Duncan and C. W. Price, 1995. Genetic and transcriptional organization of the region encoding the beta subunit of Bacillus subtilis RNA polymerase. J. Biol. Chem. 270: 20329–20336. [DOI] [PubMed] [Google Scholar]
- Bos, J., L. Smithee, B. A. Mcclane, R. F. Distefano, F. Uzal et al., 2005. Fatal necrotizing colitis following a foodborne outbreak of enterotoxigenic Clostridium perfringens type A infection. Clin. Infect. Dis. 40: e78–83. [DOI] [PubMed] [Google Scholar]
- Bryant, A. E., 2003. Biology and pathogenesis of thrombosis and procoagulant activity in invasive infections caused by group A streptococci and Clostridium perfringens. Clin. Microbiol. Rev. 16: 451–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant, A. E., and D. L. Stevens, 1997. The pathogenesis of gas gangrene, pp. 185–196 in The Clostridia: Molecular Biology and Pathogenesis, edited by J. I. Rood, B. A. Mcclane and J. G. Songer. Academic Press, London.
- Brynestad, S., and P. E. Granum, 2002. Clostridium perfringens and foodborne infections. Int. J. Food Microbiol. 74: 195–202. [DOI] [PubMed] [Google Scholar]
- Bueschel, D. M., B. H. Jost, S. J. Billington, H. T. Trinh and J. G. Songer, 2003. Prevalence of cpb2, encoding beta2 toxin, in Clostridium perfringens field isolates: correlation of genotype with phenotype. Vet. Microbiol. 94: 121–129. [DOI] [PubMed] [Google Scholar]
- Bulmer, M., 1991. The selection-mutation-drift theory of synonymous codon usage. Genetics 129: 897–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carman, R. J., 1997. Clostridium perfringens in spontaneous and antibiotic-associated diarrhoea in man and other animals. Rev. Med. Microbiol. 8: S43–S45. [Google Scholar]
- Cohan, F. M., 2002. a Sexual isolation and speciation in bacteria. Genetica 116: 359–370. [PubMed] [Google Scholar]
- Cohan, F. M., 2002. b What are bacterial species? Annu. Rev. Microbiol. 56: 457–487. [DOI] [PubMed] [Google Scholar]
- Collie, R. E., and B. A. Mcclane, 1998. Evidence that the enterotoxin gene can be episomal in Clostridium perfringens isolates associated with non-food-borne human gastrointestinal diseases. J. Clin. Microbiol. 36: 30–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comeron, J. M., M. Kreitman and M. Aguadé, 1999. Natural selection on synonymous sites is correlated with gene length and recombination in Drosophila. Genetics 151: 239–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabbs, E. R., 1984. Order of ribosomal protein genes in the Rif cluster of Bacillus subtilis is identical to that of Escherichia coli. J. Bacteriol. 159: 770–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykhuizen, D. E., and L. Green, 1991. Recombination in Escherichia coli and the definition of biological species. J. Bacteriol. 173: 7257–7268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feil, E. J., and M. C. Enright, 2004. Analyses of clonality and the evolution of bacterial pathogens. Curr. Opin. Microbiol. 7: 308–313. [DOI] [PubMed] [Google Scholar]
- Feil, E. J., E. C. Holmes, D. E. Bessen, M.-S. Chan, N. P. Day et al., 2001. Recombination within natural populations of pathogenic bacteria: short-term empirical estimates and long-term phylogenetic consequences. Proc. Natl. Acad. Sci. USA 98: 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein, J., 1974. The evolutionary advantage of recombination. Genetics 78: 737–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher, D. J., K. Miyamoto, B. Harrison, S. Akimoto, M. R. Sarker et al., 2005. Association of beta2 toxin production with Clostridium perfringens type A human gastrointestinal disease isolates carrying a plasmid enterotoxin gene. Mol. Microbiol. 56: 747–762. [DOI] [PubMed] [Google Scholar]
- Gibert, M., C. Jolivet-Reynaud and M. R. Popoff, 1997. Beta2 toxin, a novel toxin produced by Clostridium perfringens. Gene 203: 65–73. [DOI] [PubMed] [Google Scholar]
- Graur, D., and W.-H. Li, 2000. Fundamentals of Molecular Evolution. Sinauer Associates, Sunderland, MA.
- Hill, W. G., and A. Robertson, 1966. The effect of linkage on limits to artificial selection. Genet. Res. 8: 269–294. [PubMed] [Google Scholar]
- Hudson, R. R., and N. L. Kaplan, 1985. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 111: 147–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, A. L., and R. Friedman, 2004. Patterns of sequence divergence in 5 intergenic spacers and linked coding regions in 10 species of pathogenic bacteria reveal distinct recombinational histories. Genetics 168: 1795–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jepson, M., and R. Titball, 2000. Structure and function of clostridial phospholipases C. Microbes Infect. 2: 1277–1284. [DOI] [PubMed] [Google Scholar]
- Johnson, S., and D. N. Gerding, 1997. Enterotoxemic infections, pp. 117–140 in The Clostridia: Molecular Biology and Pathogenesis, edited by J. I. Rood, B. A. Mcclane and J. G. Songer. Academic Press, London.
- Kimura, M., 1980. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16: 111–120. [DOI] [PubMed] [Google Scholar]
- Kliman, R. M., and J. Hey, 1993. Reduced natural selection associated with low recombination in Drosophila melanogaster. Mol. Biol. Evol. 10: 1239–1258. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond, S. L., and S. D. W. Frost, 2005. Not so different after all: a comparison of methods for detecting amino-acid sites under selection. Mol. Biol. Evol. 22: 1208–1222. [DOI] [PubMed] [Google Scholar]
- Kumar, S., K. Tamura and M. Nei, 2004. MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief. Bioinform. 5: 150–163. [DOI] [PubMed] [Google Scholar]
- Lan, R., and P. R. Reeves, 2000. Intraspecies variation in bacterial genomes: the need for a species genome concept. Trends Microbiol. 8: 396–401. [DOI] [PubMed] [Google Scholar]
- Lan, R., and P. R. Reeves, 2001. When does a clone deserve a name? A perspective on bacterial species based on population genetics. Trends Microbiol. 9: 419–424. [DOI] [PubMed] [Google Scholar]
- Lindsay, J. A., 1996. Clostridium perfringens type A enterotoxin (CPE): more than just explosive diarrhea. Crit. Rev. Microbiol. 22: 257–277. [DOI] [PubMed] [Google Scholar]
- Long, J. R., and R. B. Truscott, 1976. Necrotic enteritis in broiler chickens. III. Reproduction of the disease. Can. J. Comp. Med. 40: 53–59. [PMC free article] [PubMed] [Google Scholar]
- Maiden, M. C., J. A. Bygraves, E. Feil, G. Morelli, J. E. Russell et al., 1998. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. USA 95: 3140–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manteca, C., G. Daube, T. Jauniaux, A. Linden, V. Pirson et al., 2002. A role for the Clostridium perfringens β2 toxin in bovine enterotoxaemia? Vet. Microbiol. 86: 191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcclane, B. A., 1996. An overview of Clostridium perfringens enterotoxin. Toxicon 34: 1335–1343. [DOI] [PubMed] [Google Scholar]
- Mcclane, B. A., 1997. Clostridium perfringens, pp. 305–326 in Food Microbiology: Fundamentals and Frontiers, edited by M. P. Doyle, L. R. Beauchat and T. J. Montville. ASM Press, Washington, DC.
- Mcvean, G. A., and B. Charlesworth, 2000. The effects of Hill–Robertson interference between weakly selected mutations on patterns of molecular evolution and variation. Genetics 155: 929–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcvean, G. A., P. Awadalla and P. Fearnhead, 2002. A coalescent-based method for detecting and estimating recombination from gene sequences. Genetics 160: 1231–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meer, R. R., and J. G. Songer, 1997. Multiplex polymerase chain reaction assay for genotyping Clostridium perfringens. Am. J. Vet. Res. 58: 702–705. [PubMed] [Google Scholar]
- Meer, R. R., J. G. Songer and D. L. Park, 1997. Human disease associated with Clostridium perfringens enterotoxin. Rev. Environ. Contam. Toxicol. 150: 75–94. [DOI] [PubMed] [Google Scholar]
- Morgan, B. A., E. Kellett and R. S. Hayward, 1984. The wild-type nucleotide sequence of the rpoBC-attenuator region of Escherichia coli DNA, and it implications for the nature of the rifd18 mutation. Nucleic Acids Res. 12: 5465–5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller, H. J., 1964. The relation of recombination to mutational advance. Mutat. Res. 106: 2–9. [DOI] [PubMed] [Google Scholar]
- Nagahama, M., M. Mukai, S. Morimitsu, S. Ochi and J. Sakurai, 2002. Role of the C-domain in the biological activities of Clostridium perfringens alpha-toxin. Microbiol. Immunol. 46: 647–655. [DOI] [PubMed] [Google Scholar]
- Naylor, C. E., J. T. Eaton, A. Howells, N. Justin, D. S. Moss et al., 1998. Structure of the key toxin in gas gangrene. Nat. Struct. Biol. 5: 738–746. [DOI] [PubMed] [Google Scholar]
- Nei, M., 1987. Molecular Evolutionary Genetics. Columbia University Press, New York.
- Nei, M., and T. Gojobori, 1986. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 3: 418–426. [DOI] [PubMed] [Google Scholar]
- Nielsen, R., and Z. Yang, 1998. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148: 929–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit, L., M. Gibert and M. R. Popoff, 1999. Clostridium perfringens: toxinotype and genotype. Trends Microbiol. 7: 104–110. [DOI] [PubMed] [Google Scholar]
- Present, D. A., R. Meislin and B. Shaffer, 1990. Gas gangrene. A review. Orthop. Rev. 19: 333–341. [PubMed] [Google Scholar]
- Rhodehamel, E. J., and S. M. Harmon, 2001. Clostridium perfringens, in Bacteriological Analytical Manual. Center for Food Safety and Applied Nutrition, U.S. Food and Drug Administration, U.S. Department of Health and Human Services (http://www.cfsan.fda.gov/∼ebam/bam-toc.html).
- Rood, J. I., 1998. Virulence genes of Clostridium perfringens. Annu. Rev. Microbiol. 52: 333–360. [DOI] [PubMed] [Google Scholar]
- Rooney, A. P., J. L. Swezey, D. T. Wicklow and M. J. Mcatee, 2005. Bacterial species diversity in cigarettes linked to an investigation of severe pneumonitis in U.S. military personnel deployed in Operation Iraqi Freedom. Curr. Microbiol. 51: 46–52. [DOI] [PubMed] [Google Scholar]
- Rozas, J., J. C. Sanchez-Delbarrio, X. Messeguer and R. Rozas, 2003. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19: 2496–2497. [DOI] [PubMed] [Google Scholar]
- Saitou, N., and M. Nei, 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4: 406–425. [DOI] [PubMed] [Google Scholar]
- Sakurai, J., M. Nagahama and M. Oda, 2004. Clostridium perfringens α-toxin: characterization and mode of action. J. Biochem. 136: 569–574. [DOI] [PubMed] [Google Scholar]
- Sarker, M. R., R. J. Carman and B. A. Mcclane, 1999. Inactivation of the gene (cpe) encoding Clostridium perfringens enterotoxin eliminates the ability of two cpe-positive C. perfringens type A human gastrointestinal disease isolates to affect rabbit ileal loops. Mol. Microbiol. 33: 946–958. [DOI] [PubMed] [Google Scholar]
- Sarker, M. R., U. Singh and B. A. Mcclane, 2000. An update on Clostridium perfringens enterotoxin. J. Nat. Toxins 9: 251–266. [PubMed] [Google Scholar]
- Schierup, M. H., and J. Hein, 2000. Consequences of recombination on traditional phylogenetic analysis. Genetics 156: 879–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp, P. M., and W.-H. Li, 1987. The rate of synonymous substitution in enterobacterial genes is inversely related to codon usage bias. Mol. Biol. Evol. 4: 222–230. [DOI] [PubMed] [Google Scholar]
- Sharp, P. M., E. Bailes, R. J. Grocock, J. F. Peden and R. E. Sockett, 2005. Variation in the strength of selected codon usage bias among bacteria. Nucleic Acids Res. 33: 1141–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu, T., K. Ohtani, H. Hirakawa, K. Ohshima, A. Yamashita et al., 2002. Complete genome sequence of Clostridium perfringens, an anaerobic flesh-eater. Proc. Natl. Acad. Sci. USA 99: 996–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shriner, D., D. C. Nickle, M. A. Jensen and J. I. Mullins, 2003. Potential impact of recombination on sitewise approaches for detecting positive natural selection. Genet Res. 81: 115–121. [DOI] [PubMed] [Google Scholar]
- Smedley III, J. G., D. J. Fisher, S. Sayeed, G. Chakrabarti and B. A. Mcclane, 2004. The enteric toxins of Clostridium perfringens. Rev. Physiol. Biochem. Pharmacol. 152: 183–204. [DOI] [PubMed] [Google Scholar]
- Songer, J. G., 1996. Clostridial enteric diseases of domestic animals. Clin. Microbiol. Rev. 9: 216–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titball, R. W., D. L. Leslie, S. Harvey and D. Kelly, 1991. Hemolytic and sphingomyelinase activities of Clostridium perfringens alpha-toxin are dependent on a domain homologous to that of an enzyme from the human arachidonic acid pathway. Infect. Immun. 59: 1872–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urwin, R., and M. C. Maiden, 2003. Multi-locus sequence typing: a tool for global epidemiology. Trends Microbiol. 11: 479–487. [DOI] [PubMed] [Google Scholar]
- Walker, N., J. Holley, C. E. Naylor, M. Flores-Díaz, A. Alape-Girón et al., 2000. Identification of residues in the carboxy-terminal domain of Clostridium perfringens alpha-toxin (phospholipase C) which are required for its biological activities. Arch. Biochem. Biophys. 384: 24–30. [DOI] [PubMed] [Google Scholar]
- Ward, T. J., L. Gorski, M. K. Borucki, R. E. Mandrell, J. Hutchins et al., 2004. Intraspecific phylogeny and lineage group identification based on the prfA virulence gene cluster of Listeria monocytogenes. J. Bacteriol. 186: 4994–5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson, G. A., 1975. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7: 256–276. [DOI] [PubMed] [Google Scholar]
- Wertz, J. E., C. Goldstone, D. M. Gordon and M. A. Riley, 2003. A molecular phylogeny of enteric bacteria and implications for a bacterial species concept. J. Evol. Biol. 16: 1236–1248. [DOI] [PubMed] [Google Scholar]
- Worobey, M., 2001. A novel approach to detecting and measuring recombination: new insights into evolution in viruses, bacteria, and mitochondria. Mol. Biol. Evol. 18: 1425–1434. [DOI] [PubMed] [Google Scholar]
- Yang, Z., and R. Nielsen, 2002. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol. Biol. Evol. 19: 908–917. [DOI] [PubMed] [Google Scholar]
- Yang, Z., W. S. Wong and R. Nielsen, 2005. Bayes empirical Bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 22: 1107–1118. [DOI] [PubMed] [Google Scholar]