

Abstract
Detailed knowledge about the rate and mode of the genetic variation is vital for understanding how HIV-1 induces disease and develops resistance as well as for studies on the molecular epidemiology and origin of the virus. To unveil the molecular clock of HIV-1 we analyzed a unique set of viruses from a known transmission history with separation times between samples of up to 25 years. The env V3 and p17gag regions of the genome were sequenced, and genetic distances were estimated by using the true tree and a nucleotide substitution model based on a general reversible Markov process with a gamma distribution to account for differences in substitution rates among sites. Linear regression analysis showed that separation times were significantly correlated with synonymous as well as nonsynonymous nucleotide distances in both V3 and p17, giving strong support for the existence of a molecular clock. The estimated rate of nucleotide substitution was 6.7 ± 2.1 × 10−3 substitutions/site per year in V3 and 2.7 ± 0.5 × 10−3 in p17. Importantly, the regression analyses showed that there was a significant genetic distance at zero divergence times. This pretransmission interval exists because the ramifications in the phylogenetic trees do not correspond to time of transmission, but rather to the coalescence time of the most recent common ancestor of the viruses carried by the transmitter and the recipient. Simulation experiments showed that neither the V3 nor the p17 clocks were overdispersed, which indicates that the introduction of nucleotide substitutions can be described adequately by a simple stochastic Poisson process.
The evolutionary rates of RNA viruses are much higher than those of their eukaryotic hosts. HIV-1 displays one of the highest evolutionary divergences detected in any life form, with genetic distances in hypervariable regions differing by at least 40% between genetic subtypes and by 10–15% within single infected individuals. This extreme variation is the result of an error-prone reverse transcriptase, high population turnover, viral proteins that accept high variation, and strong environmental selective pressures. For these reasons, it has been suggested that HIV-1 evolution more or less follows the quasispecies model, which assumes that the population size is infinite and that every possible nucleotide substitution pre-exists. According to this model, the genetic variants form an adaptive landscape that is modeled to adjust for environmental changes (1). Because all possible substitutions pre-exist according to such a deterministic model, some authors have disputed the existence of a genetic molecular clock for HIV-1 (2). However, others have argued that the fact that synonymous substitutions predominate over nonsynonymous substitutions gives support for the neutral theory of evolution, where the concept of a molecular clock is fundamental (3). More recently, it has been demonstrated that chance plays an important role in HIV-1 evolution and that the effective population size is small, which supports a stochastic, rather than deterministic, evolutionary model (4, 5).
Several authors have attempted to estimate the rate of synonymous and nonsynonymous substitutions. Li et al. (6) estimated the nonsynonymous rate at 14 × 10−3 substitutions/site per year in hypervariable regions of the envelope gene and the average synonymous rate for the whole genome at 10 × 10−3. Gojobori et al. (3) estimated the synonymous and nonsynonymous substitution rates of the gag gene at 13.08 × 10−3 and 3.92 ×10−3, respectively. More recently, synonymous and nonsynonymous rates of the V3 region were estimated at 3.7–5.1 × 10−3 and 6.6–11 × 10−3, respectively (7). However, these rate estimates must be regarded as preliminary because they were based on relatively short observation times and oversimplified models of nucleotide evolution. Thus, longer observation times are desirable when attempts are made to accurately describe the molecular clock of HIV, but because longer observation times also translates into greater genetic distances it becomes essential to accurately correct for superimposed mutational events (8). We recently reported on a unique heterosexual transmission chain in which viruses with known phylogenetic history have evolved with separation times of up to 25 years (9). Sequences from these viruses also have been used to identify models of nucleotide substitution that fit HIV-1 evolution (8). Here, we have used this knowledge to test the existence of a molecular clock and to gain detailed knowledge about its characteristics.
MATERIALS AND METHODS
Study Population and the True Tree.
Thirteen viral population sequences from nine HIV-1-infected individuals with well-characterized epidemiological relationships were studied (9). Briefly, the index case, a Swedish male (p1) became HIV-1 infected in Haiti in 1980, and subsequently infected several females (p2, p5, p7, and p8) between 1981 and 1983. In addition, samples were available from two later male sexual partners (p6 and p10) and two children (p3 and p9) of the females. Blood samples were obtained at different time points between 1986 and 1993. From some individuals more than one sample was available. The information about when the transmissions had occurred and when the samples were obtained was compiled into a tree that shows the history of the transmitted virus populations (Fig. 1). Thus, the branch lengths in the tree describe the actual time between transmissions and samplings. Reconstruction of the transmission history, by the topology of the virus history, has been presented and examined before (9, 10), as well as the nucleotide substitution pattern of the HIV-1 populations (8). All patients, except p6, displayed wild-type CCR5 genotype. Instead, p6 displayed a heterozygosity for a 32-bp deletion (wt/Δ32) in the CCR5 gene (11, 12), and other than p9, was the only patient still alive in 1998.
Figure 1.

The true transmission history of HIV-1 of the Swedish transmission chain (9). Each lineage split indicates a transmission event and each tip of a branch a sequence sample, where individual and sample numbers are given. Note that for several individuals second subsequent samples were included. The time during which the virus populations have evolved is indicated by a time scale in years at the bottom.
Sequence Data.
DNA sequences from the HIV-1 p17gag and env V3 regions of the viral genome were determined by direct population sequencing as described (13). In the earlier study of reconstructed tree topologies (9), polymorphic nucleotide positions within a sample (multistate characters) were described by using the International Union of Pure and Applied Chemistry codes (14), available under GenBank accession numbers U68496–U68521. In a second study of the nucleotide substitution pattern of the transmission history (8), the population sequences were reanalyzed to determine a majority-rule consensus sequence for each sample (these sequences are available from the authors on request). In the present study both data sets were used as described below.
Genetic Distance Estimation.
Pair-wise distances for the V3 and p17 sequences were calculated: (i) as p-distance (uncorrected proportion of differences); (ii) according to the Jukes–Cantor model (15); (iii) according to the Tamura–Nei model with gamma distributed rates among sites (16, 17); and (iv) by maximum likelihood-fitted branch lengths of the true topology, using the general reversible Markov process (REV) model including a gamma distribution (Γ) to describe rate variation among sites (8, 18). The shape parameter α of the gamma distribution was set to 0.38 for the V3 calculations and 0.25 for p17 in accordance with earlier published results (8). All pair-wise distance calculations were performed with the majority rule sequences by using the program mega (19), and branch length calculations by the maximum-likelihood method were done by using the program paml (20). Pair-wise distances also were calculated with the multistate character sequences by using a specially written computer program, popdist (21), which correctly handles all International Union of Pure and Applied Chemistry codes. The program is available from the authors on request.
Estimation of Substitution Rate.
Genetic distances were plotted against time, and a linear regression was fitted by the least-squares method. Thus, the relation between the genetic distance (d) and time (t) is represented by:
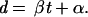 |
1 |
The slope (β) describes the substitution rate and the intercept (α) is related to two measures that we have called pretransmission interval (Δ) and ancestral divergence (Da) as discussed later and in ref. 10. Two problems arise when genetic distances are plotted against time. First, the distances are not observations of random variables (the sequences are random variables and the distances are summary statistics obtained from the sequences); and second, the distances are not independent data points. Because standard tests for significance used in regression analysis are not appropriate for comparison of genetic distances, several other methods were used to investigate the two regression parameters α and β: (i) pair-wise distances were plotted against the corresponding separation times; (ii) individual branch lengths were plotted against the corresponding times; (iii) for estimates of Da, only external branches were plotted against time; and (iv) to roughly estimate the dispersion of the substitution rate, the pair-wise distances were resampled. From one tip in the true tree (a specific sample), calculate the distances to each of the other tips, which will give 12 distance estimates for each sample. Repeat this step, starting from each of the other tips, which now will give 13 sets of 12 distances each. Next, do a linear regression for each sample set and calculate α. Use these estimates to calculate the average α for V3 and p17 (ᾱ*). However, we included only sample sets with separation times covering the full range from < 4 years up to 20–25 years to ensure that the α estimate was reasonably precise (these regressions also had higher regression constants; R2 > 0.5). There were 10 such sets for the V3 data and 11 for the p17 data. Finally, recalculate β for each of the 13 sample sets, also those excluded in step two, with α set to ᾱ* and compute the mean and SD for β from the 13 estimates.
To investigate the degree of dispersion of the observed distance estimates, we performed simulations using the program seq-gen (22). One hundred simulated sequence sets were generated each for V3 and p17 (V3 = 300 bp, p17 = 430 bp). The simulations were performed under the true tree topology with branch lengths and all parameters in the REVΓ model set to the values estimated from the real data (8). For each of the 100 simulated sets we calculated all pair-wise distances in the same way as for the real data set and plotted these distances against the corresponding separation time. The dispersion of the simulated data will follow a Poisson process under the same model as the real data (REVΓ). The dispersion of the distance measures for each simulated dataset was estimated by the correlation coefficient (R). The distribution of these correlation coefficients was determined and found to follow a normal distribution. Finally, the dispersion of the real data, as estimated by the correlation coefficient, was compared with that of the simulated data by z-statistics.
RESULTS
Evidence for the Existence of a Molecular Clock.
The genetic distances between sequences were found to be directly correlated with time, i.e., longer time interval between two sequences corresponded to greater genetic distance. The uncorrected distances (p-distance) were plotted against time for all pair-wise comparisons (Fig. 2). In both V3 and p17 a clear increase in genetic distance over time was observed. In the V3 fragment the nonsynonymous (pn) rate was higher than the synonymous (ps) rate, whereas the opposite was true in the p17 fragment, indicating differences in the selective pressure on these two gene fragments. In accordance with this result, the average ps/pn ratio was 0.78 and 3.14 in V3 and p17, respectively. More interestingly, we found that the ps rate (slope) was similar in V3 and in p17 (0.0035 t and 0.0033 t, respectively), whereas the pn rate was higher in V3 than in p17 (0.0036 t and 0.0015 t, respectively). Thus, the rates of psV3 ≈ psp17 ≈ pnV3 ≈ 2pnp17. Transitions were more common than transversions in both fragments, but p17 showed a higher transition/transversion ratio than V3, 3.97 and 1.42 (uncorrected), respectively. The REVΓ corrected ratios were 2.32 and 1.41, respectively.
Figure 2.

The proportion of nucleotide differences (p-distance) for all pair-wise comparisons of the analyzed sequences plotted against the corresponding separation times. ▵, synonymous differences; □, nonsynonymous differences; ●, total differences. The lines indicate linear regression estimates.
A problem with simple p-distances is that the estimates suffer heavily from lack of compensation for superimposed events when genetic distances increase. There exist a number of different substitution models that attempt to correct for this problem. We have shown earlier that the REVΓ model gives the best available distance estimates for HIV-1 populations (8). In accordance with this finding, the REVΓ-corrected distances showed the best correlation with separation times (V3: R2 = 0.69, and p17: R2 = 0.59). When the regression weights of four other genetic distance models (p-distance, Jukes–Cantor, Tamura–Nei with gamma, and REVΓ) were compared, the REVΓ estimates showed the strongest correspondence to time (P = 0.0007). No trend in the raw residuals of the linear regression was observed for either the V3 or the p17 data.
The Two Components of the Molecular Clock.
As described above, our data provide evidence for the existence of a molecular genetic clock in HIV-1 evolution. Thus, the slope (β) of the regression line describes the rate at which substitutions are introduced over time. Interestingly, we found an y-axis intercept (α) when we fitted a linear regression line to the data points (Eq. 1), i.e., there existed a genetic distance at zero divergence times, which was observed both when pair-wise distances and individual branch segments were plotted against time (Figs. 2 and 3). Because pair-wise distances are not independent measures of genetic distance, the significance of the intercept was investigated on the branch segment data (Fig. 3). For the V3 data, the fitted line had an intercept that was significantly separated from zero (P = 0.05). We interpret the intercept as a measure of the ancestral divergence (Da), which exists because the transmitted virus was not created at the time of transmission, but must have existed in the donor for some time before transmission, i.e., a pretransmission interval (Δ) (Fig. 3). Thus, ramifications in a phylogenetic tree derived from viral sequences do not correspond to transmission time points, but rather to the coalescence time of the most recent common ancestor of the virus carried by the donor and the recipient.
Figure 3.

(A) The pretransmission interval (Δ) describes the difference between the time of transmission and the most recent common ancestor (MRCA) of the transmitted lineage and the donor lineage. The solid line tree is the transmission history, and the dashed line tree is the evolutionary history of the transmitted virus. At t1 patient A infects patient B, and at t2 B infects C. The virus that infects B shares its MRCA with A at T1, and the virus that infects C shares its MRCA with B at T2. The pretransmission interval when A infects B is t1 − T1 = ΔAB, and t2 − T2 = ΔBC when B infects C. Because of the pretransmission interval, the transmitted lineage and the donor lineage will be separated by a genetic distance, the ancestral divergence (Da), at the time for transmission. (B) Individual branch segments from the V3 tree plotted against the corresponding time intervals. ● indicate external branches and ○ internal branches. The solid line is a linear regression line based on the external branches only, and the dotted line is based on the internal branches. From A it can be inferred that the intercept of the solid line represents an estimate of the average ancestral divergence (Da), whereas the intercept of the dotted line describes the average difference between linked ancestral divergence measures, i.e., the dispersion of Da. In both cases the slope describes the expected substitution rate.
Substitution Rate (β).
Several different methods were used to estimate the correlation between genetic distances and evolutionary times (Table 1). Reassuringly, these different methods gave very concordant results. In env V3, the substitution rate (β) was estimated at 6.7 ± 2.1 × 10−3 substitutions per nucleotide and year by using the resampled pair-wise branch distances (Table 1). In p17gag, the estimated substitution rate was 2.7 ± 0.5 × 10−3 substitutions per nucleotide and year. Thus, the substitution rate was three times faster in V3 than in p17. It is important to stress that our estimates of substitution rates in p17 and V3 cannot be directly compared with previously published estimates for these gene fragments because we used a model (REVΓ) that corrects much more effectively for superimposed substitutional events than other models.
Table 1.
Different estimates of the ancestral divergence and the substitution rate
| Gene fragment | Pairwise* | Branch† | Resample‡ | External§ |
|---|---|---|---|---|
| Ancestral divergence, Da | ||||
| env V3 | 29.6 | na | 27.4 ± 7.0 | 21.3 |
| p17 gag | 7.7 | na | 6.6 ± 4.2 | 4.3 |
| Substitution rate, β | ||||
| env V3 | 6.9 | 7.0 | 6.7 ± 2.1 | 6.6 |
| p17 gag | 2.5 | 2.8 | 2.7 ± 0.5 | 2.9 |
As discussed in the text, our best estimates are in bold. na, not applicable. Values are based on × 10−3.
Value based on a linear regression line fitted to all pair-wise branch distances (Fig. 4).
Value based on a linear regression line fitted to all individual branch segments (Fig. 3B).
Value based on resampled pair-wise distances as described in Materials and Methods.
Value based on a linear regression line fitted to external branch segments only (Fig. 3B).
Ancestral Divergence (Da) and Pretransmission Interval (Δ).
In env V3, the intercept (α) was estimated at 59.1 × 10−3 substitutions per nucleotide by using pair-wise estimates from maximum likelihood-fitted branch lengths in the true tree according to the REVΓ model. The resampling method gave a similar estimate of α, but in addition provided an estimate of the dispersion, i.e., 54.8 ± 14.0 × 10−3. In p17gag, α was estimated at 15.4 × 10−3 and 13.2 ± 8.4 × 10−3 substitutions per nucleotide by these two methods, respectively. When pair-wise distances are plotted against separation times they always will be influenced by two pretransmission intervals. Hence, in these plots the ancestral divergence will be equal to the intercept divided by two, Da = α/2. In contrast, when individual branch lengths are plotted against time, external branches will be affected by Δ only on the inside end (at the coalescence point). Therefore, the intercept in these plots provides a direct estimate of Da (Fig. 3 and Table 1). Internal branches, on the other hand, will be affected by a Δ on both ends and thus provide a measure of the difference between linked Das. By this relation, Da was estimated at 21.3 × 10−3 and 4.3 × 10−3 substitutions per nucleotide for V3 and p17, respectively. In other words, at the time of transmission the average genetic distance between the transmitted lineage and the donor lineage was approximately 2% in V3 and 0.4% in p17.
Lineage-Specific Rate Variation. The evolutionary distances were estimated for each of the 24 branch segments of the true tree topology. The expected (average) distance is indicated by the regression line in Fig. 4, and it is clear that for any given time interval the observed genetic distance deviated from the expected. Such dispersion could be the result of lineage-specific differences in nucleotide substitution rates. In line with this observation, it has been reported that nonsynonymous substitutions accumulate faster in HIV-1-infected individuals with a slow rate of disease progression compared with individuals with a rapid disease progression, which suggests that immune selection may drive HIV-1 evolution (7, 23). However, because mutations are stochastic events and our sequences are not infinitely long, it is expected that the observed substitution rate will vary from lineage to lineage even in the absence of diverse biological pressures. Furthermore, under such a simple stochastic model it is expected that the sampling variance will accumulate over time and consequently that the deviation between observed and expected distances will increase with time. This effect is known as a Poisson error.
Figure 4.

The molecular genetic clock of HIV-1. Pair-wise genetic distances from the V3 and p17 trees were plotted against the corresponding times (●). The linear regression line displays the expected distance between two sequence samples for a given separation time. The highlighted area shows the expected dispersion from 95% of simulated data points after a Poisson process under an identical situation as the real data.
To investigate whether the dispersion in our data could be explained by a simple stochastic variation or if other biological processes may have caused overdispersion (more variation than expected from a Poisson process) we performed simulation experiments. The simulations were carried out with identical settings as those used for the real data and the true tree (topology, branch lengths, and REVΓ parameters) for both the V3 and p17 regions. Fig. 4 summarizes the results from the simulations. The real data points fell well within the expected dispersion of 95% of the simulated data in both V3 and p17. More precisely, the mean and SD of the correlation coefficient (R) of the simulated data was 0.740 ± 0.082 for V3, and 0.689 ± 0.086 for p17, whereas RV3 = 0.766 and Rp17 = 0.765 for the real data. Hence, the dispersion of the V3 and p17 clocks fell among 38% and 19% of the least dispersed simulated sets (1 − z = 0.6841 and 0.1111), respectively. Thus, the variance of these two clocks was not greater than that expected from a stochastic process. This behavior indicates that the evolution of HIV-1 in different patients, on average throughout infection, is described adequately by a constant molecular clock.
We also investigated the effect of genetic bottlenecks during transmission by dividing all possible pair-wise comparisons into four categories based on the number of transmissions that separated the sequences. Interestingly, we found that the number of transmissions did not have any measurable effect on the estimated substitution rate (β) (Table 2). We observed small rate differences when branch segments belonging to specific patients were compared. However, the study was not designed to investigate this issue because most patients were represented by only one or very few branch segments. For the same reason it was not meaningful to correlate nucleotide substitution rates to clinical data.
Table 2.
Substitution rate in relation to number of transmissions
| Number of transmissions | Number of observations | Substitution
rate, (×10−3 substitutions/site per year)
|
|
|---|---|---|---|
| env V3 | p17 gag | ||
| 1 | 9 | 6.64 ± 3.80 | 3.41 ± 3.44 |
| 2 | 13 | 6.98 ± 3.03 | 3.41 ± 2.54 |
| 3 | 11 | 7.43 ± 2.92 | 2.82 ± 0.92 |
| 4 | 4 | 7.73 ± 1.35 | 2.17 ± 0.36 |
DISCUSSION
The objective of this study was to investigate the relationship between nucleotide substitutions and time in evolving HIV-1 populations, i.e., if a molecular clock exists or not, and if so how the clock should be described. With this aim we investigated a unique set of samples for which the evolutionary history is exactly known (8, 9), which allowed direct correlation between the genetic distances and the time that separated the samples. Two important conclusions could be drawn from the study. First, we show that the concept of a molecular clock fits HIV-1 evolution well. Second, we demonstrate that a significant genetic distance at zero divergence time exists, a finding we show depends on a pretransmission interval.
Evolving HIV-1 populations display very high nucleotide substitution rates. During the evolution some sites will fluctuate rapidly in their nucleotide state whereas other sites will display substitutions that are stable over longer time intervals. Under these circumstances fixation of the nucleotide state can be defined in a number of ways, including rise over the background misincorporation level of the reverse transcriptase or rise over the detection limit of the assay system. However, the important point is whether a substitution becomes dominant in the replicating population (defined by the effective population size) and thereby is carried on to all individuals in the next generation. There are also substitutions that do not become fixed, i.e., substitutions that fluctuate frequently in the population. Several factors may contribute to these fluctuations within a single host, such as changes in immune pressure, differences in cell tropism, and other types of compartmentalization of virus variants. In addition, many fluctuating substitutions may be evolutionary neutral (24), especially those in third codon positions (silent/synonymous substitutions). Rapidly fluctuating substitutions easily are underestimated when sequences separated by long time intervals are compared. In practice, each site in a gene fragment will have its own substitution rate, and it is important to account for these rate differences because superimposed events will be more common in sites with high substitution rates (8). The substitution model (here REVΓ) attempts to correct the estimated distance for the fast type of substitutions by knowledge of the amount of slow type (fixed) of substitutions. If many slow type substitutions have occurred an even greater number of fast type substitutions must have occurred, and the estimated genetic distance will be corrected more dramatically. Thus, the corrected genetic distance attempts to estimate the total number of substitutions, and it is this distance that is related to time.
We found a significant distance at zero separation times, estimated by the y-axis intercept, which indicates that the time at which sequences coalesce to a common ancestor predates the time for transmission. This finding is explained by the fact that a transmitted virus is not born at the moment of transmission, but rather at some time before the transmission, and we refer to this finding as the pretransmission interval (Δ) (10). The pretransmission interval would have little influence on distance estimates if intrapatient genetic distances were small in the donor, i.e., if the donor population could be explained by a small effective population size. However, HIV-1 populations are known to be very heterogeneous, except possibly during primary infection and end-stage AIDS. In accordance with this, we found that the average genetic distance between the transmitted lineage and the donor lineage, i.e., the ancestral divergence (Da), was approximately 2% in V3 and 0.4% in p17. Unfortunately, we cannot directly estimate the pretransmission interval in units of time by extrapolation of the average substitution rate in the population because the rate at which individual members of a virus population are replaced is likely to be much higher than the rate at which the population moves through sequence space. In classical population biology a similar phenomenon is referred to as the Red Queen’s Hypothesis (25), where species/populations are competing in a zero-sum game against each others, i.e., forced to run constantly to maintain nearly equal fitness. An analogous problem to the pretransmission interval is the relationships between gene trees and species trees in other organisms (26). One may consider the transmission events as speciation events in which the transmitted genes are orthologs of some lineages in the donor, and similarly, all the various lineages within a host are paralogs. The pretransmission interval therefore resembles some of the pitfalls in the interpretation of regular gene evolution. Moreover, Fitch (27) has mentioned that viral phylogenies may not necessarily correspond to viral transmission histories. However, the pretransmission interval and the problems it creates has not attracted attention previously in HIV research, which may in part explain why our estimates of the evolutionary rate of HIV-1 are lower than those of other investigators (3, 6, 7). In this context it is worth noting that this difference would have been even greater had it not been counteracted by the fact that we estimated genetic distances by a more realistic evolutionary model (i.e., the REVΓ model).
The effects of genetic bottlenecks are particularly interesting in the context of a molecular clock. Genetic bottlenecks may be caused by transmission from one host to another, and by definition involve few virus variants or even a single one. Although the frequency at which bottlenecks occur within single hosts are not known, Kimura (24) showed that if substitutions are neutral the substitution rate is independent of population size. This independence is because the decreased probability of fixation of a new mutant in a large population (1/N) is exactly balanced by the increase in the rate at which new mutants arise (Nμ), so that the expected number of substitutions simply is (1/N)(Nμ) = μ. For this to be true in the case of transmission, mutants also need to be neutrally transmitted. To investigate this case we examined whether number of transmission steps influenced the distance estimates. Interestingly, we found no correlation between the number of transmission events and genetic distances (Table 2), which suggests that transmission is a neutral event, i.e., if selection exists it does not influence the overall substitution rate. Bottlenecks also may be encountered within a host, for instance as a result of immune selection or drug treatment, and fixed substitutions survive transmissions and the transmitted virus therefore must be drawn from the dominant and replicating population. Note that this does not mean that the dominant average sequence (master sequence) will be transmitted (it may not even exist in the population), but that the transmitted form shares many features with the dominant and replicating population in the donor. In agreement with our findings Zhang et al. (28) recently reported a similar rate of accumulation in divergence within and between individuals, i.e., transmission did not reset the molecular clock. An important consequence of this finding is that attempts to estimate the age of HIV are not likely to be affected by the number of transmissions involved.
The neutral theory of genetic evolution (24) predicts that the molecular clock is stochastic rather than metronomic, which means that the probability of change is constant, but that there will be variation around the expected rate of change. Already in the original molecular clock hypothesis, proposed by Zuckerkandl and Pauling (29), the observed rates of evolution were suggested to be approximately described by a simple Poisson process. Thus, this model predicts that the variance in the number of substitutions should be equal to the mean. However, many authors have reported the evolutionary rates of other organisms than HIV to be more dispersed than predicted by a Poisson process (overdispersion) (30–32), i.e., there is more variation in branch lengths in a tree than expected from a Poisson process. Several potential explanations for overdispersion have been proposed. It has been suggested that overdispersion may exist because mutant sites do not evolve independently (32). Another hypothesis invokes a generation-time effect, i.e., that the number of mutational errors will increase with increasing number of replication cycles. Interestingly, our simulation experiments indicated that neither the V3 clock nor the p17 clock were overdispersed (Fig. 4), which means that HIV-1 evolution can be described adequately by a neutral evolutionary model in which nucleotide substitutions are introduced by a stochastic process. Although positive and purifying (negative) selection events appear rare compared with neutral or nearly neutral substitutions they influence the evolution. Both positive and purifying selection instead may cause underdispersion, i.e., less variation than expected. If certain substitutions are not allowed then the variance will decrease. For instance, stop codons will not be tolerated in the middle of a gene, thus eliminating that genetic option. This process is observed as purifying selection and will be the same for every generation. In addition, positive selection will work as a dynamic force (not identical in every generation) that also restricts the genetic possibilities.
In this study, we have unveiled the molecular clock of HIV, which provides possibilities to explore important questions about the evolution of HIV, such as the origin and age of the virus. However, the true phylogenetic relationships between human and other primate lentiviruses must be investigated further. For instance, cross-species transmissions may have changed the substitution rates and genetic recombination may have obscured the evolutionary history. Finally, the molecular clock also can help determine the evolutionary possibilities and constraints for multidrug resistance and the possibilities for the virus to invade new ecological niches.
Acknowledgments
We thank F. Granath for helpful suggestions, and R. Sokal and two anonymous reviewers for valuable comments. This study was supported by grants from the Swedish Research Council for Engineering Sciences, the National Institutes of Health-Department of Energy interagency agreement (3-Y01-AI-70001–13), and the Swedish Medical Research Council.
ABBREVIATION
- REV
reversible Markov process
Footnotes
References
- 1.Holland J J, De La Torre J C, Steinhauer D A. Curr Top Microbiol Immunol. 1992;176:1–20. doi: 10.1007/978-3-642-77011-1_1. [DOI] [PubMed] [Google Scholar]
- 2.Coffin J M. Science. 1995;267:483–489. doi: 10.1126/science.7824947. [DOI] [PubMed] [Google Scholar]
- 3.Gojobori T, Moriyama E N, Kimura M. Proc Natl Acad Sci USA. 1990;87:10015–10018. doi: 10.1073/pnas.87.24.10015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nijhuis M, Boucher C A B, Schipper P, Leitner T, Schuurman R, Albert J. Proc Natl Acad Sci USA. 1998;95:14441–14446. doi: 10.1073/pnas.95.24.14441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leigh Brown A J. Proc Natl Acad Sci USA. 1997;94:1862–1865. doi: 10.1073/pnas.94.5.1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li W-H, Tanimura M, Sharp P M. Mol Biol Evol. 1988;5:313–330. doi: 10.1093/oxfordjournals.molbev.a040503. [DOI] [PubMed] [Google Scholar]
- 7.Lukashov V V, Kuiken C L, Goudsmit J. J Virol. 1995;69:6911–6916. doi: 10.1128/jvi.69.11.6911-6916.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leitner T, Kumar S, Albert J. J Virol. 1997;71:4761–4770. doi: 10.1128/jvi.71.6.4761-4770.1997. , and correction (1998) 72, 2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leitner T, Ecanilla D, Franzén C, Uhlén M, Albert J. Proc Natl Acad Sci USA. 1996;93:10864–10869. doi: 10.1073/pnas.93.20.10864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leitner T, Fitch W M. In: Molecular Evolution of HIV. Crandall K A, editor. Baltimore: Johns Hopkins Univ. Press; 1999. pp. 315–345. [Google Scholar]
- 11.Samson M, Libert F, Doranz B J, Rucker J, Liesnard C, Farber C M, Saragosti S, Lapoumeroulie C, Cognaux J, Forceille C, et al. Nature (London) 1996;382:722–725. doi: 10.1038/382722a0. [DOI] [PubMed] [Google Scholar]
- 12.Dean M, Carrington M, Winkler C, Huttley G A, Smith M W, Allikmets R, Goedert J J, Buchbinder S P, Vittinghoff E, Gomperts E, et al. Science. 1996;273:1856–1862. doi: 10.1126/science.273.5283.1856. [DOI] [PubMed] [Google Scholar]
- 13.Leitner T, Halapi E, Scarlatti G, Rossi P, Albert J, Fenyö E M, Uhlén M. BioTechniques. 1993;15:120–126. [PubMed] [Google Scholar]
- 14.International Union of Pure and Applied Chemistry. Biochemistry. 1966;5:1445–1453. [Google Scholar]
- 15.Jukes T H, Cantor C R. In: Mammalian Protein Metabolism. Munro H N, editor. Vol. 3. New York: Academic; 1969. pp. 21–132. [Google Scholar]
- 16.Tamura K, Nei M. Mol Biol Evol. 1993;10:512–526. doi: 10.1093/oxfordjournals.molbev.a040023. [DOI] [PubMed] [Google Scholar]
- 17.Kumar S, Tamura K, Nei M. Comput Appl Biosci. 1994;10:189–191. doi: 10.1093/bioinformatics/10.2.189. [DOI] [PubMed] [Google Scholar]
- 18.Yang Z. J Mol Evol. 1994;39:105–111. doi: 10.1007/BF00178256. [DOI] [PubMed] [Google Scholar]
- 19.Kumar S, Tamura K, Nei M. mega: Molecular Evolutionary Genetics Analysis. University Park: Pennsylvania State Univ.; 1993. [Google Scholar]
- 20.Yang Z. paml: Phylogenetic Analysis Using Maximum Likelihood. Pennsylvania State Univ., University Park: Institute of Molecular Evolutionary Genetics; 1995. [Google Scholar]
- 21.Kumar S, Leitner T, Albert J. popdist:Population Distance. Stockholm: Swedish Institute for Infectious Disease Control; 1995. [Google Scholar]
- 22.Rambaut A, Grassly N. seq-gen:Sequence Generator. Oxford: Oxford Univ. Press; 1996. [Google Scholar]
- 23.Delwart E L, Sheppard H W, Walker B D, Goudsmit J, Mullins J I. J Virol. 1994;68:6672–6683. doi: 10.1128/jvi.68.10.6672-6683.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kimura M. The Neutral Theory of Molecular Evolution. Cambridge: Cambridge Univ. Press; 1983. [Google Scholar]
- 25.Van Valen L. Evol Theory. 1973;1:1–30. [Google Scholar]
- 26.Pamilo P, Nei M. Mol Biol Evol. 1988;5:568–583. doi: 10.1093/oxfordjournals.molbev.a040517. [DOI] [PubMed] [Google Scholar]
- 27.Fitch W M. Mol Phylogenet Evol. 1996;5:247–258. doi: 10.1006/mpev.1996.0018. [DOI] [PubMed] [Google Scholar]
- 28.Zhang L, Diaz R S, Ho D D, Mosely J W, Busch M P, Mayer A. J Virol. 1997;71:2555–2561. doi: 10.1128/jvi.71.3.2555-2561.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zuckerkandl E, Pauling L. In: Evolving Genes and Proteins. Bryson V, Vogel H J, editors. New York: Academic; 1965. pp. 97–166. [DOI] [PubMed] [Google Scholar]
- 30.Otha T, Kimura M. J Mol Evol. 1971;1:18–25. doi: 10.1007/BF01659391. [DOI] [PubMed] [Google Scholar]
- 31.Langley C H, Fitch W M. J Mol Evol. 1974;3:161–177. doi: 10.1007/BF01797451. [DOI] [PubMed] [Google Scholar]
- 32.Takahata N. Genetics. 1987;116:169–179. doi: 10.1093/genetics/116.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]