

Abstract
We report a complex set of scaling relationships between mutation and reproduction in a simple model of a population. These follow from a consideration of patterns of genetic diversity in a sample of DNA sequences. Five different possible limit processes, each with a different scaled mutation parameter, can be used to describe genetic diversity in a large population. Only one of these corresponds to the usual population genetic model, and the others make drastically different predictions about genetic diversity. The complexity arises because individuals can potentially have very many offspring. To the extent that this occurs in a given species, our results imply that inferences from genetic data made under the usual assumptions are likely to be wrong. Our results also uncover a fundamental difference between populations in which generations are overlapping and those in which generations are discrete. We choose one of the five limit processes that appears to be appropriate for some marine organisms and use a sample of genetic data from a population of Pacific oysters to infer the parameters of the model. The data suggest the presence of rare reproduction events in which ∼8% of the population is replaced by the offspring of a single individual.
DNA sequences are variable within species because mutations have occurred between the present day and the time of the most recent common ancestor (TMRCA) at most genetic loci. Mutation rates are quite small: on the order of 10−10 per base pair per replication event in eukaryotes and 10−6–10−10 in microbes (Drake et al. 1998), while mutation rates measured from sequence differences between species range from ∼10−8 to ∼10−10 per base pair per generation (Li 1997). The abundance of genetic variation within most species implies that a great number of generations must have elapsed since the MRCA. The occurrence of the MRCA at a locus results from the birth and death of individuals in a population (hereafter synonymous with species). Over time, some genetic lineages are lost and others leave many descendants. Other things being equal, TMRCA should be greater in a large population than in a small one. If we take 10−5 as a typical mutation rate per locus per generation, then to see any genetic variation, TMRCA must be roughly on the order of the inverse of this, or ∼100,000 generations.
What, then, do we expect the relationship to be between TMRCA and the population size N? The bulk of work has focused on just one possibility: that TMRCA should be a constant multiple of N generations. Then if N is very large, and the inverse of the mutation rate 1/μ is also large, the level and pattern of genetic diversity in a sample of DNA sequences will depend only on the product Nμ. For example, in the neutral haploid Wright–Fisher model (Fisher 1930; Wright 1931) there is a chance 1/N that two sequences are descended from a common ancestor in the previous generation and a chance 1 − (1 − μ)2 ≈ 2μ that there is a mutation between them in that generation. The expected number of differences between a pair of sequences is E[K] ≈ 2Nμ, which is simply the product of the expected number of generations to the common ancestor (N) and the expected number of mutations per generation on the two genetic lineages (≈2μ).
A rigorous formulation of these ideas yields the coalescent (Kingman 1982a,b; Hudson 1983; Tajima 1983), which is a continuous-time stochastic process for the ancestry of a sample from the present back to the MRCA. In the limit N → ∞, and with time measured in units proportional to N generations, each pair of ancestral lines reaches a common ancestor, or coalesces, with rate 1. Independently, each ancestral line undergoes mutation with rate θ/2. This holds when Nμ is constant in the limit as N tends to infinity. In the Wright–Fisher model above 2Nμ → θ, so that E[K] = θ, as N → ∞. This scaling between mutation and population size is shared by the various extensions of the coalescent that are reviewed in Nordborg (2001). The discovery of the coalescent greatly expanded the ways in which genetic data can be used to make inferences about historical events and the characteristics of populations (Tavaré 2004). Although the coalescent is a continuous-time process that exists in the limit as N → ∞ with time rescaled by N, it is used to approximate the behavior of gene genealogies in a wide range of species whose population sizes are very large.
It is important to note that the scaling relationship between N and μ in the coalescent is the consequence of a key assumption: that the variance 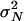 of the number of offspring among individuals in the population is not too large. Specifically, in the original proof of the coalescent Kingman (1982a,b) assumed that this variance converged to a constant σ2 in the limit N → ∞. This assumption has the additional consequence that only binary mergers of ancestral lines occur in the limit, so the gene genealogy of the sample is a bifurcating tree. Recently, a broader class of ancestral processes has been described in which this assumption about the distribution of offspring number is relaxed. While Kingman's coalescent is robust to many deviations from its assumptions (Möhle 1998, 1999), a major shift in the behavior of the ancestral process occurs when the variance of offspring number is large. The general ancestral process allows for multiple mergers of ancestral lines and happens on a timescale that is faster than that of Kingman's coalescent (Pitman 1999; Sagitov 1999; Schweinsberg 2000; Möhle and Sagitov 2001).
of the number of offspring among individuals in the population is not too large. Specifically, in the original proof of the coalescent Kingman (1982a,b) assumed that this variance converged to a constant σ2 in the limit N → ∞. This assumption has the additional consequence that only binary mergers of ancestral lines occur in the limit, so the gene genealogy of the sample is a bifurcating tree. Recently, a broader class of ancestral processes has been described in which this assumption about the distribution of offspring number is relaxed. While Kingman's coalescent is robust to many deviations from its assumptions (Möhle 1998, 1999), a major shift in the behavior of the ancestral process occurs when the variance of offspring number is large. The general ancestral process allows for multiple mergers of ancestral lines and happens on a timescale that is faster than that of Kingman's coalescent (Pitman 1999; Sagitov 1999; Schweinsberg 2000; Möhle and Sagitov 2001).
We consider a simple neutral population model that can exhibit multiple-mergers behavior in the limit as the population size tends to infinity. Depending on parameter values, it can also converge to Kingman's coalescent. Through analysis and simulations, we address two main questions. First, what are the possible behaviors of such a model with respect to the scaling relationship between mutation rate and population size? This question stems from the desire to explain genetic diversity. In short, the model must predict nonzero levels of genetic variation if the coalescent with multiple mergers is to be a viable alternative to Kingman's coalescent for many species. Second, what are the differences between the coalescent with multiple mergers and Kingman's coalescent with respect to predictions about patterns of genetic variation in a sample? Kingman's coalescent is the standard for interpreting genetic variation, but it is not uncommon to reject this null model, even using simple tests (Tajima 1989; Fu and Li 1993). For many species, the coalescent with multiple mergers might be a better null model than Kingman's coalescent.
The variance of offspring number does appear to be very high in some species, in particular those with type III survivorship curves, which produce very large numbers of offspring in the face of high mortality early in life (Hedgecock 1994). This strategy is common among marine organisms but it also occurs in terrestrial species that have large reproductive potential, such as some plants and fungi. Hedgecock (1994) proposed that very large 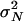 , or Vk (Crow and Kimura 1970), is the primary reason that levels of genetic variation in many species are much lower than predictions based on their population sizes (and the assumption that θ ∝ Nμ). Estimates of Ne based on temporal variation of allele frequencies or on samples of genetic data from a single time point are often much lower than the estimates, made independently, of the actual population size N. Small values of the ratio Ne/N are taken as evidence of large Vk since Ne/N ≈ 1/Vk in many models (Crow and Kimura 1970; Hedrick 2005). For example, Hedgecock (1994) estimated the ratio Ne/N to be between 10−5 and 10−6 in a population of the Pacific oyster (Crassostrea gigas). Turner et al. (2002) estimated Ne/N to be <10−3 in a commercially important fish, the red drum (Sciaenops ocellatus). Hedgecock (1994) also cites the case of the American lobster (Homarus americanus) whose effective population size is estimated to be ∼104 while some 107 lobsters are harvested annually. Árnason (2004) estimated Ne/N to be 10−5–10−6 in the Atlantic cod (Gadus morhua).
, or Vk (Crow and Kimura 1970), is the primary reason that levels of genetic variation in many species are much lower than predictions based on their population sizes (and the assumption that θ ∝ Nμ). Estimates of Ne based on temporal variation of allele frequencies or on samples of genetic data from a single time point are often much lower than the estimates, made independently, of the actual population size N. Small values of the ratio Ne/N are taken as evidence of large Vk since Ne/N ≈ 1/Vk in many models (Crow and Kimura 1970; Hedrick 2005). For example, Hedgecock (1994) estimated the ratio Ne/N to be between 10−5 and 10−6 in a population of the Pacific oyster (Crassostrea gigas). Turner et al. (2002) estimated Ne/N to be <10−3 in a commercially important fish, the red drum (Sciaenops ocellatus). Hedgecock (1994) also cites the case of the American lobster (Homarus americanus) whose effective population size is estimated to be ∼104 while some 107 lobsters are harvested annually. Árnason (2004) estimated Ne/N to be 10−5–10−6 in the Atlantic cod (Gadus morhua).
We suggest that the multiple-mergers coalescent processes might resolve many of the questions raised by Hedgecock (1994) and others. These ancestral processes are radically different from Kingman's coalescent: the relationship between θ and the population size N is less than linear, gene genealogies include multifurcations, and these processes have no effective population size in the usual sense (see the discussion). We identify one such multiple-mergers coalescent process, of five that are possible under the model we propose in the next section, and we apply it to a sample of genetic data from Pacific oysters in British Columbia (Boom et al. 1994). The model includes the possibility that the offspring of a single individual replace a substantial fraction of the population and yet still predicts that some genetic variation should be observed. This addresses the point made by Árnason (2004) in his study of Atlantic cod, that “large” reproduction events cannot be too frequent or there would be no genetic variation. For Pacific oysters in British Columbia, we find that the individuals who win Hedgecock's (1994) reproduction “sweepstakes” replace ∼8% of the population.
METHODS AND RESULTS
We consider the idealized model in Figure 1. At each discrete time step, exactly one individual reproduces and is the parent of U − 1 new individuals. The parent persists, while its offspring replace U − 1 individuals who die. The total population size is N and the N − U other individuals simply persist, until the next time step when they might be chosen to die or to reproduce. We assume that there are no fitness differences among individuals in the population. Thus, the parent is chosen at random, uniformly from the population, and so are the individuals who will die, with the exception that the parent does not die in the same time step that it reproduces. Mutations can occur to any of the U − 1 offspring when they are produced by the parent, while the parent and the N − U other individuals cannot mutate. This is a generalization of a well-studied model in population genetics, which was introduced by Moran (1958, 1962). In the usual Moran model, U is always 2, while in the general model it is a random variable that can be any number from 2 to N.
Figure 1.

A modified Moran model, in which individuals can have many offspring.
Mathematical analysis of ancestral limit processes:
Consider a sample of size n taken without replacement from the population. The gene genealogy traces the ancestral lines of the sample back to their MRCA. Time is measured backward into the past, with the time of sampling defined to be time zero. We use the term “x-merger” to denote the event that x ancestral lines are descended from a single member of the population (the parent above) in a single time step. If an x-merger occurs when there are i ancestral lines, then the number of ancestral lines changes from i to i − x + 1 in that time step, and x = 2, 3,…, i. The probability of an x-merger is given by
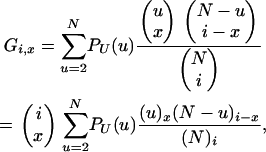 |
(1) |
in which PU(u) is the probability function, or distribution, of the random variable U, and the notation (r)j is for the descending factorial, r(r − 1) ⋯ (r − j + 1) with (r)0 = 1. The events x = 0 and x = 1 are also possible, and we have 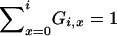 , but events in which x < 2 do not lead to mergers. The assumption of a single reproduction event per time step excludes the possibility of simultaneous mergers (Schweinsberg 2000; Möhle and Sagitov 2001).
, but events in which x < 2 do not lead to mergers. The assumption of a single reproduction event per time step excludes the possibility of simultaneous mergers (Schweinsberg 2000; Möhle and Sagitov 2001).
A key quantity in assessing convergence to a continuous-time limit process is what Möhle (1998) has called the coalescence probability, which he denoted cN, but which in our notation is G2,2. From Equation 1, we have
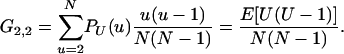 |
(2) |
The coalescence probability must tend to zero as N → ∞ for a continuous-time limit process to exist. One unit of time in the limit process is typically taken to be 1/G2,2 steps in the discrete-time model. The requirement that G2,2 → 0 as N → ∞ excludes certain distributions of U, namely those that have too much weight on values of U of order N. As in Kingman's coalescent, we seek a limit process for use as an approximation to the behavior of gene genealogies in a large population.
Let μ be the probability of mutation for each of the U − 1 offspring in each single time step. Mutations to the offspring occur independently, but neither the parent nor the N − U individuals who simply live through each time step can mutate. To capture the fact that mutation rates are very small, we let μ → 0 as N → ∞, although for the moment we refrain from specifying its rate of approach to zero. With μ infinitesimally small, genetic variation cannot be explained by mutations that co-occur with mergers because only a finite number of mergers occur in the ancestry of any (finite) sample. If the model is to predict realistic (neither zero nor infinite) levels of genetic variation, then in one time step the probability
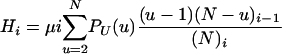 |
(3) |
that one of the i lines is an offspring, and it mutates, must be of the same order of magnitude as Gi,x for x = 2, 3,…, i. The probability Hi is similar to μGi,1, but requires that the single line is one of the u − 1 offspring and not the parent itself.
The usual way to measure time in these models is to scale it by the inverse of the coalescence probability, so that one unit of time in the limit process is equal to 1/G2,2 steps in the discrete-time model (Pitman 1999; Sagitov 1999; Möhle and Sagitov 2001; Birkner et al. 2005). However, as is illustrated below, we find that this choice of timescale makes it difficult to interpret the relative sizes of gene genealogies. Therefore, we scale time by 1/G2,2 times a constant ψ2, which derives from the simple model for PU(u) that we adopt in Equation 7 below. We emphasize that the predictions of the model concerning patterns of genetic variation do not depend on which of these timescales is used.
After scaling by ψ2/G2,2 the rate of x-mergers becomes
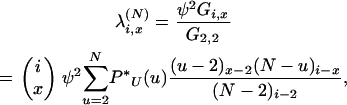 |
(4) |
where x = 2, 3,…, i, and
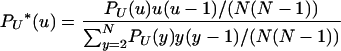 |
(5) |
is the rescaled distribution of U in which each value is weighted by the corresponding probability of coalescence. Of course, 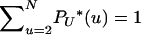 . The limit process follows from the existence of limits
. The limit process follows from the existence of limits 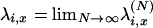 (Pitman 1999; Sagitov 1999). Note that PU*(u) corresponds to the measure Λ invoked in other works (Pitman 1999; Sagitov 1999; Möhle and Sagitov 2001; Birkner et al. 2005).
(Pitman 1999; Sagitov 1999). Note that PU*(u) corresponds to the measure Λ invoked in other works (Pitman 1999; Sagitov 1999; Möhle and Sagitov 2001; Birkner et al. 2005).
Whether a limit process of our model predicts reasonable levels of genetic variation will depend on the value of the scaled rate of mutation per ancestral line,
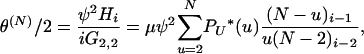 |
(6) |
in the limit N → ∞. In particular, we wish to distinguish cases in which 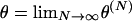 must be zero from those in which θ could be greater than zero. As discussed above, we assume that μ → 0 as N → ∞. Therefore, for θ to be greater than zero, the value of the sum in Equation 6 must grow with N.
must be zero from those in which θ could be greater than zero. As discussed above, we assume that μ → 0 as N → ∞. Therefore, for θ to be greater than zero, the value of the sum in Equation 6 must grow with N.
For the remainder of this work, we adopt the following simple model for the distribution of U in our modified Moran model. We assume that
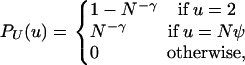 |
(7) |
in which ψ is a constant between 0 and 1, and γ ≥ 0. In seeking a continuous-time limit process, we require that G2,2 → 0 and this further restricts us to γ > 0. In words, most of the time (with probability 1 − N−γ) the parent has the usual number of Moran-model offspring, but occasionally (with probability N−γ) the parent and its offspring replace a fraction ψ of the population. In the usual Moran model, where PU(2) = 1, coalescence occurs on a timescale of 1/G2,2 = N(N − 1)/2 steps. Thus, if γ > 2 we expect Nψ-reproduction events to be too infrequent to shift the ancestral process from Kingman's coalescent. In contrast, the parameter range 0 < γ ≤ 2, in which large (U = Nψ) reproduction events occur at least as frequently as regular (U = 2) reproduction events, will be of particular interest. The model requires that Nψ is an integer, and we assume implicitly that this is true.
The constant ψ is a parameter of the limit process and it has a clear biological interpretation. It is the scaled family size, or the scaled number of offspring, of a large reproduction event, measured as a fraction of the total population. In comparison, work on general multiple-mergers coalescent processes occurs in a more abstract mathematical setting (Pitman 1999; Sagitov 1999; Birkner et al. 2005). More easily interpreted models include the power-law distribution function for family sizes that yields the beta-coalescent (Schweinsberg 2003; Birkner et al. 2005) and the models of recurrent selective sweeps (Gillespie 2000) that are best approximated by a coalescent with simultaneous multiple mergers (Durrett and Schweinsberg 2005). Note that our assumption in Equation 7, that the family size of large families is on the order of the population size, is required to produce an ancestral process that is different from Kingman's coalescent given our modified Moran model; see Möhle and Sagitov (2001, p. 1552).
We show in the appendix that five different limit processes of our modified Moran model are possible as N tends to infinity, depending on the value of γ > 0. These are summarized in Table 1. Consideration of λi,x alone uncovers three different behaviors in the limit. If γ < 2, the result is a multiple-mergers coalescent process, because the rate of Nψ-reproduction events is much greater than the rate of 2-reproduction events. In this case, 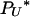 (Nψ) → 1 as N → ∞, and the limit process is of the type described by Pitman (1999) and Sagitov (1999) with Λ = δψ, i.e., the δ-function at the point ψ. Again, note that our timescale differs from theirs by the factor ψ2. If γ = 2, then the two types of reproduction events occur on the same timescale, and
(Nψ) → 1 as N → ∞, and the limit process is of the type described by Pitman (1999) and Sagitov (1999) with Λ = δψ, i.e., the δ-function at the point ψ. Again, note that our timescale differs from theirs by the factor ψ2. If γ = 2, then the two types of reproduction events occur on the same timescale, and 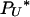 (Nψ) → ψ2/(2 + ψ2) as N → ∞. This corresponds to the case where Λ has mass 2/(2 + ψ2) at 0 and mass ψ2/(2 + ψ2) at ψ. Note that the mass at 0 affects only the rate of binary mergers, which is the only possible type of merger when a 2-reproduction event occurs. If γ > 2, then
(Nψ) → ψ2/(2 + ψ2) as N → ∞. This corresponds to the case where Λ has mass 2/(2 + ψ2) at 0 and mass ψ2/(2 + ψ2) at ψ. Note that the mass at 0 affects only the rate of binary mergers, which is the only possible type of merger when a 2-reproduction event occurs. If γ > 2, then 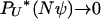 in the limit, and the ancestral limit process is Kingman's coalescent, as expected.
in the limit, and the ancestral limit process is Kingman's coalescent, as expected.
TABLE 1.
Five different possible ancestral limit processes for the modified Moran model
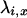 |
Leading term of 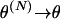
|
Case | ||
|---|---|---|---|---|
|
2ψ(1 − ψ)i−1μ → 0 | 0 < γ < 1 | ||
|
 |
γ = 1 | ||
|
2Nγ–1μ → θ > 0 | 1 < γ < 2 | ||
|
|
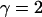 |
||
|
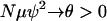 |
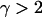 |
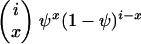
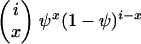
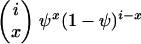
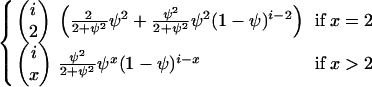
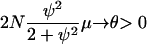
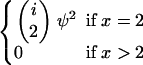
In the first case above (0 < γ < 2), Nψ-reproduction events are responsible for all mergers in the limit because 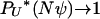 and
and 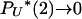 as N → ∞. Our consideration of mutation and genetic diversity subdivides 0 < γ < 2 into three different cases. The key point here is that, despite the fact that
as N → ∞. Our consideration of mutation and genetic diversity subdivides 0 < γ < 2 into three different cases. The key point here is that, despite the fact that 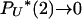 , these infrequent 2-reproduction events can generate many mutations in the ancestry of the sample if
, these infrequent 2-reproduction events can generate many mutations in the ancestry of the sample if 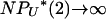 as
as 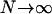 (see Equation A4 in the appendix). In the first two cases in Table 1, the rate of Nψ-reproduction events is too large (
(see Equation A4 in the appendix). In the first two cases in Table 1, the rate of Nψ-reproduction events is too large (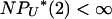 ) and only a finite number of mutations will occur before the sample reaches its MRCA. The scaled mutation parameter becomes a constant times μ, and this will be so small that the predicted level of genetic diversity is zero. In the third case, 1 < γ < 2, multiple mergers can occur, but it is also reasonable to expect some genetic variation to be observed. The scaled mutation parameter is μ times a strictly increasing function of N, so θ could be appreciable if the population size is large enough. In the final two cases in Table 1, the scaled mutation parameter is a linear function of N, which is the case typically in population genetics.
) and only a finite number of mutations will occur before the sample reaches its MRCA. The scaled mutation parameter becomes a constant times μ, and this will be so small that the predicted level of genetic diversity is zero. In the third case, 1 < γ < 2, multiple mergers can occur, but it is also reasonable to expect some genetic variation to be observed. The scaled mutation parameter is μ times a strictly increasing function of N, so θ could be appreciable if the population size is large enough. In the final two cases in Table 1, the scaled mutation parameter is a linear function of N, which is the case typically in population genetics.
Among the five possible limit processes, we suggest that the case 1 < γ < 2 might be a good null model for many organisms, namely those with very skewed offspring number distributions and very large population sizes. A large variance in offspring number leads to an ancestral process of coalescence that includes multiple mergers, while a very large population size is needed because the mutation parameter θ scales less than linearly with N. Specifically, θ = 2Nγ−1μ and 0 < γ − 1 < 1, so depending on the value of γ, N might have to be very large for the level of genetic variation to be appreciable.
Consider a sample of two DNA sequences at some genetic locus, and let K be the number of mutations on their gene genealogy. If mutations occur according to the infinitely many sites model without recombination (Watterson 1975), then every mutation results in a polymorphic site or in a difference between the two sequences at some site. For this limit process with 1 < γ < 2, we have λ2,2 = ψ2, and E[TMRCA] = 1/ψ2. Then, since the rate of mutation is θ/2 for each of the two ancestral lines, we have E[K] = θ/ψ2 and
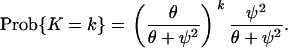 |
This agrees with intuition from the discrete model, which says that the level of genetic variation in a sample from a population with a larger value of ψ should be smaller than the level of genetic variation in a sample from a population with a smaller value of ψ, all other parameters being equal. Note that under the usual time scaling for multiple-mergers coalescent processes (Pitman 1999; Sagitov 1999; Möhle and Sagitov 2001), E[TMRCA] = 1 and by analogy with Kingman's coalescent the mutation parameter is defined to be θ(N) = 4Nγ−1μ/ψ2, so that E[K] = θ.
Properties of multiple-mergers genealogies in simulations:
The level and pattern of variation in a sample depends on the sample size n, the mutation parameter θ, and the family-size parameter ψ. A program, written in C, to simulate the ancestral process for the case 1 < γ < 2 is available from the authors upon request. The program simulates the ancestry, or gene genealogy, of a sample, including the tree relating the members of the sample and all the branch lengths, or coalescent times. It also implements the inference method described in the next section.
Figure 2 shows estimates of the expected total length of the gene genealogy, Ttot, which is the sum of the lengths of all ancestral lines back to the MRCA, as ψ ranges from 0.05 to 0.95. The result for our timescale is given in Figure 2a, while Figure 2b shows the same results when time is measured using the usual scaling (Pitman 1999; Sagitov 1999). Figure 2 should be interpreted as a comparison of different populations, which have the same values of N and γ, but different values of ψ. Under our timescale, of two populations that experience Nψ-reproduction events with probability N−γ per time step, the population with the larger value of ψ will have shorter gene genealogies. Under the usual timescale, in Figure 2b, the average total tree length increases with ψ. This is because, as ψ increases to 1, every sample will likely reach its MRCA at the first Nψ-reproduction event in the past, so that E[TMRCA] = 1 under the usual timescale, regardless of sample size, and E[Ttot] = n. The predictions about levels of polymorphism are the same under both timescales due to the different definitions of θ.
Figure 2.

The expected total length of the gene genealogy of a sample of size n = 10, computed as the average over 1 million simulation replicates, as a function of ψ. (a) 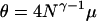 . (b)
. (b) 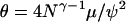 . a and b correspond to the two ways of measuring time in the limit process discussed in the text.
. a and b correspond to the two ways of measuring time in the limit process discussed in the text.
It is also of interest to know how E[Ttot] depends on the sample size n. Under Kingman's coalescent, 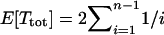 , so the dependence on n is logarithmic. The weak dependence on n when n is large under Kingman's coalescent, and the associated “diminishing return” on further sampling, has shaped discussions of sampling strategies for the measurement of sequence polymorphism (Pluzhnikov and Donnelly 1996). Figure 3 compares the dependence of E[Ttot] on n in simulations of the current model with 1 < γ < 2 for a range of values of ψ. The logarithmic dependence under Kingman's coalescent is shown for reference (Figure 3, thick curve). When ψ is small, the dependence on n is close to that under Kingman's coalescent, but becomes dramatically different (linear) as ψ increases to 1. To emphasize the dependence on n, rather than the effect of ψ that is shown in Figure 2, the values of Ttot in Figure 3 are normalized by the values at n = 5 for each ψ.
, so the dependence on n is logarithmic. The weak dependence on n when n is large under Kingman's coalescent, and the associated “diminishing return” on further sampling, has shaped discussions of sampling strategies for the measurement of sequence polymorphism (Pluzhnikov and Donnelly 1996). Figure 3 compares the dependence of E[Ttot] on n in simulations of the current model with 1 < γ < 2 for a range of values of ψ. The logarithmic dependence under Kingman's coalescent is shown for reference (Figure 3, thick curve). When ψ is small, the dependence on n is close to that under Kingman's coalescent, but becomes dramatically different (linear) as ψ increases to 1. To emphasize the dependence on n, rather than the effect of ψ that is shown in Figure 2, the values of Ttot in Figure 3 are normalized by the values at n = 5 for each ψ.
Figure 3.

The expected total length of the gene genealogy (the average of 100,000 replicates) as a function of the sample of size n and for five different values of ψ. To emphasize the dependence on n, for each ψ the values are normalized by the values at the smallest (leftmost point) sample size, n = 5. The thick solid line shows the predictions for Kingman's coalescent.
The shapes of gene genealogies can also be very different under the current model than under Kingman's coalescent. For example, when ψ is large, gene genealogies will tend to be star shaped, with all ancestral lines emanating from the MRCA. One way to measure the shape of a gene genealogy is to compute the total length of all branches that are ancestral to 1, 2,…, n − 1 members of the sample. Let Ti be the sum of the lengths of all branches in the gene genealogy that have i descendants in the sample. The tests of Tajima (1989) and Fu and Li (1993), which are often described as tests of selective neutrality, in fact simply detect deviations from the predictions of Kingman's coalescent about Ti, under the additional assumption of the infinitely many sites mutation (Watterson 1975).
Figure 4 shows the dependence of E[Ti] on ψ, estimated from simulations for a sample of size n = 4. The values are given as fractions of the expected total tree length E[Ttot], so that they sum to one for each value of ψ. When ψ is small, the allocation to different kinds of branches is similar to that under Kingman's coalescent, in which case E[T1]/E[Ttot] = 0.55, E[T2]/E[Ttot] = 0.27, and E[T3]/E[Ttot] = 0.18 when n = 4. As ψ grows, the genealogy becomes dominated by external branches, and this is of course true for samples of any size. For small samples it is possible to generate analytical predictions for E[Ti] or other quantities by enumerating all possible gene genealogies. The lines in Figure 4 show the predictions for n = 4 derived in the appendix. Although Figure 4 implies that ψ needs to be relatively large for the differences from Kingman's coalescent to become apparent, the application to data below indicates a greater sensitivity to ψ for larger samples.
Figure 4.

The expected unfolded site frequencies as a fraction of the total length of the gene genealogy for a sample of size n = 4, computed as the average over 1 million simulation replicates, as a function of ψ.
Application to Pacific oyster data:
We used the program described above as the basis for a method of inferring θ and ψ from samples of genetic data. As noted already, Pacific oysters may have a population structure in which many or most individuals leave few offspring, or none at all, while others may even replace the entire population if conditions are favorable (Hedgecock 1994). Our model is a simplified version of this, in which reproduction events are nonoverlapping in time and where large reproduction events are of a single type (an individual replaces a fraction ψ of the population). These Nψ-reproduction events occur with probability N−γ at each reproduction event and, in basing our method of inference on the program above, we also assume that 1 < γ < 2. Figures 2 and 4 imply that we should be able to estimate θ and ψ on the basis of information about T1, T2,…, Tn−1 (and/or Ttot). Note that, just as it is impossible to disentangle N and μ in Kingman's coalescent without independent knowledge of one or the other, we cannot estimate γ, but only the composite parameter θ = 2Nγ−1μ.
We use the data of Boom et al. (1994), which are the result of a restriction-enzyme digest of mtDNA on a sample of size n = 141 individuals. These data were previously analyzed under a conceptually related model in which some fraction of a Wright–Fisher population produced all the offspring every generation and the other fraction produced no offspring at all (Wakeley and Takahashi 2003). We adopt the same framework for inference and fit the two parameters of our model by a computational maximum-likelihood method on the total number of segregating sites S and the number of singleton polymorphisms η1. Under the infinite-sites mutation model, these are identical to the total number of mutations on the gene genealogy and the total number of mutations on the external branches of the gene genealogy. Then, given a gene genealogy, S − η1 and η1 are independent and Poisson distributed with parameters θ(Ttot − T1)/2 and θT1/2, respectively. As above, Ttot is the total branch length of the genealogy and T1 is the total length of external branches. At each point in a grid of values of θ and ψ we estimated the log-likelihood of the data as the average over a large number of simulated genealogies.
The data are S = 50 and η1 = 31 in the sample of size n = 141, and a contour plot of the log-likelihood surface is shown in Figure 5. We estimated the surface by simulating 10,000 gene genealogies for each point in a grid composed of 80 values of ψ and 100 values of θ. Within the constraint of this grid, there were two maximum-likelihood points, whose approximate positions are marked with a single x in the figure. The points are adjacent on the grid and differ only in their values of ψ, which were 0.075 and 0.0775, while θ = 0.0308 at both points. We estimated E[S] and E[η1] at these two points using simulations and obtained average values 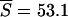 and
and 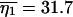 at the point (ψ = 0.075, θ = 0.0308) and
at the point (ψ = 0.075, θ = 0.0308) and 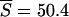 and
and 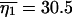 at the point (ψ = 0.0775, θ = 0.0308). In contrast, Kingman's coalescent, with its single parameter θ, cannot generate expected values close to S = 50 and η1 = 31. For example, if we estimate θ using Watterson's (1975) moment method, we obtain
at the point (ψ = 0.0775, θ = 0.0308). In contrast, Kingman's coalescent, with its single parameter θ, cannot generate expected values close to S = 50 and η1 = 31. For example, if we estimate θ using Watterson's (1975) moment method, we obtain 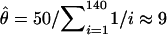 . Under Kingman's coalescent, the expected number of singletons is E[η1] = θ = 9, which is much smaller than the observed value η1 = 31.
. Under Kingman's coalescent, the expected number of singletons is E[η1] = θ = 9, which is much smaller than the observed value η1 = 31.
Figure 5.

The log-likelihood surface for the Pacific oyster data of Boom et al. (1994), estimated over a grid of points for ψ and θ using simulations. The maximum likelihood occurred at two adjacent points, which are covered by a single x. Contour lines are drawn every two log-likelihood units from the maximum.
DISCUSSION
It is not known what fraction of species conform to the assumptions of Kingman's coalescent. We have used a simple model to show that the dynamics of small-mutation-rate loci in large populations can display a number of interesting behaviors, depending on the distribution of offspring number among individuals. We focused on one limit process in which gene genealogies result from a multiple-mergers coalescent process (Pitman 1999; Sagitov 1999), but still some genetic variation should be observed if the population size is large enough. This ancestral process may be appropriate for many marine organisms (Hedgecock 1994) as it predicts a less-than-linear dependence of heterozygosity on actual population size and can account for the large numbers of low-frequency polymorphisms (e.g., η1 above) observed in some data. Even using a simple method of inference it is possible to estimate the parameters of the model from a sample of genetic data. The results suggest that the ancestral process in the Pacific oyster is a multiple-mergers coalescent in which a single individual may replace a significant fraction (8% by our estimate) of the population with its offspring.
Our results hold for a modified Moran model in which there is a chance that the parent has a large number of offspring. An important feature of this model is that generations are overlapping. Many organisms have overlapping generations, although we do not claim that the details of our model are true for any particular species. In contrast, most work in population genetics is done under the Wright–Fisher model of reproduction (Fisher 1930; Wright 1931), which is an idealized model of nonoverlapping generations. Under the standard assumptions, the Wright–Fisher model and the Moran model have the same ancestral limit process, and that is Kingman's coalescent. Interestingly, analysis of a modified Wright–Fisher model that is comparable to our modified Moran model yields a different range of ancestral limit processes. Consider a Wright–Fisher-type model in which most generations proceed according to the usual dynamics, but where occasionally (with probability N−α each generation) there is a single highly fecund individual. This special individual has chance ψ of being the parent of each individual in the next generation, while the other N − 1 individuals share the remaining fraction 1 − ψ of reproduction events according to the usual Wright–Fisher sampling process.
We show in the appendix that the range of ancestral processes under this modified Wright–Fisher model is similar, but not identical to those under our modified Moran model. This is due to the fact that in the modified Wright–Fisher model all individuals die and are replaced by offspring every generation (and thus all N have the potential to mutate), whereas in the modified Moran model only a fraction of individuals are replaced by offspring every time step. Consequently, there is no range of α > 0 in the modified Wright–Fisher model equivalent to 0 < γ ≤ 1 in the modified Moran model, where the population scaled mutation rate must tend to zero as N → ∞. The behavior of the modified Wright–Fisher model corresponds to that of the modified Moran model with γ > 1 if α = γ − 1. When γ > 2 and α > 1, the difference in opportunities for mutation in the two models is perfectly compensated for by the difference in probabilities of coalescence. Thus, consideration of large variances in offspring number uncovers a fundamental difference between models with overlapping vs. models with nonoverlapping generations.
As with any idealization, there are probably many aspects of our modified Moran model that would be unrealistic for a given species. Among other things, one might question whether the population size has been constant over time, whether all genetic variation is selectively neutral, whether the population is well mixed, and whether the age distribution is close to what our model would predict. Given the difference between our model and the modified Wright–Fisher model discussed above, it would be risky to extend the well-known robustness of Kingman's coalescent (Möhle 1998, 1999) to multiple-mergers coalescent processes. For example, the model we considered looks superficially similar to Wright–Fisher models with periodic, extreme bottlenecks or with periodic selective sweeps. However, in both these cases the limit process would include simultaneous multiple mergers—see Durrett and Schweinsberg (2005) for an analysis of periodic selective sweeps—rather than asynchronous multiple mergers (Sagitov 1999) as we have here.
Robustness results in population genetics are usually described in terms of effective population size. This term has been defined loosely to be the size of an ideal, Wright–Fisher population that would have the same “rate of genetic drift” as the population under consideration. The rate of genetic drift can be defined in several different ways (Ewens 1982), but the essential idea is that the dynamics of a complicated model can in some cases be shown to be identical to those of a simpler model via an effective population size alone. In other cases, the dynamics of a more complicated model cannot be reduced to those of a simpler model, and then there is no effective population size. For example, the well-known result that the effective size of a population whose size fluctuates over time is equal to the harmonic mean of the population sizes over time requires that the fluctuations are not too large and that the average population size does not change over time.
Sjödin et al. (2005) recently formalized the concept of the coalescent effective population size, which they argue should supplant all other definitions. The existence of a coalescent effective size means that the ancestral process for a sample from the population converges to Kingman's coalescent in the limit as the actual population size tends to infinity, so that all aspects of genetic variation in samples should conform to the predictions of Kingman's coalescent. In the limit process we apply to the Pacific oyster data of Boom et al. (1994), in which we have assumed 1 < γ < 2, the ancestral process is drastically different from Kingman's coalescent, so the coalescent effective size does not exist. Other definitions of effective size are similarly inapplicable and uninformative because the dynamics of genetic diversity in the population both forward and backward in time are in no sense equivalent to those of the idealized Wright–Fisher model. From the forward-time perspective, the presence of the Nψ-reproduction events would produce jumps in allele frequencies that would invalidate the usual diffusion approximation (Ewens 2004). The modified Moran model and the modified Wright–Fisher model considered here have effective population sizes in the usual sense only when γ > 2 and α > 1 and the ancestral limit process is Kingman's coalescent.
Acknowledgments
We thank Martin Möhle and Jason Schweinsberg for their insights and two anonymous reviewers for their comments. J.W. was supported by a Presidential Early Career Award for Scientists and Engineers (DEB-0133760) from the National Science Foundation.
APPENDIX
Ancestral limit processes with mutation:
Here we consider the limits of Equation 4 and Equation 6 as N → ∞, under the assumption that the number of ancestral lines i is finite and treating the parameters ψ and γ as constants, with 0 < ψ < 1 and γ > 0. We rewrite Equation 4 and Equation 6 as
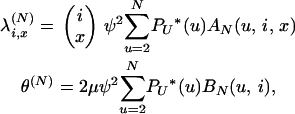 |
in which we define the functions AN(u, i, x) = (u − 2)x−2(N − u)i−x/(N − 2)i−2 and BN(u, i) = (N − u)i−1/(u(N − 2)i−2), and where
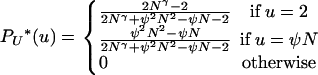 |
is obtained from Equations 5 and 7. We point out that we have not dealt explicitly with the fact that 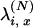 should be the rescaled rate of an x-merger and no mutation, but we note that the correction would simply be to multiply
should be the rescaled rate of an x-merger and no mutation, but we note that the correction would simply be to multiply 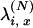 by 1 + O(μ).
by 1 + O(μ).
We consider the limits of PU*(u), AN(u, i, x), and BN(u, i) as N → ∞. For PU*(u), depending on the value of γ,
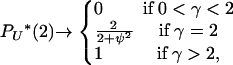 |
(A1) |
and of course 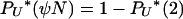 . Next, we have AN(2, i, 2) = 1, and AN(2, i, x) = 0 for x > 2, while
. Next, we have AN(2, i, 2) = 1, and AN(2, i, x) = 0 for x > 2, while
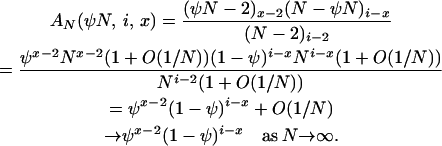 |
Using these results for PU*(2) and AN(u, i, x), if x = 2, then
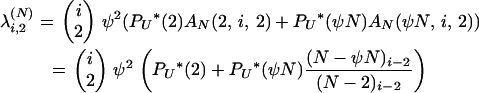 |
and
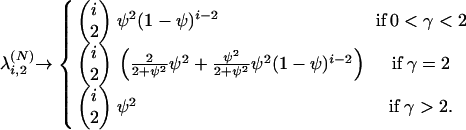 |
(A2) |
If x > 2, then
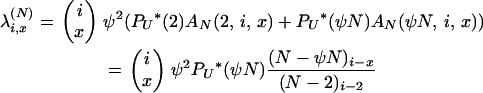 |
and
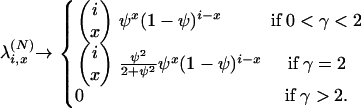 |
(A3) |
Equations A2 and A3 give the first column of Table 1.
Now consider BN(u, i) as N → ∞. We have the pair of equations
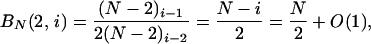 |
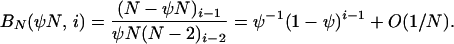 |
The scaled mutation parameter becomes
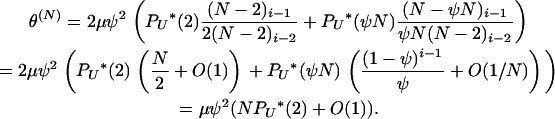 |
(A4) |
We assume that μ → 0 and N → ∞, which means that 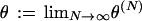 can be nonzero only if
can be nonzero only if 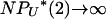 as N → ∞ and if we further assume that μ ∝ 1/(
as N → ∞ and if we further assume that μ ∝ 1/(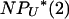 ). Therefore, to explore the full range of γ, it is necessary to know the rates at which
). Therefore, to explore the full range of γ, it is necessary to know the rates at which 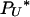 (2) and
(2) and 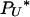 (ψN) approach their limits given in Equation A1. Analysis of the first line of Equation A4 reveals
(ψN) approach their limits given in Equation A1. Analysis of the first line of Equation A4 reveals
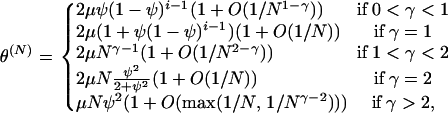 |
(A5) |
which, together with assumptions about how μ scales with N, gives column 2 of Table 1. It is interesting to note that, in the first two cases above, θ(N) depends on the number of ancestral lines, i. When 0 < γ ≤ 1, mutations on the different lines are not independent because they occur at ψN-reproduction events, where it is possible that several lines mutate at once.
Expected lengths of i-branches:
Here we derive the total length of all branches that have i = 1, 2, 3 descendants in a sample of size four. Let qi,x be the probability of an x-merger among i ancestral lines given that a merger has occurred. Thus,
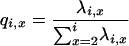 |
(A6) |
and 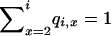 . With respect to the site frequencies, or the total length of branches in the ancestry of the sample that have j = 1, 2,…, i − 1 descendants in the sample, there are only five possible gene genealogies of a sample of size four, and these are defined by the series of events that takes the sample from the present time back to the MRCA.
. With respect to the site frequencies, or the total length of branches in the ancestry of the sample that have j = 1, 2,…, i − 1 descendants in the sample, there are only five possible gene genealogies of a sample of size four, and these are defined by the series of events that takes the sample from the present time back to the MRCA.
Let 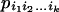 be the probability of a gene genealogy in which a series of k mergers takes the sample back to its MRCA, and where ij is equal to the number of ancestral lines present between the (j − 1)st and the jth mergers. The probabilities of the five possible gene genealogies of a sample of size four are
be the probability of a gene genealogy in which a series of k mergers takes the sample back to its MRCA, and where ij is equal to the number of ancestral lines present between the (j − 1)st and the jth mergers. The probabilities of the five possible gene genealogies of a sample of size four are 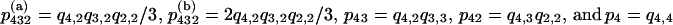 . The first two are the two possible kinds of rooted binary trees for a sample of size four, which differ in the number of tips at either side of the root: 2 and 2 in (a) vs. 3 and 1 in (b).
. The first two are the two possible kinds of rooted binary trees for a sample of size four, which differ in the number of tips at either side of the root: 2 and 2 in (a) vs. 3 and 1 in (b).
When there are i ancestral lines, the expected time back to the next merger is equal to 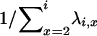 . The structure of the gene genealogy determines how many branches in the interval are ancestral to one, two, or three members of the sample and thus would contribute to either T1, T2, or T3, respectively. For example, all the branches in the star tree, which has probability p4, are included in T1. Considering each of the five possible trees, we have
. The structure of the gene genealogy determines how many branches in the interval are ancestral to one, two, or three members of the sample and thus would contribute to either T1, T2, or T3, respectively. For example, all the branches in the star tree, which has probability p4, are included in T1. Considering each of the five possible trees, we have
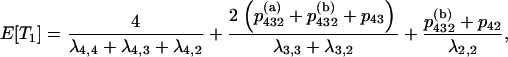 |
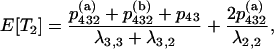 |
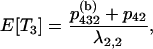 |
which gives
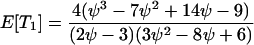 |
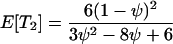 |
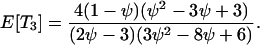 |
The expected total length of the gene genealogy of a sample of size four, E[Ttot], is equal to the sum of these, and Figure 4 plots E[Ti]/E[Ttot] for i = 1, 2, 3.
Overlapping vs. nonoverlapping generations:
Here we compare the results for a sample of size two under the modified Moran model to those under the modified Wright–Fisher model. We consider the probability of coalescence G2,2 and the expected number of pairwise differences E[K].
For the modified Moran model, from Equation 2 and Equation 7 in the main text, we have
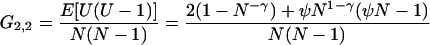 |
and this gives
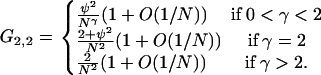 |
(A7) |
Further, when μ is small, and considering the different cases in which a mutation can occur, yields the following recursion for the expected number of pairwise differences:
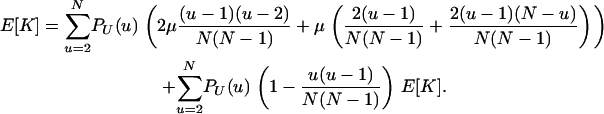 |
Upon rearrangement, this gives
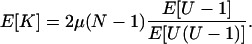 |
We note that the limit condition θ > 0 is identical to the condition 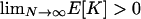 . Given the distribution in Equation 7 in the main text, this becomes
. Given the distribution in Equation 7 in the main text, this becomes
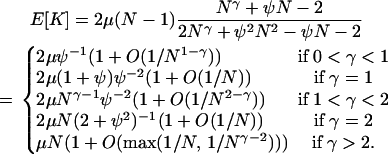 |
(A8) |
The discrepancy between the first two cases above and the first two cases in Equation A5 is attributable to the fact that mutations on different lines are not independent.
Compare these results to those for the modified Wright–Fisher model, in which all adults die each generation and are replaced by offspring, all of which can mutate. We assume that with probability N−α each generation, where α > 0, a single adult has probability ψ of being the parent of each individual in the next generation. If this happens, then each of the other N − 1 adults has chance (1 − ψ)/(N − 1) of being the parent of each individual in the next generation. With probability 1 − N−α each generation, the standard Wright–Fisher model holds, in which each adult has chance 1/N of being the parent of each individual in the next generation. Under this model,
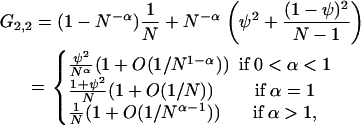 |
(A9) |
and we can compare this to Equation A7. Analysis of the expected number of differences between the two samples when μ is small, or E[K] = 2μ/G2,2, gives
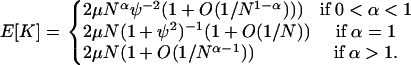 |
(A10) |
This verifies that the limit process for the modified Wright–Fisher model is simpler than that of the modified Moran model in the sense that Equation A10 contains no cases in which E[K] must tend to 0 if μ → 0 as N → ∞. One can check that the limit process for the modified Wright–Fisher model is a multiple-mergers process in the case 0 < α ≤ 1, rather than a Kingman coalescent, by showing that 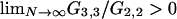 (Möhle and Sagitov 2001). Note that, similarly to the case 1 < γ < 2 in the modified Moran model, it is necessary to assume that the mutation rate scales less than linearly with population size in the modified Wright–Fisher model when 0 < α < 1 if the model is to predict any genetic variation.
(Möhle and Sagitov 2001). Note that, similarly to the case 1 < γ < 2 in the modified Moran model, it is necessary to assume that the mutation rate scales less than linearly with population size in the modified Wright–Fisher model when 0 < α < 1 if the model is to predict any genetic variation.
References
- Árnason, E., 2004. Mitochondrial cytochrome b variation in the high-fecundity Atlantic cod: trans-Atlantic clines and shallow gene genealogy. Genetics 166: 1871–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkner, M., J. Blath, M. Capaldo, A. Etheridge, M. Möhle et al., 2005. Alpha-stable branching processes and beta-coalescents. Electron. J. Probab. 10: 303–325. [Google Scholar]
- Boom, J. D. G., E. G. Boulding and A. T. Beckenbach, 1994. Mitochondrial DNA variation in introduced populations of Pacific oyster, Crassostrea gigas, in British Columbia. Can. J. Fish. Aquat. Sci. 51: 1608–1614. [Google Scholar]
- Crow, J. F., and M. Kimura, 1970. Introduction to Population Genetics Theory. Harper & Row, New York.
- Drake, J. W., B. Charlesworth and D. Charlesworth, 1998. Rates of spontaneous mutation. Genetics 148: 1667–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrett, R., and J. Schweinsberg, 2005. A coalescent model for the effect of advantageous mutations on the genealogy of a population. Stoch. Proc. Appl. 115: 1628–1657. [Google Scholar]
- Ewens, W. J., 1982. On the concept of effective size. Theor. Popul. Biol. 21: 373–378. [Google Scholar]
- Ewens, W. J., 2004. Mathematical Population Genetics I. Theoretical Introduction. Springer-Verlag, Berlin.
- Fisher, R. A., 1930. The Genetical Theory of Natural Selection. Clarendon, Oxford.
- Fu, X.-Y., and W.-H. Li, 1993. Statistical tests of neutrality of mutations. Genetics 133: 693–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie, J. H., 2000. Genetic drift in an infinite population: the pseudo-hitchhiking model. Genetics 155: 909–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedgecock, D., 1994. Does variance in reproductive success limit effective population sizes of marine organisms?, pp. 1222–1344 in Genetics and Evolution of Aquatic Organisms, edited by A. Beaumont. Chapman & Hall, London.
- Hedrick, P. W., 2005. Large variance in reproductive success and the Ne/N ratio. Evolution 59: 1596–1599. [PubMed] [Google Scholar]
- Hudson, R. R., 1983. Testing the constant-rate neutral allele model with protein sequence data. Evolution 37: 203–217. [DOI] [PubMed] [Google Scholar]
- Kingman, J. F. C., 1982. a The coalescent. Stoch. Proc. Appl. 13: 235–248. [Google Scholar]
- Kingman, J. F. C., 1982. b On the genealogy of large populations. J. Appl. Probab. 19A: 27–43. [Google Scholar]
- Li, W.-H., 1997. Molecular Evolution. Sinauer Associates, Sunderland, MA.
- Möhle, M., 1998. Robustness results for the coalescent. J. Appl. Probab. 35: 438–447. [Google Scholar]
- Möhle, M., 1999. Weak convergence to the coalescent in neutral population models. J. Appl. Probab. 36: 446–460. [Google Scholar]
- Möhle, M., and S. Sagitov, 2001. A classification of coalescent processes for haploid exchangeable population models. Ann. Appl. Probab. 29: 1547–1562. [Google Scholar]
- Moran, P. A. P., 1958. Random processes in genetics. Proc. Camb. Philos. Soc. 54: 60–71. [Google Scholar]
- Moran, P. A. P., 1962. Statistical Processes of Evolutionary Theory. Clarendon Press, Oxford.
- Nordborg, M., 2001. Coalescent theory, pp. 179–212 in Handbook of Statistical Genetics, edited by D. J. Balding, M. J. Bishop and C. Cannings. John Wiley & Sons, Chichester, UK.
- Pitman, J., 1999. Coalescents with multiple collisions. Ann. Probab. 27: 1870–1902. [Google Scholar]
- Pluzhnikov, A., and P. Donnelly, 1996. Optimal sequencing strategies for surveying molecular genetic diversity. Genetics 144: 1247–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagitov, S., 1999. The general coalescent with asynchronous mergers of ancestral lines. J. Appl. Probab. 36: 1116–1125. [Google Scholar]
- Schweinsberg, J., 2000. Coalescents with simultaneous multiple collisions. Electron. J. Probab. 5: 1–50. [Google Scholar]
- Schweinsberg, J., 2003. Coalescent processes obtained from supercritical Galton-Watson processes. Stoch. Proc. Appl. 106: 107–139. [Google Scholar]
- Sjödin, P., I. Kaj, S. Krone, M. Lascoux and M. Nordborg, 2005. On the meaning and existence of an effective population size. Genetics 169: 1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, F., 1983. Evolutionary relationship of DNA sequences in finite populations. Genetics 105: 437–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavaré, S., 2004. Ancestral inference in population genetics, pp. 1–188 in École d'Été de Probabilités de Saint-Flour XXXI–2001 (Lecture Notes in Mathematics, Vol. 1837), edited by O. Cantoni, S. Tavaré and O. Zeitouni. Springer-Verlag, Berlin.
- Turner, T. F., J. P. Wares and J. R. Gold, 2002. Genetic effective size is three orders of magnitude smaller than adult census size in an abundant, estuarine-dependent marine fish (Sciaenops ocellatus). Genetics 162: 1329–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeley, J., and T. Takahashi, 2003. Gene genealogies when the sample size exceeds the effective size of the population. Mol. Biol. Evol. 20: 208–213. [DOI] [PubMed] [Google Scholar]
- Watterson, G. A., 1975. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7: 256–276. [DOI] [PubMed] [Google Scholar]
- Wright, S., 1931. Evolution in Mendelian populations. Genetics 16: 97–159. [DOI] [PMC free article] [PubMed] [Google Scholar]