

Abstract
Geneticists seeking to understand HIV-1 evolution among human hosts generally assume that hosts represent a panmictic population. Social science research demonstrates that the network patterns over which HIV-1 spreads are highly nonrandom, but the effect of these patterns on the genetic diversity of HIV-1 and other sexually transmitted pathogens has yet to be thoroughly examined. In addition, interhost phylogenetic models rarely account explicitly for genetic diversity arising from intrahost dynamics. This study outlines a graph-theoretic framework (exponential random graph modeling, ERGM) for the estimation, inference, and simulation of dynamic partnership networks. This approach is used to simulate HIV-1 transmission and evolution under eight mixing patterns resembling those observed in empirical human populations, while simultaneously incorporating intrahost viral diversity. Models of parametric growth fit panmictic populations well, yielding estimates of total viral effective population on the order of the product of infected host size and intrahost effective viral population size. Populations exhibiting patterns of nonrandom mixing differ more widely in estimates of effective population size they yield, however, and reconstructions of population dynamics can exhibit severe errors if panmixis is assumed. I discuss implications for HIV-1 phylogenetics and the potential for ERGM to provide a general framework for addressing these issues.
UNDERSTANDING genetic variation in human immunodeficiency virus (HIV)-1 is a crucial public health issue, since its high mutation rate creates myriad genetic forms differing in infectivity, virulence, and susceptibility to treatment. Geneticists have spent considerable effort both in describing the distribution of this variation and in uncovering the functional differences that these forms create. These tasks are complicated by the fact that HIV-1 forms a metapopulation with different evolutionary forces operating at two levels. Each infected host contains her own complex pathogen population, in which genetic diversity is a function of familiar population genetic forces including mutation, drift, and selection. Individual hosts' viral pools are then linked epidemiologically and phylogenetically into a single metapopulation, where the forces shaping genetic diversity include these concepts as well as the patterns of host behavior transmitting the pathogen. Considerable work on the genetics of metapopulations exists (e.g., Chesser et al. 1993; Hedrick and Gilpin 1997; Beerli and Felsenstein 2001; Wakeley and Aliacar 2001); however, the metapopulation dynamics of HIV-1 are unique in many ways (new demes founded at rates determined by patterns of human sexual or injection drug behavior, demes isolated once founded, and guaranteed extinction of demes ∼5–20 years after their founding) and require an exploration incorporating these unique features.
In particular, common experience and a large body of social science research (e.g., Laumann et al. 1994) tell us that partner selection patterns for the behavior transmitting HIV-1 are highly nonrandom. Such substructure is known to introduce irregularities into the growth patterns of an epidemic and consequently into the genetic diversity within that epidemic. Quantifying the effects of realistic behavioral models by finding analytical solutions in a population genetics framework is generally quite difficult, if not impossible, however. Much work has now been done, relating patterns of HIV-1 genetic diversity to host population history when the latter is either stable or growing in some parametric trajectory (e.g., Grassly et al. 1999; Holmes et al. 1999; Pybus et al. 2000). This literature discusses the fact that nonrandom mixing patterns may substantially affect the outcome of these models and tests simple models of social subdivision, but otherwise does not seek to quantify these potential effects.
Previous work also tends to equate the estimates of viral effective population size it derives with the effective number of hosts through time, thus assuming that intrahost viral dynamics contribute little to genetic diversity occurring between viral samples from different hosts (e.g., Grassly et al. 1999). However, viral diversity emerges rapidly within an infected host, and anyone who infects multiple others late in infection may do so with viral sequences that are separated from a common ancestor by many years. This is certain to increase the effective size of the interhost viral population above simply the number of hosts. An enormous amount of research has examined these intrahost dynamics, but the modeling of inter- and intrahost dynamics has remained largely unintegrated. This is not surprising, since work on each alone has proven immensely challenging. However, network structure affects the distribution of waiting times to transmission, sometimes called the “pretransmission interval” (Leitner and Albert 1999), which in turn determines the size of the effect that intrahost genetic diversity has on interhost genetic diversity.
In this article I present a graph-theoretic framework for quantifying and simulating arbitrarily complex patterns of structured (i.e., nonrandom) host mixing, using a general class of probability models, known as exponential random graph models (ERGM). These models are then applied to the simulation of populations undergoing panmixis and to seven types of structured mixing whose general forms are informed by the literature on community-level sexual networks. HIV-1 transmission and evolution are then simulated within these populations using an approach that incorporates both intrahost and interhost viral evolution. Phylogenetic trees are constructed using a sample of hosts from each run, which are analyzed using skyline plot methods to derive estimates for current effective population size and growth rate under models of exponential, logarithmic, and “expansion” growth (Pybus et al. 2000). These estimates are then compared to known census population size and growth rate.
BACKGROUND
HIV-1 interhost phylogenetics:
Applications of phylogenetic methods to HIV-1 focused initially on determining genetic distance between samples of sequences where the patterns of relationships were themselves of primary interest (e.g., Ou et al. 1992 for a dental practice, Leigh Brown et al. 1997 for risk groups and cities in the British Isles, and McCutchan et al. 1992 for major HIV-1 subtypes). A more recent application involves reconstructing the population dynamics of the infected human population, including the size and growth rate of some subsection of the epidemic through time, from a small sample of hosts in that subsection. Grassly et al. (1999) use mismatch distributions and a likelihood approximation to compare the population history of HIV-1 subtypes A and B under the assumption of exponential growth. Holmes et al. (1999) uses a technique developed earlier (Nee et al. 1994, 1995) to compare the reconstructed number of phylogenetic lineages through time to that expected under constant population size, linear growth, and exponential growth. Pybus et al. (2000) and Strimmer and Pybus (2001) extend this method to include the generation of maximum-likelihood estimates of the effective population size of the virus (Ne) at all points in the past using phylogenetic trees. This uses the size of the interval between each pair of coalescent events in a reconstructed tree to estimate the harmonic mean of Ne during the interval, which in most scenarios is close to the arithmetic mean and thus to the maximum-likelihood estimate. This allows them to obtain maximum-likelihood values for parameters of specific population growth patterns as well as compare nested hypotheses of parametric growth scenarios.
Ne is the effective population size, the size of the hypothetical (“Wright–Fisher”) population that would yield an observed level of genetic diversity if all members had an equal chance of contributing offspring to the next generation. “Equal chance” requires a lack of selection and implies certain outcomes—among them, that the number of offspring left in a subsequent generation by each member of the prior is Poisson and that no correlation in offspring number exists for successive generations of a lineage. Genetic diversity in this type of population is generated in well-understood ways. Of course few real populations fit every assumption of the Wright–Fisher model; the framework's value comes from numerous formulas relating Ne to census population size for various violations. This provides a common metric for comparing populations and for testing hypotheses about their demographic history or exposure to selection. HIV-1 metapopulation dynamics share general features with some of these models, and they can thus provide us with expectations as to how these patterns will generally diverge from panmixis. In the framework of source-sink models (Gaggiotti 1996), for instance, newly HIV-infected individuals show some of the characteristics of a “sink” and infecting individuals those of a “source.” Unfortunately, existing models cannot capture all of the complex partnership patterns we see empirically in the human sexual/injection networks that result in HIV-1 phylogenies. For instance, assortative mixing by partner number (i.e., highly active people tending to have highly active partners) implies a correlation in offspring number among successive viral generations by lineage purely for network reasons, a phenomenon that has received little attention in the genetics literature. In addition, most theoretical work on effective population size has been developed for diploid organisms with sexual reproduction, while HIV-1 is a haploid virus (albeit one that experiences a form of recombination); the concept of Ne still holds here, but is based solely on variation in reproductive success among lineages rather than on degrees of relatedness within breeding pairs. Because of these limitations, most work on HIV-1 phylogenetics thus far has invoked the reasonable assumption that the genetic signatures of network irregularities will not be great, so Ne is likely to be a good approximation of the number of infected hosts. One exception is Grassly et al. (1999), who compare mismatch data to models of population panmixis and binary subdivision. They found that at the global level, the improved fit to the subdivision model was not sufficiently large to warrant the increase in model complexity.
The existing literature on interhost phylogenetics and HIV population dynamics also assumes that the effects of intrahost viral diversity are negligible. Each infected person harbors a viral population containing on the order of 108 different genetic sequences (Vartanian et al. 1992), which changes constantly through drift, mutation, and selection. However, estimates of Ne during the long latency period are more on the order of 103 (Leigh Brown 1997; Nijhuis et al. 1998; Rodrigo et al. 1999; but see Rouzine and Coffin 1999), presumably since only a small fraction of infected cells replicate. This diversity appears within a few months of infection and is maintained throughout the many years of clinical latency. Given this, the sequences that a host contributes to each of the other people he infects (or to sequence analysis) may be separated from each other by years of evolution, plenty of time for HIV-1 to develop substantial genetic distance. The actual amount of time until the most recent common ancestor for two such lineages is determined both by intrahost viral dynamics and by the expected waiting time between getting infected and infecting another, which is a function of partnership network structure.
For n infected hosts, each with its own effective viral pool of size Nei (for “intrahost Ne”), the total viral effective population size across hosts (Ne) should be on the order of nNei, which can be thought of as the interhost viral census size. (This is not strictly true, of course, since Nei is the effective intrahost size. For the purposes of this article, however, these intrahost dynamics are conditioned upon, and the phrase “interhost viral census size” is considerably less cumbersome than are more precise alternatives.)
Although in diploid organisms subdivided metapopulations display values of Ne that are considerably lower than census population size, the reason for this (subdivision increases the expected degree of relatedness within mating pairs compared to panmixis) does not apply for haploid populations, in which the concept of Ne is not based on inbreeding. We might expect Ne to differ from nNei under host panmixis, however, due to the common practice of sampling a single lineage from each participant in an interhost viral phylogenetic analysis, which prevents similar sequences found in individual viral pools from being jointly sampled. This effect should be pronounced only when the number of hosts studied is a sizeable portion of the total host population under investigation.
Violations of host panmixis observed in practice generally involve greater-than-Poisson variance in the number of sexual or injection partners. On the whole these should reduce Ne below nNei, possibly by a considerable amount. Comparing 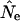 obtained from phylogenetic methods to nNei allows us to see the combined effect of departures from the panmictic demographic model under consideration, isolation of the hosts' viral pools from each other, and the strategies of sampling sequences by host, but does not allow us to separate these effects out from one another. However, by comparing panmictic populations (in which the latter two phenomena also appear) to violations of panmixis, we can isolate the additional effect of structured host mixing on
obtained from phylogenetic methods to nNei allows us to see the combined effect of departures from the panmictic demographic model under consideration, isolation of the hosts' viral pools from each other, and the strategies of sampling sequences by host, but does not allow us to separate these effects out from one another. However, by comparing panmictic populations (in which the latter two phenomena also appear) to violations of panmixis, we can isolate the additional effect of structured host mixing on 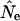 .
.
Network epidemiology:
The methods used here to model transmission are drawn from social network analysis, an outgrowth of both graph theory and social theory in which analysis focuses on the network of relationships between pairs of agents. Within this framework I focus on a probabilistic model class known as ERGM or p* modeling, first developed in the spatial statistics literature (Besag 1974; Frank and Strauss 1986; Strauss and Ikeda 1990) and introduced to social network analysis by Wasserman and Pattison (1996). This approach derives a model for partnership formation by defining probabilities for each possible graph (or “network”) containing n actors. Let xij represent the value of the tie between nodes i and j; if, as here, relationships are binary (xij = 1 if a tie is present or 0 if a tie is absent) and nondirected (xij = xji ∀ i, j), then a graph x is defined by its 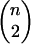 tie values
tie values 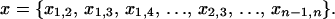 In its general form, the model represents the probability that a random graph X will take on value x as
In its general form, the model represents the probability that a random graph X will take on value x as
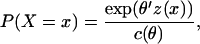 |
(1) |
where z(x) is a vector of network statistics, θ is a vector of parameters, and c(θ) is a normalizing constant ensuring that the probabilities sum to one over all graphs with n nodes. Examples of commonly used z-statistics include the number of ties in the graph, the number of nodes with a certain number of ties, or the number of triads (sets of three nodes that possess all pairwise ties). Equation 1 can be reformulated in terms of the log-odds of a single tie,
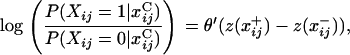 |
where 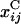 represents the complement of xi,j,
represents the complement of xi,j, 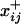 represents the network with xi,j = 1, and
represents the network with xi,j = 1, and 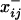 represents the network with xi,j = 0. This formulation removes the normalizing constant while also highlighting the potentially recursive nature of tie probabilities, since z-statistics may depend on values of other ties. The presence of c prevents the probability of any graph or the marginal probability of any tie from being calculated directly. Instead, Markov chain Monte Carlo (MCMC) is used to draw samples from the proper distribution.
represents the network with xi,j = 0. This formulation removes the normalizing constant while also highlighting the potentially recursive nature of tie probabilities, since z-statistics may depend on values of other ties. The presence of c prevents the probability of any graph or the marginal probability of any tie from being calculated directly. Instead, Markov chain Monte Carlo (MCMC) is used to draw samples from the proper distribution.
This representation is so general as to include any possible probability model based on network statistics (Besag 1974); it allows for various social structures to be represented in a common statistical framework, but also emphasizes the need for guidance in choosing parameterizations. This can be provided by the growing number of studies that have collected network data on sexual relationships, as well as by modeling work showing the effect of network structure on HIV-1 transmission patterns (e.g., Martin 1987; Orubuloye et al. 1991; Klovdahl et al. 1994; Garnett et al. 1996; Morris et al. 1996; Friedman et al. 1997; Rothenberg et al. 1998). Assortative mixing by social attributes (race, age, location, and occupation) is commonly observed and can greatly affect the dynamics of disease spread (Morris and Dean 1994), as can the existence of a “core group” that is not only highly active but preferentially mixes with other highly active people (Garnett et al. 1996). Watts and May (1992) and Morris and Kretzschmar (1997) demonstrated that the timing of partnerships (concurrent vs. serial) is a strong determinant of prevalence. Another important pattern includes “bridges,” or individuals who serve as epidemiological links between groups that otherwise would have no interaction, as when men have both commercial and noncommercial female sex partners (Morris et al. 1996). Unfortunately, none of these studies has sequenced HIV-1 from seropositive subjects, which could allow for a simultaneous examination of network structure and phylogenetics in a real population.
METHODS
The above qualitative observations of the social networks literature suggested eight mixing patterns under which to simulate HIV-1 evolution: one panmictic host population (random pattern), four populations divided into equally active subgroups with internal preferential mixing (assortative patterns), two populations with a small highly active subgroup (core patterns), and one bridges pattern. This last pattern consists of husbands, wives, and commercial sex workers (CSWs); each husband/wife pair maintains its relationship throughout the course of the simulation, while husbands may have simultaneous ties with CSWs; initial infection always occurs in a CSW. Details of individual mixing patterns are listed in Table 1 and depicted schematically in Figure 1, while the statistics and parameters necessary to create them are in Table 2. Parameter values were calculated using the likelihood approach of Strauss and Ikeda (1990). (Note that Strauss and Ikeda refer to the result of this method as a pseudolikelihood; however, in each model here the probability of each tie is independent of the existence of all other ties, and their method yields the true likelihood.) For each mixing pattern 100 runs were simulated, each for 10 years. The exception is random mixing, which served as a basis of comparison for other patterns in some analyses, and for which 10,000 runs were simulated. Each run contains 200 actors sharing an expected 200 partnerships at any moment, although the number fluctuates with probabilities determined by the model parameterization. Equal activity levels imply that systematic differences in outcome should relate to the pattern, not the magnitude, of partnerships. Populations are small since computing needs for network simulation scale with the square of population size, a continuing limitation on network-based methods.
TABLE 1.
Population mixing models
| Model | Abbreviation | Description |
|---|---|---|
| Random | Random | All partnerships equally likely |
| Two subgroups, weak assortativity | 2-strong | Population divided into two groups of 100; 150 intragroup ties and 50 intergroup ties expected |
| Two subgroups, strong assortativity | 2-weak | Population divided into two groups of 100; 195 intragroup ties and 5 intergroup ties expected |
| Eight subgroups, weak assortativity | 8-strong | Population divided into eight groups of 25; 150 intragroup ties and 50 intergroup ties expected |
| Eight subgroups, weak assortativity | 8-weak | Population divided into eight groups of 25; 195 intragroup ties and 5 intergroup ties expected |
| Core/periphery, strong assortativity | Core-strong | Population divided into active group of 25 and periphery of 175; 100 intracore ties, 95 intraperiphery ties, 5 core-periphery ties expected |
| Core/periphery, weak assortativity | Core-weak | Population divided into active group of 25 and periphery of 175; 30 intracore ties, 150 intraperiphery ties, 20 core-periphery ties expected |
| Bridges | Bridges | Population divided into 95 husbands, 95 wives, and 10 commercial sex workers. Spouses remain married throughout, 105 ties between husbands and other females expected |
In each assortative population (2-strong, 2-weak, 8-strong, 8-weak), individuals chose partners preferentially from within their own cluster, but all clusters have equal activity. In the core populations, core members are more active than periphery members and also disproportionately choose other core members as partners.
Figure 1.

Graphical representation of the eight mixing models: (a) random, (b) 2-weak, (c) 2-strong, (d) core-weak, (e) core-strong, (f) 8-weak, (g) 8-strong, and (h) bridges. In all diagrams, the size of the dots represents their relative expected partner number. In a–g clusters of dots represent groups that choose each other as partners more than they choose members of other clusters; the distance between the clusters represents their relative probability of choosing across cluster. In h there are no clusters; joined pairs represent married couples who remain partnered throughout; the larger members of the pair are husbands, who enter into partnerships with commercial sex workers, shown in the center.
TABLE 2.
Graph statistics and parameters for each mixing model
| Model | Z | θ |
|---|---|---|
| Random | No. of ties | −4.590 |
| 2-strong | No. of ties | −3.907 |
| No. of intergroup ties | −3.693 | |
| 2-weak | No. of ties | −4.174 |
| No. of intergroup ties | −1.119 | |
| 8-strong | No. of ties | −2.426 |
| No. of intergroup ties | −4.895 | |
| 8-weak | No. of ties | −2.708 |
| No. of intergroup ties | −2.387 | |
| Core-strong | No. of ties | −5.071 |
| No. of core–periphery ties | −1.703 | |
| No. of intracore ties | 4.377 | |
| Core-weak | No. of ties | −4.610 |
| No. of core–periphery ties | −0.773 | |
| No. of intracore ties | 2.413 | |
| Bridges | No. of male–CSW ties | −2.085 |
| No. of male–male ties | −∞ | |
| No. of female–female ties | −∞ |
CSW, commercial sex worker.
The basic framework used in this investigation comprises three steps: (1) simulation of dynamic social networks, (2) simulation of HIV-1 transmission within these networks, and (3) simulation of mutation along a 500-base viral sequence among infected hosts. All three were implemented in a single software package, EvolveNet. This is available for download at http://faculty.washington.edu/goodreau, along with documentation for simulating networks according to user-defined mixing patterns, simulating viral transmission and evolution within those networks, and outputting to common tree formats (PHYLIP and MEGA).
Dynamic network simulation:
Dynamic networks of social relationships are modeled with the ERGM formulation in Equation 1, using an MCMC algorithm adopted for network data (Gilks et al. 1996; Snijders 2002). Each iteration of the algorithm comprises six steps:
Select two nodes i, j randomly.
Calculate Δzij(x), the amount by which the vector of z-statistics changes when xij is toggled from its current state to the opposite.
- Calculate the acceptance probability ratio (which avoids calculating c):
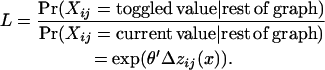
Select a random number r from a uniform (0, 1) distribution.
If L > r then accept the toggle for the updated state; otherwise retain the original value as the updated state.
Record the updated state of the network and increase the current time by Exponential ∼ (λs).
The acceptance rule in step 5 guarantees that the stationary distribution of the chain equals the probability distribution of Equation 1 (Metropolis et al. 1953). ERGMs with MCMC are generally used to simulate static networks from a probability model of network structure. However, since networks at consecutive steps in the chain are either identical or differ by one tie, this approach also provides a simple way to approximate dynamic networks (implemented in step 6) while retaining the instantaneous probability distribution of the static model. (In general there is no guarantee that this approach will yield a chain that resembles a real dynamic network process on the local scale. However, the relatively simple model parameterizations used here should at least ensure that the chain mixes well locally. More realistic methods for dynamic network modeling in the ERGM framework are still in development.) All models share the value of λs (13.63) that corresponds to a mean uncensored relationship duration of 4 years in these populations.
The first chain begins with no ties and is run through a 1-million-iteration burn-in before to virtually eliminate dependence on initial conditions. Snapshots are then taken every 100,000 steps and used as seed networks for each of the 10-year dynamic simulations. The outcome of this process is a set of 100 runs for each mixing pattern (10,000 for random), consisting of a set of relations and their starting and ending times.
One additional constraint was added for programming convenience; once a tie had formed between two actors and was then broken, it could not reform during the same 10-year simulation. Although this means that the resulting networks are not drawn perfectly from the stated probability distribution, the differences are small enough in sparse networks of this duration that the effects are assumed to be minimal.
Viral transmission:
In this simulation a constant, universal probability of transmission within each serodiscordant couple is used. This ignores many sources of heterogeneity, including time since infection of first partner, number of acts within partnership, and actor attributes such as genetic resistance to infection. However, this makes it possible to state that differences in outcome are due solely to mixing pattern. Estimates for mean infectivity per partnership per unit time do not appear in the literature; instead, a number (0.0007 infections/serodiscordant partnership/day) was selected because it yielded seroincidence in a simulated panmictic population approximating that found in various United States communities (Moss et al. 1994; Holmberg 1996).
Independent, constant infectivity implies that each serodiscordant partnership has a waiting time until transmission of Exponential ∼ (λt), where λt = 1/0.0007 days. The expected waiting time until the first transmission among all serodiscordant couples is thus Exp ∼ (sλt), where s is the number of serodiscordant partnerships. The quantity s changes every time a partnership forms or ends and every time a transmission event occurs. Transmission is thus modeled in each network by infecting one member randomly and then iterating the following steps beginning at time t0:
Calculate the number of serodiscordant partnerships s and the time until the next partnership forms or dissolves (tp).
Draw a random number r from Exponential ∼ (sλt).
If r < tp, add r to the current time, randomly select a serodiscordant partnership, and infect its seronegative member. Otherwise, advance the current time by tp.
Viral evolution:
Each infected host is represented in the final analysis by a single 500-base viral sequence, referred to as the host's sampled lineage; generating the genetic distance between an individual's sampled lineage and that of those they infect requires accounting for intrahost dynamics, ideally without modeling each host's entire viral pool. This is accomplished by using a model of intrahost dynamics to define the probability distribution for the timing of the sequences' most recent common ancestor. The model used here entails Ne = 1 at infection, followed by rapid expansion and contraction over the first 60 days and a subsequent steady state of Ne = 103 (Leigh Brown et al. 1997). For a constant-sized population, time back to the most recent coalescence event among n sequences in a population of size N follows an exponential distribution with parameter
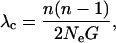 |
(2) |
where G is generation length (Rodrigo and Felsenstein 1999). Here Ne = 103 and G = 1.5 days, an average of the 1.2-day estimate of Rodrigo et al. (1999) and the 1.8-day estimate they cite from personal communication with Perelson. Any two sequences within a single host that have not coalesced by the beginning of the steady state are assumed to coalesce at the point of infection. Thus, the following steps are completed for each infection event (where actor i infects actor j), beginning with the most recent:
Determine the number of potentially coalescing lineages within actor i (ni). These include the sampled lineages of i, j and anyone else i has infected more recently than j but who has not coalesced in a previous step.
- Determine the most recent coalescence time t using Equation 2.
- If t is less recent than another infection event involving i, no coalescence occurs yet.
- If t is <60 days more recent than i's infection date, all ni lineages coalesce at i's infection date.
- Otherwise, two lineages coalesce. If ni > 2, the two coalescing lineages are selected randomly, ni is reduced by one, and step 2 is repeated for the reduced set of lineages.
HIV-1 mutation is simulated using Leitner and Albert's (1999) mean mutation rate for the env gene, 6.7 × 10−3 substitutions/base/year, or 1.8 × 10−5 substitutions/base/day. It also includes the env nucleotide frequencies and transition matrix from Leitner et al. (1997), both shown in Table 3, and gamma-distributed intersite mutation rates, using their point estimate α = 0.384 for env. The instantaneous mutation rate for site x (μx) equals the product of the overall mutation rate, the relative mutation rate of the nucleotide at x, and x's gamma mutation factor. (Note that the expected value of both the second and third factor is 1.) The effects of selection and recombination are ignored.
TABLE 3.
Allele frequencies and transition matrix for env
| A. Allele frequencies | ||||
| A | 0.4627 | |||
| C | 0.1474 | |||
| G | 0.1598 | |||
| T | 0.2302 | |||
| A | C | G | T | |
| B. Transition matrix | ||||
| A | −0.8351 | 0.2519 | 0.4599 | 0.1233 |
| C | 0.7909 | −1.3932 | 0.0574 | 0.5449 |
| G | 1.3318 | 0.0529 | −1.5406 | 0.1558 |
| T | 0.2477 | 0.3488 | 0.1081 | −0.7047 |
Data are from Leitner et al. (1997).
Simulation of mutation begins when the first person in the population is infected from an outside source and proceeds forward in time. The initial sequence for this person is generated randomly with nucleotide probabilities given in Table 3B. Mutations occur along this lineage by repeatedly calculating the current mutation rate μx for each site x, drawing a time until the next mutation tx for each site from Exponential ∼ (μx), and selecting min{t1, … , t500}. The new nucleotide is selected with probabilities derived from the corresponding row in Table 3B. This process is repeated for every infected actor, with the initial sequence in their sampled lineage matching that of the sequence to which it coalesces.
The sampled sequences from each host are used to generate mismatch distributions for the population. For each run in which ≥25 hosts are infected, 25 hosts are drawn randomly to contribute sampled lineages to a phylogenetic tree, built using an ultrametric Fitch–Margoliash distance method with power 2 (implemented in PHYLIP as KITSCH). Distance matrices are derived using the Kimura two-parameter model (implemented in PHYLIP as DNADIST), with a transition/transversion ratio (1.42) derived from the given mutation matrix and a coefficient of variation (1.614) corresponding to the Jin–Nei gamma parameter of 0.384 used in the simulation. The classic skyline plot method of Pybus et al. (2000) is then used to generate maximum-likelihood parameter estimates for the reconstructed trees under the assumptions of exponential growth, logistic growth, and “expansion growth” (exponential growth beginning from an arbitrary population size in the infinite past). All three of these are reasonable models for the process under investigation: the initial stages of an epidemic generally see exponential increases in prevalence; saturation of small populations like these may on the other hand yield logistic growth; while beginning with a single host whose viral pool jumps to 1000 shortly after infection suggests expansion growth may be most appropriate. Skyline plot analysis was conducted with Genie 2.0 (Pybus and Rambaut 2002a), using the Powell algorithm for maximum-likelihood (ML) estimation. Because the latter two methods are generalizations of exponential growth, the fit of each can be compared to that of the exponential model using a log-likelihood-ratio test. [Although one can also use a Kolmogorov–Smirnov test to examine fit for each model individually, the assumptions of this approach are rarely met in these populations (Pybus and Rambaut 2002b) and the power of this test varies widely over the range of sample sizes in this study (results not shown).]
Estimates obtained from Genie are expressed in terms of mutation events; rescaling by the mutation rate yields 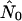 (estimate of current effective interhost viral population size) and
(estimate of current effective interhost viral population size) and 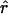 (estimate of the exponential growth rate of the effective interhost viral population size).
(estimate of the exponential growth rate of the effective interhost viral population size).
Note that in this analysis, the sampled population fraction of the host population used for phylogenetic analysis is much larger than one would usually have in practice (100% for the mismatch analysis and 25 of 25–130 for the phylogenetic trees). Coalescent methods require sample sizes to be much smaller than the population from which they are drawn for the approximations on which they are based to hold. However, each host represents an effective viral pool of 103, so the viral sample is much smaller than the viral population, which is the one under investigation.
RESULTS
Figure 2 compares HIV-1 prevalence across mixing patterns. Summary statistics for these distributions are contained in Table 4, including the results of a Kullback–Leibler distance test between each distribution and that of the random mixing pattern. [P-values were obtained by sampling 100 runs from the 10,000 runs of the random pattern, calculating the Kullback–Leibler (KL) distance for this sample from the distribution of all 10,000, and repeating 1000 times]. Subdivision into two groups shows no significant effect on prevalence, even when strongly isolated. All other patterns have prevalence distributions differing significantly from the random pattern, with clustering patterns lower and core and bridges patterns higher. These results are consistent with previous work described above, demonstrating network effects on HIV-1 transmission.
Figure 2.

Distribution of infected population across 100 runs of each model.
TABLE 4.
Distribution of infected hosts across 100 runs of each mixing pattern
| Model | Mean | SD | KL distance | P |
|---|---|---|---|---|
| Random | 27.5 | 24.7 | — | — |
| 2-strong | 26.9 | 20.5 | 0.260 | 0.37 |
| 2-weak | 25.6 | 22.0 | 0.228 | 0.65 |
| 8-strong | 14.0 | 7.7 | 1.327 | <0.001 |
| 8-weak | 23.5 | 19.6 | 0.361 | 0.01 |
| Core-strong | 42.7 | 9.6 | 1.992 | <0.001 |
| Core-weak | 52.7 | 28.2 | 0.594 | <0.001 |
| Bridges | 102.9 | 21.9 | 3.348 | <0.001 |
Kullback–Leibler (KL) distances are between the given distribution and 10,000 runs of the random mixing pattern. See text for calculation of P-values.
Figure 3 examines the relation between infected host size and the mean of the mismatch distribution for each mixing pattern. Here 10,000 runs of the random pattern are used to provide sufficient numbers for comparison at every host population size. Lines show the mean and 2.5 and 97.5% quantiles for random runs at each size. Points indicate runs for the relevant mixing pattern. For most patterns the observed level of genetic variation falls within the range observed in random runs most or all of the time; the exceptions are the core-strong and 8-strong patterns. In these cases mean genetic diversity tends to be higher than that in random populations of the same size, presumably because these patterns see rapid initial spread in the core or single subgroup, with little later on.
Figure 3.

Mismatch mean by infected host size, for each mixing pattern in comparison to 10,000 random runs.
More interesting differences appear in the analysis using full phylogenetic information. Table 5 shows the number of runs by mixing pattern for which each growth model provided the best fit. (For no run did both the logistic and the expansion models fit significantly better than exponential). Examples of logistic growth are found in all mixing patterns, but far more frequently in core-strong than in any other and far less in the 2-weak and bridges patterns. Just as with mismatch means, seeing logistic growth with the core-strong pattern is not surprising, since transmission is likely to spread rapidly in the core and then much more slowly into the periphery, inducing a marked leveling off of new infections. Why the 2-weak and bridges patterns are less likely than panmixis to follow a logistic growth model is not as apparent. Expansion models are not common with any pattern except 8-strong, where the number of runs is too small to draw strong conclusions. Figure 4, a–c, provides one example wherein each parametric growth model fits best.
TABLE 5.
Frequency of parametric models yielding best fit by likelihood-ratio test
| No. of runs with n ≥ 25 | No. with best-fitting model as
|
|||
|---|---|---|---|---|
| Model | Exponential (%) | Logistic (%) | Expansion (%) | |
| Random | 41 | 31 (76) | 7 (17) | 3 (7) |
| 2-strong | 48 | 38 (79) | 8 (17) | 2 (4) |
| 2-weak | 42 | 39 (75) | 2 (5) | 1 (2) |
| 8-strong | 8 | 6 (75) | 1 (13) | 1 (13) |
| 8-weak | 37 | 31 (84) | 6 (16) | 0 (0) |
| Core-strong | 100 | 56 (56) | 43 (43) | 1 (1) |
| Core-weak | 81 | 67 (83) | 10 (12) | 4 (5) |
| Bridges | 100 | 98 (98) | 2 (2) | 0 (0) |
Figure 4.

Examples of runs fit by different parametric growth models. Thick line, skyline plot; thin line, best-fitting model; dashed line, ML exponential model (for runs not fit best by exponential). (a) A 2-weak run fit best by the exponential model. (b) A core-strong run fit best by logistic growth. (c) A random run fit best by expansion growth. (d) A bridge run in which exponential yields a population estimate vastly larger than the census population, but for which no other model fits significantly better.
Figure 5 graphs the ratio of 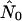 from the best-fitting model for each run against nNei for that run.
from the best-fitting model for each run against nNei for that run. 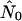 is in fact on the order of nNei for most runs and never on the order of n, the number of hosts alone. All but one run of the random pattern see N0 within one order of magnitude of nNei, and the same is true for most other mixing patterns as well. In all these cases the relative overestimate by the exponential model is largely independent of population size. In the range of values we see (N0r = ∼102–103), Pybus et al. (2000) in fact report an upward bias of
is in fact on the order of nNei for most runs and never on the order of n, the number of hosts alone. All but one run of the random pattern see N0 within one order of magnitude of nNei, and the same is true for most other mixing patterns as well. In all these cases the relative overestimate by the exponential model is largely independent of population size. In the range of values we see (N0r = ∼102–103), Pybus et al. (2000) in fact report an upward bias of 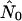 by roughly a factor of 2 [b(θ) = 1 in their terminology]. For the core-strong pattern, however,
by roughly a factor of 2 [b(θ) = 1 in their terminology]. For the core-strong pattern, however, 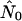 is often two to three orders of magnitude larger than nNei (and than
is often two to three orders of magnitude larger than nNei (and than 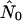 for a panmictic population of the same size.) The bridges pattern sees many runs up to two orders of magnitude larger than nNei, a phenomenon that varies strongly with seroprevalence. Figure 4d illustrates a case where all three models yield a very high
for a panmictic population of the same size.) The bridges pattern sees many runs up to two orders of magnitude larger than nNei, a phenomenon that varies strongly with seroprevalence. Figure 4d illustrates a case where all three models yield a very high 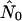 and neither logistic nor expansion growth fits better than exponential, even though the shape of the curve might suggest logistic growth. Similar patterns are seen in most of the runs for which
and neither logistic nor expansion growth fits better than exponential, even though the shape of the curve might suggest logistic growth. Similar patterns are seen in most of the runs for which 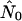 is high.
is high.
Figure 5.

Ratio of n0 from best-fitting model to nNei, by infected population size. Note that core-strong has an additional point at (26, 2.1 × 105).
Expansion growth is a reasonable model since Nei of the sole infected host jumps to 103 almost initially in the simulation. It is thus interesting to see what estimate for initial population size (that is, Ne in the infinite past) is obtained across all runs for which expansion growth is the best-fitting model. A boxplot of these values is shown in Figure 6, demonstrating that this approach does in fact center around 103.
Figure 6.

Boxplot of estimates for initial population size across all runs best fit by expansion growth.
Exponential growth and expansion growth both provide an estimate of the exponential growth rate 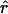 that can be compared to one calculated directly for the observed host population size. The viral population 2133 generations before present (3650 days/1.5 days per generation = 2133 generations) is 1, although it jumps to 103 shortly thereafter. Consider the r implied by a population growing exponentially from N2133 = 1 to N0 = 1000n or from N2133 = 1000 to N0 = 1000n; the values for r are simply
that can be compared to one calculated directly for the observed host population size. The viral population 2133 generations before present (3650 days/1.5 days per generation = 2133 generations) is 1, although it jumps to 103 shortly thereafter. Consider the r implied by a population growing exponentially from N2133 = 1 to N0 = 1000n or from N2133 = 1000 to N0 = 1000n; the values for r are simply
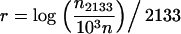 |
(3) |
as implied by the formula for exponential growth. (Note that there is no negative in the formula since time is scaled backward.) Figure 7a shows the ratio of 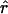 obtained from the best-fitting model (square for exponential, circle for expansion) to the value of r from Equation 3 with N2133 = 1; Figure 7b repeats this for N2133 = 1000. Interestingly,
obtained from the best-fitting model (square for exponential, circle for expansion) to the value of r from Equation 3 with N2133 = 1; Figure 7b repeats this for N2133 = 1000. Interestingly, 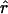 tends to be slightly smaller than the former and slightly larger than the latter, and there is little variation among mixing patterns. Note that in this range of values for N0r Pybus et al. (2000) found very little bias in
tends to be slightly smaller than the former and slightly larger than the latter, and there is little variation among mixing patterns. Note that in this range of values for N0r Pybus et al. (2000) found very little bias in 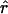 .
.
Figure 7.

Ratio of r obtained from skyline plot model to r assuming exponential growth with N0 = 1000n and (a) N2133 = 1 and (b) N2133 = 1000. Boxes, mean ±1 SD; whiskers, 2 SD; circles, outliers.
DISCUSSION
HIV-1 research is unique for the large role played by population genetics in its practical applications; accurate estimates of viral population size and dynamics, both within and between hosts, are important for applications as wide ranging as reconstructing general patterns of viral spread, understanding differences in transmissibility of viral genotypes, and testing hypotheses for the transmission of antiviral resistance. The work here was a first step to fill a critical gap in HIV population genetics: our understanding of how the network of behaviors that spread HIV-1 in the first place can affect the relationship between census size and effective population size at the community level.
Parametric models for estimating the growth rate of the epidemic through phylogenetic analysis seem to be highly robust for all populations regardless of mixing pattern. These parametric models also yielded estimates for current viral effective population size (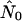 ) close to the product of infected host size (n) and intrahost viral effective population size (Nei) when the hosts are panmictic. That such accurate estimates could be obtained from small samples is a very promising sign. It is important to realize, though, that our ability to estimate the effective number of hosts in the section of the epidemic from which our sample of sequences is drawn is thus confounded with our ability to accurately estimate Nei. Although multiple studies have now obtained estimates for Nei on the order of 103, this is hardly a settled debate.
) close to the product of infected host size (n) and intrahost viral effective population size (Nei) when the hosts are panmictic. That such accurate estimates could be obtained from small samples is a very promising sign. It is important to realize, though, that our ability to estimate the effective number of hosts in the section of the epidemic from which our sample of sequences is drawn is thus confounded with our ability to accurately estimate Nei. Although multiple studies have now obtained estimates for Nei on the order of 103, this is hardly a settled debate.
The degree to which estimates of N0 change with violation of panmixis varies considerably on the basis of the type of violation. Most mixing patterns saw estimates of N0 in the same range as panmixis, although patterns with a highly segregated core and those resembling commercial sex worker networks often yielded values of Ne that were orders of magnitude greater than census size. Both of these are likely to be common patterns in populations in which HIV-1 is circulating, and the latter in particular is one that bears little resemblance to existing population genetic models for population substructure. Moreover, populations with different modes of transmission (and different viral subtypes) will generally display different network patterns; the bridges pattern, for instance, is observed in some heterosexual populations, while mixing patterns with cores and subgroups are likely to appear in many homosexual or intravenous drug-using populations. Thus it is reasonable to believe that a large part of the observed difference in N0 between viral subtypes A and B (approximately two orders of magnitude for gag and approximately one for env; Pybus et al. 2000) may be due to differences induced by different departures from panmixis rather than differences in census size. In both cases, the absolute numbers should not be taken to represent the effective number of hosts, but the effective combined viral pool, with the effective number of hosts considerably lower than this.
In fact, any attempt to use phylogenetic methods to identify pathogen population size and growth rate through time, to date the timing of past epidemic events, predict the future trajectory of host size on the basis of the past, or compare the past transmission routes of subtypes, will need to account for both intrahost dynamics and the details of the social networks spreading the pathogen, since each can affect Ne by multiple orders of magnitude. As we use simulation to develop more insight into the distribution of Ne expected through time under different transmission modes and rates, this approach may begin to contribute greatly to the current debate on the relative importance of sexual and iatrogenic transmission in the early history of the HIV-1 epidemic in Africa and elsewhere (Gisselquist and Potterat 2003).
This study has purposefully avoided many forms of complexity requiring consideration in future work. Chief among these are the birth/death dynamics of real populations and heterogeneity in infectivity over time and by partnership type and duration. The former is certain to have reduced the rate of new infections in the population toward the end of simulation and thus perhaps to have induced similarities to logistic growth that might not exist in dynamic populations. The expected effect of ignoring the latter form of heterogeneity is to underestimate the importance of short relationships as sources of viral spread. Evidence is now growing that infectivity is concentrated during the first few months after infection when viral load is high (Leynaert et al. 1998; Shiboski and Padian 1998). If this is true, then the model here will have underestimated the frequency with which new infections occur before the infector has reached the Nei steady state, making the relationships between Ne and nNei murkier. This will have its greatest effect in populations where short partnerships are relatively important, such as those involving commercial sex workers. Ignoring recombination may also overemphasize the contribution of intrahost diversity to Ne, possibly to a considerable extent (Wain-Hobson et al. 2003).
This study focused on population sizes equivalent to an at-risk group in a small community. Many applications of HIV interhost phylogenetics study much larger populations, including whole subtypes of HIV-1 or the entire epidemic. Mixing is nonrandom within the community and is also certainly nonrandom at higher levels as well (by geographical distance, at the very least). Whether these higher levels of nonpanmictic behavior will also have significant effects on patterns of genetic variation or whether our methods are in fact robust to them is not addressed in this study. Further work (analytical or simulation-based) should identify how the effects observed here scale up to more global populations.
This work demonstrated that host mixing structure may have strong effects on population genetics of HIV-1, and these may not always be accountable by existing parametric models for population growth. This does not imply that no parsimonious parametric models can be used to describe such populations. For example, a four-parameter model combining the features of logistic growth and expansion growth (i.e., logistic growth beginning at an arbitrary population size in the infinite past) may have fit some of the runs here that were not otherwise well fit by any of the three tested models, since both leveling off toward the present and starting from a population of size 1000 occurred. Other models that allow for multiple plateaus with logistic growth between each may best describe some populations with strongly segregated clusters.
More research on social network structure in HIV-transmitting populations (ideally integrating genetic data, which has not yet been done in any of the large-scale studies of complete sexual network data in a population) should help us hone in on the full range of patterns found across risk groups and allow us to generalize our phylogenetic models to incorporate all of these possibilities. Since ERGM provides a means for parameterizing any possible network structure, it seems a strong candidate for the framework in which the two steps of this process are bridged. By doing so we will finally gain the ability to understand the genetics of human sexually transmitted pathogens in a way that truly reflects the unique processes that spread them.
Acknowledgments
I thank Martina Morris, Ken Weiss, Mark Handcock, Jim Wood, Andy Clark, Anne Buchanan, Steve Koester, Anna Barón, John Potterat, Simon Frost, Oliver Pybus, Jamie Jones, and the anonymous reviewers. This research was funded by graduate fellowships from the National Science Foundation and the Population Council and by a research grant from the National Institute of Drug Abuse (7R01DA012831) and a training grant from the National Institute of Allergy and Infectious Diseases (5T32AI007140).
References
- Beerli, P., and J. Felsenstein, 2001. Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc. Natl. Acad. Sci. USA 98: 4563–4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besag, J., 1974. Spatial interaction and the statistical analysis of lattice systems. J. R. Stat. Soc. Ser. B 36: 192–236. [Google Scholar]
- Chesser, R. K., O. E. Rhodes, Jr., D. W. Sugg and A. Schnabel, 1993. Effective sizes for subdivided populations. Genetics 135: 1221–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank, O., and D. Strauss, 1986. Markov graphs. J. Am. Stat. Assoc. 81: 832–842. [Google Scholar]
- Friedman, S. R., A. Neaigus, B. Jose, R. Curtis, M. Goldstein et al., 1997. Sociometric risk networks and risk for HIV infection. Am. J. Public Health 87: 1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaggiotti, O. E., 1996. Population genetic models of source-sink metapopulations. Theor. Popul. Biol. 50: 178–208. [DOI] [PubMed] [Google Scholar]
- Garnett, G. P., J. P. Hughes, R. M. Anderson, B. P. Stoner, S. O. Aral et al., 1996. Sexual mixing patterns of patients attending sexually transmitted diseases clinics. Sex. Transm. Dis. 23: 249–257. [DOI] [PubMed] [Google Scholar]
- Gilks, W. R., S. Richardson and D. J. Spiegelhalter, 1996. Markov Chain Monte Carlo in Practice. Chapman & Hall, London.
- Gisselquist, D., and J. J. Potterat, 2003. Heterosexual transmission of HIV in Africa: an empiric estimate. Int. J. STD AIDS 14: 162–173. [DOI] [PubMed] [Google Scholar]
- Grassly, N. C., P. H. Harvey and E. C. Holmes, 1999. Population dynamics of HIV-1 inferred from gene sequences. Genetics 151: 427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick, P. W., and M. E. Gilpin, 1997. Genetic effective size of a metapopulation, pp. 165–181 in Metapopulation Biology: Ecology, Genetics and Evolution, edited by I. Hanski and M. E. Gilpin. Academic Press, New York.
- Holmberg, S. D., 1996. The estimated prevalence and incidence of HIV in 96 large US metropolitan areas. Am. J. Public Health 86: 642–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes, E. C., O. G. Pybus and P. H. Harvey, 1999. The molecular population dynamics of HIV-1, pp. 177–207 in The Evolution of HIV, edited by K. A. Crandall. Johns Hopkins University Press, Baltimore.
- Klovdahl, A. S., J. J. Potterat, D. E. Woodhouse, J. B. Muth, S. Q. Muth et al., 1994. Social networks and infectious disease: the Colorado Springs study. Soc. Sci. Med. 38: 79–88. [DOI] [PubMed] [Google Scholar]
- Laumann, E. O., J. H. Gagnon, R. T. Michael and S. Michaels, 1994. The Social Organization of Sexuality: Sexual Practices in the United States. University of Chicago Press, Chicago.
- Leigh Brown, A. J., 1997. Analysis of HIV-1 env gene sequences reveals evidence for a low effective number in the viral population. Proc. Natl. Acad. Sci. USA 94: 1862–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh Brown, A. J., D. Lobidel, C. M. Wade, S. Rebus, A. N. Phillips et al., 1997. The molecular epidemiology of human immunodeficiency virus type 1 in six cities in Britain and Ireland. Virology 235: 166–177. [DOI] [PubMed] [Google Scholar]
- Leitner, T., and J. Albert, 1999. The molecular clock of HIV-1 unveiled through analysis of a known transmission history. Proc. Natl. Acad. Sci. USA 96: 10752–10757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitner, T., S. Kumar and J. Albert, 1997. Tempo and mode of nucleotide substitutions in gag and env gene fragments in human immunodeficiency virus type 1 populations with a known transmission history. J. Virol. 71: 4761–4770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leynaert, B., A. M. Downs and I. De Vincenzi, 1998. Heterosexual transmission of human immunodeficiency virus: variability of infectivity throughout the course of infection. Am. J. Epidemiol. 148: 88–96. [DOI] [PubMed] [Google Scholar]
- Martin, J. L., 1987. The impact of AIDS on gay male sexual behavior patterns in New York City. Am. J. Public Health 77: 578–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutchan, F. E., B. L. Ungar, P. Hegerich, C. R. Roberts, A. K. Fowler et al., 1992. Genetic analysis of HIV-1 isolates from Zambia and an expanded phylogenetic tree for HIV-1. J. AIDS 5: 441–449. [PubMed] [Google Scholar]
- Metropolis, N., A. W. Rosenbluth, M. N. Rosenbluth, A. H. Teller and E. Teller, 1953. Equation of state calculations by fast computing machines. J. Chem. Phys. 21: 1087–1092. [Google Scholar]
- Morris, M., and L. Dean, 1994. Effect of sexual behavior change on long-term human immunodeficiency virus prevalence among homosexual men. Am. J. Epidemiol. 140: 217–232. [DOI] [PubMed] [Google Scholar]
- Morris, M., and M. Kretzschmar, 1997. Concurrent partnerships and the spread of HIV. AIDS 11: 641–648. [DOI] [PubMed] [Google Scholar]
- Morris, M., C. Podhisita, M. J. Wawer and M. S. Handcock, 1996. Bridge populations in the spread of HIV/AIDS in Thailand. AIDS 10: 1265–1271. [DOI] [PubMed] [Google Scholar]
- Moss, A. R., K. Vranizan, R. Gorter, P. Bacchetti, J. Watters et al., 1994. HIV seroconversion in intravenous drug users in San Francisco, 1985–1990. AIDS 8: 223–231. [DOI] [PubMed] [Google Scholar]
- Nee, S., R. M. May and P. H. Harvey, 1994. The reconstructed evolutionary process. Philos. Trans. R. Soc. Lond. 344: 305–311. [DOI] [PubMed] [Google Scholar]
- Nee, S., E. C. Holmes, A. Rambaut and P. H. Harvey, 1995. Inferring population history from molecular phylogenies. Philos. Trans. R. Soc. Lond. 349: 25–31. [DOI] [PubMed] [Google Scholar]
- Nijhuis, M., C. A. Boucher, P. Schipper, T. Leitner, R. Schuurman et al., 1998. Stochastic processes strongly influence HIV-1 evolution during suboptimal protease-inhibitor therapy. Proc. Natl. Acad. Sci. USA 95: 14441–14446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orubuloye, I. O., J. C. Caldwell and P. Caldwell, 1991. Sexual networking in the Ekiti District of Nigeria. Stud. Fam. Plann. 22: 61–73. [PubMed] [Google Scholar]
- Ou, C. Y., C. A. Ciesielski, G. Myers, C. I. Bandea, C. C. Luo et al., 1992. Molecular epidemiology of HIV transmission in a dental practice. Science 256: 1165–1171. [DOI] [PubMed] [Google Scholar]
- Pybus, O. G., and A. Rambaut, 2002. a GENIE: estimating demographic history from molecular phylogenies. Bioinformatics 18: 1404–1405. [DOI] [PubMed] [Google Scholar]
- Pybus, O. G., and A. Rambaut, 2002. b GENIE v3.0 User Manual. University of Oxford (http://evolve.zoo.ox.ac.uk/software/Genie).
- Pybus, O. G., A. Rambaut and P. H. Harvey, 2000. An integrated framework for the inference of viral population history from reconstructed genealogies. Genetics 155: 1429–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigo, A. G., and J. Felsenstein, 1999. Coalescent approaches to HIV population genetics, pp. 233–272 in The Evolution of HIV, edited by K. A. Crandall. Johns Hopkins University Press, Baltimore.
- Rodrigo, A. G., E. G. Shpaer, E. L. Delwart, A. K. Iversen, M. V. Gallo et al., 1999. Coalescent estimates of HIV-1 generation time in vivo. Proc. Natl. Acad. Sci. USA 96: 2187–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenberg, R. B., C. Sterk, K. E. Toomey, J. J. Potterat, D. Johnson et al., 1998. Using social network and ethnographic tools to evaluate syphilis transmission. Sex. Transm. Dis. 25: 154–160. [DOI] [PubMed] [Google Scholar]
- Rouzine, I. M., and J. M. Coffin, 1999. Linkage disequilibrium test implies a large effective population number for HIV in vivo. Proc. Natl. Acad. Sci. USA 96: 10758–10763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiboski, S. C., and N. S. Padian, 1998. Epidemiological evidence for time variation in HIV infectivity. J. AIDS Hum. Retrovirol. 19: 627–635. [DOI] [PubMed] [Google Scholar]
- Snijders, T. A. B., 2002. Markov chain Monte Carlo estimation of exponential random graph models. JoSS. 3: 1–40. [Google Scholar]
- Strauss, D., and M. Ikeda, 1990. Pseudolikelihood estimation for social networks. J. Am. Stat. Assoc. 85: 204–212. [Google Scholar]
- Strimmer, K., and O. G. Pybus, 2001. Exploring the demographic history of DNA sequences using the generalized skyline plot. Mol. Biol. Evol. 18: 2298–2305. [DOI] [PubMed] [Google Scholar]
- Vartanian, J. P., A. Meyerhans, M. Henry and S. Wain-Hobson, 1992. High-resolution structure of an HIV-1 quasispecies: identification of novel coding sequences. AIDS 6: 1095–1098. [DOI] [PubMed] [Google Scholar]
- Wain-Hobson, S., C. Renoux-Elbe, J. P. Vartanian and A. Meyerhans, 2003. Network analysis of human and simian immunodeficiency virus sequence sets reveals massive recombination resulting in shorter pathways. J. Gen. Virol. 84: 885–895. [DOI] [PubMed] [Google Scholar]
- Wakeley, J., and N. Aliacar, 2001. Gene genealogies in a metopopulation. Genetics 159: 893–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman, S., and P. Pattison, 1996. Logit models and logistic regression for social networks: I. An introduction to Markov graphs and p*. Psychometrika 60: 401–426. [Google Scholar]
- Watts, C. H., and R. M. May, 1992. The influence of concurrent partnerships on the dynamics of HIV/AIDS. Math. Biosci. 108: 89–104. [DOI] [PubMed] [Google Scholar]