

Abstract
Mitochondrial myopathy, associated with muscle weakness and progressive external ophthalmoplegia, is caused by mutations in mitochondria oxidative phosphorylation genes including the heart–muscle isoform of the mitochondrial adenine nucleotide translocator (ANT1). To develop therapies for mitochondrial disease, we have prepared a recombinant adeno-associated viral vector (rAAV) carrying the mouse Ant1 cDNA. This vector has been used to transduce muscle cells and muscle from Ant1 mutant mice, which manifest mitochondrial myopathy. AAV-ANT1 transduction resulted in long-term, stable expression of the Ant1 transgene in muscle precursor cells as well as differentiated muscle fibers. The transgene ANT1 protein was targeted to the mitochondrion, was inserted into the mitochondrial inner membrane, formed a functional ADP/ATP carrier, increased the mitochondrial export of ATP and reversed the histopathological changes associated with the mitochondrial myopathy. Thus, AAV transduction has the potential of providing symptomatic relief for the ophthalmoplegia and ptosis resulting from paralysis of the extraocular eye muscles cause by mutations in the Ant1 gene.
Keywords: AAV, mitochondria, myopathy, adenine nucleotide translocator (ANT), oxidative phosphorylation, ophthalmoplegia
Introduction
Mitochondrial myopathy, generally defined as progressive muscle weakness associated with ragged red fiber muscle fibers (RRF)1 and the proliferation of subsarcolemmal mitochondria, can be caused by an assortment of defects in mitochondrial oxidative phosphorylation (OXPHOS) genes.2 Severe cases of mitochondrial myopathy can present with progressive external ophthalmoplegia (PEO), caused by paralysis of the extraocular eye muscles.3 One of the best characterized causes of mitochondrial PEO is mutations in the nuclear DNA (nDNA)-encoded gene for the heart–muscle isoform of the adenine nucleotide translocator (Ant1) gene.4
The ANTs export ATP generated by OXPHOS out across the mitochondrial inner membrane in exchange for cytosolic ADP, and thus are the main modulators of energy flux in the aerobic cell. ANT is an integral mitochondrial inner membrane protein that forms functional homodimers or -tetramers.5 ANTs work as electrogenic pumps6 and account for about 10% of mitochondrial inner membrane protein in tissues with high ATP requirements such as the heart and muscle. Efficient export of ATP also plays an important role in tissue development and the ANTs are thought to participate in the regulation of the mitochondrial permeability transition pore (mtPTP), and thus to play an important role in apoptosis.7 Humans have three different ANT isoform genes while mice have only two. However, the heart/muscle-specific isoform (ANT1) is common to both species.8
A mouse model of mitochondrial myopathy with RRFs was created by the genetic inactivation of the Ant1 gene. This recessive mutant results in a massive proliferation of muscle mitochondria and the hyperinduction of mitochondrial enzymes along with increased mitochondrial DNA (mtDNA) rearrangements.9 Hence, the ANT1-deficient mouse provides an excellent model for exploring therapeutic approaches for mitochondrial myopathy.
While there is a growing awareness of the importance of myopathy in the morbidity of disease, few therapeutic options are currently available to ameliorate the attendant muscle symptoms. Myoblast injection10 and direct DNA transformation11 have been explored, but met with only modest success. By contrast, virus-mediated gene transduction, particularly using adeno-associated viral (AAV) vectors12 has proven more successful. The non-pathogenic AAV has a relatively high affinity for muscle13 and can efficiently infect both dividing and nondividing muscle cells. Infection with recombinant AAV (rAAV) virus results in both a persistent episomal form as well as integration into the host genome.14 Hence, the AAV system has permitted long-term expression of transgenes, thus enhancing its attractiveness for treating muscle disorders.
Although most muscles are affected in mitochondrial myopathy, the primary concern of these patients is weakness of the extraocular eye muscles resulting in ophthaloplegia and ptosis. Since the extraocular eye muscles are small, very few muscle cells may need to be treated to provide significant symptomatic relief.
The rAAV system has already been successfully used to transduce mitochondrial-targeted transgenes. The mtDNA ATP6 gene with a corrected genetic code was allotypically expressed from the nucleus and the ATP6 polypeptide successfully imported into the mitochondria by coupling it to a mitochondrial targeting peptide.15 Furthermore, the yeast NADH:Coenzyme Q10 oxidoreductase gene has been transduced into the cells of a Leigh syndrome patient with nDNA-encoded complex I-deficiency.16 While these studies are unlikely to have immediate therapeutic applications, they demonstrate that the rAAV system can be used to transduce small OXPHOS polypeptides into human cells. Hence, the AAV system should be ideal for transduction of the 300 amino-acid Ant1 gene, thus permitting treatment of the mitochondrial myopathy and PEO resulting from mutations in the Ant1 gene.
In the present study, we report preparation of an rAAV harboring the mouse Ant1 cDNA and the use of this virus to transduce the nuclei of skeletal muscle fibers in ANT1-deficient mice. This resulted in the successful introduction of functional ANT1 protein into the skeletal muscle mitochondria and the amelioration of the biochemical and histopathological effects of the Ant1 genetic defect.
Results
Creation and production of the Ant1 transgene vector
The 1.2 kb Ant1 cDNA was introduced into the AAV 2 vector pTR-UF117 by substituting the GFP gene, transcribed from the CMV promoter, with the mouse Ant1 cDNA. The resulting pAAV-ANT1 vector was sequence verified and viral vector stocks were produced and purified to yield multiplicities of infection (MOI) of 9 × 1012 per cell.
Transduction of Ant1 into cultured myoblasts and myotubes: expression and cytotoxicity
Primary myoblastoid cell lines were prepared from 1-month-old Ant1+/+ (wild-type) and Ant1−/− (ANT1-deficient) mice. The undifferentiated myoblasts were transduced with AAV-ANT1 and AAV-GFP vectors and the transduction efficiency, the intracellular fate of the ANT1 transgene protein and the cell survival were monitored using the AAV-GFP as an internal control. Cell lines overexpressing ANT1 were then evaluated for expression of the ANT1 cDNA before and after differentiation of the myoblasts into myotubes. Parallel experiments were conducted using the immortalized myoblast cell line C2C12 (ATCC® No. CRL-1772).
The rAAV-GFP transduction efficiency for cultured mature myotubes, derived from ANT1-deficient mice, was evaluated by GFP expression and was found to be 10–15% lower than transduction of myotubes from wild-type tissue, perhaps due to problems with viral internalization in ANT1-deficient cells. On the other, hand, Ant1+/+ and −/− myoblasts showed no significant differences in transduction efficiency. Consequently, we routinely transduced cultured myoblasts and then induced them to differentiate into myotubes for further investigation.
Primary myoblastoid cell lines generated from ANT1-deficient and wild-type mice were analyzed for cell growth rate by measuring their increase in cell number after 3 days of growth with or without AAV-ANT1 transduction (Figure 1). Myoblasts lacking ANT1 increased in cell number about ⅓ as fast as wild-type myoblasts, indicating the importance of ANT for myoblast physiology. AAV-ANT1 transduction did not significantly inhibit cell proliferation of either the Ant1+/+ or −/− myoblasts after 3 days in culture. However, transduction of the ANT1-deficient myoblasts with AAV-ANT1 did not restore normal growth rate either. Therefore, increased ANT1 transduction did not negatively affect the growth of cells with ANT1, but did not stimulate the growth of cells without ANT1 (Figure 1).
Figure 1.
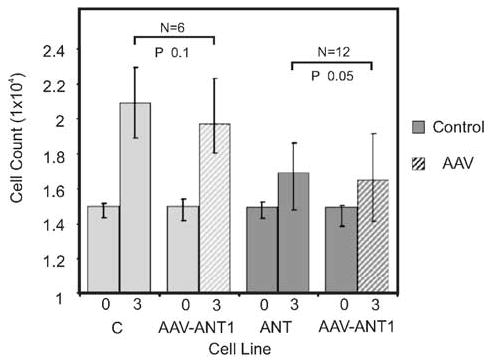
Growth rates of primary myoblast cultures from wild-type and ANT1-deficient muscle, with and without AAV-ANT1 transduction. Myoblasts were generated from wild-type (C) and ANT1−/− (ANT) mice, and either left untransduced (solid bars) or were transduced with rAAV-ANT1 (AAV-ANT1, hatched bars). Cell numbers were counted at day 0 (0) and day 3 (3) after plating.
The persistence of the AAV-ANT1 vector in transduced primary cultured myoblasts was monitored by PCR. Vector-specific sequence amplicons as well as ANT1 transcripts (detected by reverse transcription PCR (RT-PCR)) were detectable at 1 week postinfection in myoblasts and at 2 weeks postinfection in the myotubes, demonstrating that the transgenic vector can be retained over an extended period of time.
Myoblasts express both ANT1 and ANT2 isoforms, whereas differentiated myotubes or adult muscle (1 month of age) expresses only ANT1.18
By using isoform-specific antibodies, we detected the ANT1 protein by Western blots in the AAV-ANT1-transduced ANT1-deficient myoblasts and throughout their differentiation process into myotubes (Figure 2a). Thus, the AAV-ANT1 vector is able to transduce the Ant1 cDNA into ANT1-deficient cells and result in the expression of ANT1 mRNA and protein in both myoblasts and myotubes, with minimal effect on the viability of the muscle cells.
Figure 2.

ANT1 expression in pAAV-ANT1-transduced myoblasts, myotubes and skeletal muscle. (a) Detection of ANT1 protein in myoblasts and myotubes. Western blot analysis of ANT1 in wild-type (WT) and ANT1−/− (ANT1-) myoblasts 5 days after transduction with AAV-ANT1 (tWTb, tANT1-b) and in myotubes 7 days after induction of differentiation (tWTd, tANT1-d), probed with ANT1 and COXIV-specific antibodies. (b) Mitochondrial localization of ANT1 transgenic protein in myoblasts. Western blot analysis of subcellular fractions obtained from wild-type (WT), untransduced ANT1−/− (ANT1-) and AAV-ANT1-transduced ANT1−/− (tANT1-) myoblastoid cell lines. Whole-cell lysates (LYS), cytoplasmic fractions (CYT) and mitochondrial fractions (MIT) were probed with antibodies specific for Hsp70, ANT1 and COX IV. The inset shows the localization of ANT1 in tANT- mitochondria in the mitochondrial lysate (MLYS) and inner (IM), but not the outer mitochondrial membrane (OM) or the mitochondrial matrix (M). (c) Immunofluorescence microscopy of myocytes: Wild-type (WT), ANT1−/− (ANT1-) and ANT1−/− myocytes transduced with AAV-ANT1 (tANT1-) were stained with an antibody specific for ANT (green fluorescence). Mitochondria were counterstained with Mitotracker CMX-ROS (red fluorescence). (d) Detection of rAAV-ANT1 DNA in transduced skeletal muscle. Agarose gel electrophoresis of PCR- fragments obtained from left (L) and right (R) gastrocnemius muscle and liver (I) of 1-month-old mice. DNA was extracted and amplified with oligonucleotides specific for the transgene Ant1 cDNA. MW: 50 bp ladder; C: control; P: pAAV =AAV vector; WT =wild-type control; tWT and tANT1- =wild-type and ANT1−/− tissues injected with the transgene. (e): Detection of transgenic Ant1 mRNA. Agarose gel electrophoresis of transgene-specific RT-PCR fragments (cANT), from untransduced WT and tissues transduced with either the full viral dose (tANT1-) and half-dose (tANT1-1/2). Utilization of comparable levels of RNA was confirmed by RT-PCR coamplification of the GADPH mRNA. Abbreviations as in (d), except P− =AAV vector without transgene, P+ =AAV containing the Ant1 transgene. (f) Detection of ANT1 protein in skeletal muscle. Western blot analysis of ANT1 in mouse tissues 4 weeks after injection, probed with ANT1 and COXIV-specific antibodies. WT/G =control gastrocnemius muscle; tANT1- =ANT1-deficient mice injected with rAAV-ANT1 in the lower left hind limb; tANT1-/V =vastus lateralis, tANT1-/S =semitendinosus muscle, tANT1-/T =tibialis, tANT1-/G =gastrocnemius muscle. MW =31 kDa molecular weight marker.
Mitochondrial localization of the ANT1 transgene protein
To determine if the ANT1 transgene protein was properly integrated into the mitochondrial inner membrane, mitochondria were purified by density gradient from ANT1-deficient myoblasts 14 days after transduction with AAV-ANT1. The transgene ANT1 protein, detected by Western blots using ANT1-specific antibody, colocalized with the mitochondrial inner membrane protein cytochrome c oxidase subunit IV (COXIV) (Figure 2b). Subfractionation of the AAV-ANT1-transduced Ant1−/− mitochondria into mitochondrial membrane and matrix fractions confirmed that the transduced ANT1 protein was inserted into the mitochondrial inner membrane (Figure 2b, insert).
The mitochondrial localization of the ANT1 protein was further confirmed by staining AAV-ANT1-transduced ANT1-deficient myotubes with a general ANT antibody. The ANT1 immunofluorescence colocalized with a mitochondria-specific counterstain (Figure 2c). Therefore, the ANT1 transgene is expressed in both myoblasts and myotubes and the transgene protein is specifically targeted to the mitochondrial inner membrane.
AAV-ANT1 transduction of mouse muscle
The AVV-ANT1 vector was next used to transduce ANT1 activity into the skeletal muscle of ANT1-deficient mice. AAV-ANT1 vector was injected into the gastrocnemius muscles in the left hind limb of ANT1-deficient newborn mice, the right leg serving as a control. The animals were killed at different times and the muscles assessed for AAV-ANT1 gene content and ANT1 transgene expression. The AAV-ANT1 viral DNA was found to be primarily limited to the injected muscle groups, although a weak signal was also found in the liver, suggesting minor systemic distribution of the viral vector through the bloodstream (Figure 2d).
Detection of the Ant1 transcript by RT-PCR revealed strong expression in the left injected hind limb. This expression was roughly proportional to the amount of vector injected (Figure 2e).
The ANT1 protein expression in injected skeletal muscle was also assessed by Western blot, and found to correspond with the ANT1 transcript levels. The injected left gastrocnemius muscle was found to be positive for ANT1, while other muscle groups were uniformly negative (Figure 2f).
Transgene distribution in mouse muscle
ANT1 protein expression was also observed by immunohistochemistry in the transduced left hind limb muscle tissue. A general ANT antibody was used here, since ANT2 expression is very low in muscle and confined to endothelial and connective tissue and mononuclear cells.
In adult Ant1−/− mice, the ANT1 transgene protein was observed in the immediate vicinity of the injected muscle, as confirmed by coinjecting the AAV-ANT1 virus with the AAV-GFP vector and by tissue damage caused by the oversized needle used. This is consistent with previous studies, which have shown that the AAV vector can only penetrate five to 10 fiber layers in adult muscle tissue.18 The expression of the transgenic ANT1 protein in individual fibers of adult mice gave a particulate staining pattern similar to that obtained for ANT1 in wild-type muscle (Figure 3a), confirming that transgenic ANT1 protein was correctly targeted to the mitochondrion.
Figure 3.
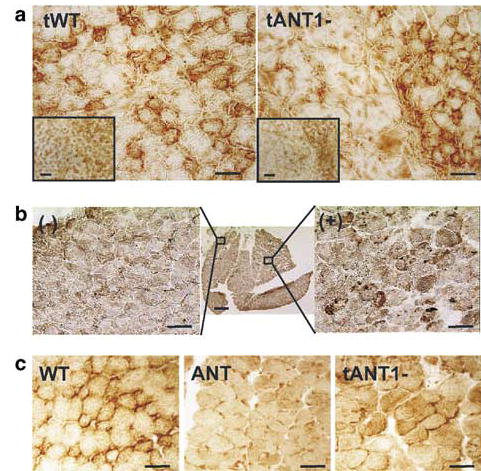
Ant1 expression in gastrocnemius muscle detected by immunohistochemical stain (DAB) for ANT. (a) Transgene Ant1 expression in wild-type (tWT) and ANT1−/− (tANT1-) mice injected with the transgene at 3 months of age. (b) Ant1 expression in a 1-month-old ANT1−/− mice injected with rAAV-ANT1 as a neonate (center), showing transduction of upper right quadrant of the section (bar scale: 20 μm). Flanking pictures show untransduced (−) (left) and transduced (+) (right) areas of the muscle (bar scale: 50 μm). (c) Ant1 expression in a 1-year-old wild-type (WT) and ANT1−/− mouse (ANT) injected with rAAV-ANT1 (tANT1-) as a neonate (bar scale: 20 μm).
Injection of AAV-ANT1 virus into neonatal ANT1-deficient muscle resulted in transgene expression in a much greater number of muscle fibers (Figure 3b). This may reflect the broader distribution of the vector upon injection into neonatal tissue as well as the initial infection of not only muscle fibers but also a larger number of muscle precursor cells present in neonatal tissue.19 These transduced precursor cells could subsequently proliferate and differentiate into mature fibers expressing the transgene.
Comparison of the immunohistological staining for ANT1 in 1- and 3-month-old animals versus 1-year-old animals revealed a continued expression of the Ant1 transgene, even though the level of expression declined somewhat with age (Figure 3c).
ANT1 activity in AAV-ANT1-transduced muscle
To assess the functionality of the transgenic ANT1 protein within the mitochondrial inner membrane, we assessed the ATP-binding and ATP-transport capacities in the AAV-ANT1-transduced tissues. First, we examined the level of ANT protein in tissue homogenates of AAV-ANT1-transduced gastrocnemius muscle using the fluorescent ATP analogue naphthoyl-atractyloside (N-ATR), which bind to the ATP binding site of the ANTs. Changes in N-ATR fluorescence by displacement with the irreversible inhibitor C-ATR permitted quantification of the number of ANT molecules.
AAV-ANT1 transduction of the left gastrocnemius muscle of ANT1-deficient mice increased the level of ANT1 protein from undetectable to 5–30% of wild-type ANT1 levels (Figure 4a). Injection of wild-type muscle with AAV-ANT1 vector did not significantly increase ANT1 binding capacity. Hence, AAV-ANT1 transduction partially restored N-ATR binding capacity, and thus was able to generate functional ANT1 protein in the previously ANT1-deficient muscle mitochondria. Moreover, addition of the transgenic Ant1 did not adversely alter the maximum level of ANT1 protein in wild-type muscle, suggesting that chromosomal Ant1 expression may be regulated.
Figure 4.
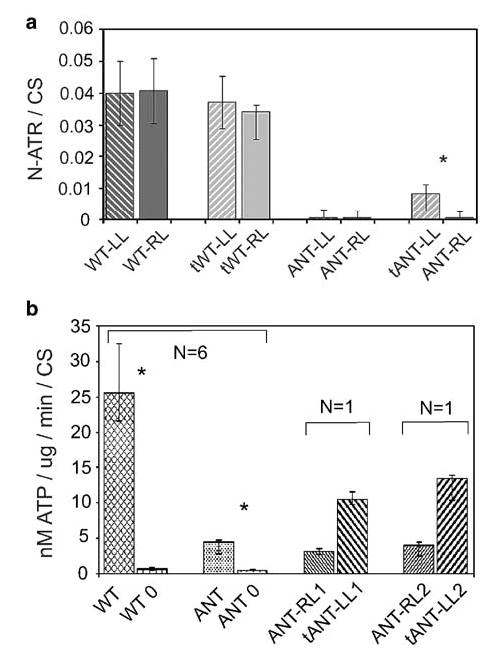
Demonstration of active ANT1 in transduced skeletal muscle. (a) Quantification of transgenic ANT1 protein by binding of the ATP analogue N-ATR in mitochondrial preparations from 3-month-old mouse right (RL) and left (LL) hind limb muscles. Neonates were injected with either saline (WT-, ANT-) or rAAV-ANT1 (tWT-, tANT1-). ATP binding capacities were quantified by the increase in N-ATR fluorescence following competitive release with C-ATR, normalized to CS activities. Six animals were tested per group. *P≤0.01. (b) ATP excreted from transduced skeletal muscle mitochondria. Mitochondria were isolated from 1-year-old mouse hind limb muscles, injected with saline (ANT-RL) or with rAAV-ANT1 (tANT-LL) as neonates. ATP production per mg mitochondrial protein was measured in triplicate and normalized to CS activity. ATP export in wild-type (WT) and ANT1−/− mitochondria (ANT), could be blocked with CATR (WT-0, ANT-0). Six animals tested per group. Two ANT1−/− mice (1 and 2) were transduced with rAAV-ANT1 as neonates, the left hind limb treated with virus (LL) and the right hind limb treated with saline vehicle (RL). Mitochondria from each limb were isolated and analyzed for ATP secretion. *P≤0.005.
To confirm that the transgene ANT1 molecules were able to transport ATP, we measured the rate of ATP export from coupled, respiring mitochondria isolated from 1-year-old ANT1-deficient mice, transduced with AAV-ANT1 as neonates. Mitochondria from AAV-ANT1-transduced muscle secreted up to 45% of the ATP produced from wild-type mitochondria in multiple assays from two independent mice (Figure 4b). This indicates that the N-ATR binding activity in transduced tissues was in fact functional ANT1.
Interestingly, mitochondria from the ANT1-deficient mouse muscle secreted about 5–10% of the ATP secreted by wild-type muscle mitochondria. This residual ATP secretion could be due to the leakage of ATP through damaged mitochondrial membranes. Alternatively, it could be due to the presence of a small portion of mitochondria from nonmuscle cells, which contaminated the muscle of the ANT1-deficient hind limb. These mitochondria would still express ANT2, and hence would secrete ATP. This later alternative seems the more probable since addition of the irreversible ATP-inhibitor C-ATR reduced ATP secretion to <2% in both Ant1−/− and Ant1+/+ mitochondria.
Whatever permitted the secretion of some ATP from the ANT1-deficient mitochondrial preparations, the amount of ATP secreted by the AAV-ANT1-transduced hind limb muscle mitochondria was much greater than that of the untransduced mitochondria. This confirms that AAV-ANT1 transduction was able to reconstitute mitochondrial ATP secretion in the ANT1-deficient mouse skeletal muscle.
Histopathological analysis of AAV-ANT1-transduced ANT1-deficient muscle
AAV-ANT1 transduction of the muscles of ANT1-deficient mice also significantly ameliorated the pathological features of mitochondrial myopathy. In wild-type mice, the soleus is composed predominantly of slow-twitch, oxidative, Type I fibers, whereas the gastrocnemius is predominantly fast-twitch, glycolytic, Type IIa/b fibers.20 In the ANT1-deficient mice, these muscle groups exhibited the classical features of mitochondrial myopathy including altered fiber size and number, central nuclei, altered fiber type characteristics and RRFs.21 In addition, the majority of the Type II and IIa fibers in the ANT1-deficient gastrocnemius muscle take on a histological appearance more similar to the oxidative, Type I fibers, as determined by ATPase activity and increased levels of the mitochondrial enzymes NADH-dehydrogenase (NADH-DH), succinate dehydrogenase (SDH) and cytochrome oxidase (COX). Still, the size of these muscle fibers and the MHC expression pattern remain unchanged.
In the AAV-ANT1-transduced muscle, some muscle tissue damage was observed at the injection site, possibly resulting from endomysial infiltration of mononucleic cells resulting in the formation of connective scar tissue. However, these same effects were observed following injection of the AAV-GFP vector, and thus were not a consequence of Ant1 cDNA expression. In both adult and newborn ANT1-deficient muscle, the acute tissue injury observed following AAV-ANT1 injection resolved in about 2 weeks (Figure 5a1 and a2).
Figure 5.
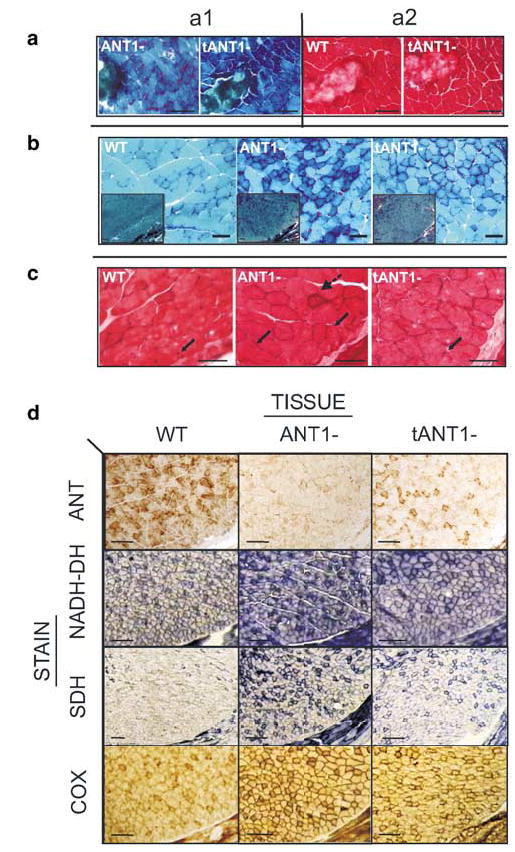
Reversal of mitochondrial myopathy following rAAV-ANT1 transduction of adult ANT1-deficient mouse muscle. (a) Frozen sections of gastrocnemius muscles stained with modified Gomori-Trichrome (a1) or H&E (a2) 2 weeks after transverse injection of 3-month-old wild-type (WT) and ANT1−/− (ANT1-) mice with an 18-gauge needle. (b) Gomori-Trichrome-stained frozen sections of soleus muscles from 1-month-old wild-type (WT), ANT1−/− (ANT1-) mice, ANT1−/− mice transduced with rAAV-ANT1 as neonates. Insets show lower magnification of muscle area (bar scales: 20 μm; insets: 50 μm). (c) H&E staining of gastrocnemius muscles from the same animals. Solid arrows indicate localization of peripheral myonuclei in wild-type (WT), central nuclei in ANT1−/− (ANT1-) and peripheral in transduced ANT1−/− animals (tANT1-). Broken arrow indicates endomysial inflammation in ANT1−/− tissues (Bar scales: 10 μm). (d) Reversal of hyperinduction of mitochondrial OXPHOS enzymes by AAV-ANT1 transduction. Immunohistochemical stain for ANT1 and histochemical stains for NADH-DH, SDH and COX of transverse-sectioned gastrocnemius muscle of 3-month-old wild-type (WT), ANT1−/− (ANT1-) and ANT1−/− mice injected with AAV-ANT1 as neonates (tANT1-) (bar scale: 50 μm).
By contrast, transduction of ANT1-deficient muscle with the AAV-ANT1 vector resulted in significant reductions in the histopathological features associated with mitochondrial myopathy, which were not seen in muscle injected with the control vector. The most dramatic therapeutic effect of ANT1-AAV transduction was the reduction in RRFs in the soleus and/or gastrocnemius Type I muscle fibers of ANT1-deficient mice (Figure 5b). However, reversal of the RRF pathological was not complete since some evidence of subsarcolemmal red staining was retained.
Following AAV-ANT1 transduction, the rounded pathologic aspect of the muscle fibers of the ANT1-deficient muscle were restored to the more angular structure of normal fibers. Moreover, the number of fibers with central nuclei was clearly reduced in the AAV-ANT1-injected areas of the gastrocnemius. Finally, the endomysial inflammation pattern visible in the highly oxidative fibers of the soleus muscle of Ant1−/− mice was reduced in the treated muscles (Figure 5c).
In addition to the reduction in RRFs, AAV-ANT1 transduction of the ANT1-deficient gastrocnemius muscle resulted in a striking reduction in the levels of the OXPHOS enzymes NADH-DH, SDH and COX, with the histochemical staining approaching normal levels (Figure 5d). However, AAV-ANT1 transduction had no apparent influence on myofiber size or MHC expression pattern (data not shown).22 Therefore, transduction of Ant1 into ANT1-deficient muscle dramatically improved the muscle pathology associated with this ANT1 enzyme defect.
Discussion
We have successfully treated the mitochondrial myopathy of the ANT1-deficient mouse through AAV-assisted gene transfer of the Ant1 cDNA. The Ant1 cDNA was transcribed from a CMV promoter, and transduced myoblasts and myotubes synthesized the Ant1 mRNA and protein.
Following an initial phase, when the ANT1 protein levels were highest, the level of ANT1 expression achieved a relatively constant level in both cultured myoblasts and in differentiated myotubes. The initial higher level of ANT1 expression was most likely the product of the transient presence of episomal copies of the rAAV-ANT1 vector.
As expected, developing and regenerating muscle tissue showed higher transduction efficiencies than differentiated myofibers.23 The slightly reduced transduction levels of ANT1-deficient myotubes might be the result of changes in the efficiency of receptor-mediated endocytosis or due to other ATP requiring processes necessary for efficient AAV transduction.24
An initial concern was that viral transduction with the Ant1 gene would result in excessively high expression levels of ANT1 and that this would induce apoptosis. The ANTs interact with the mtPTP and are known to regulate its activation by Ca2+ and ANT ligands leading to apoptosis.25 Reduction of ANT1 activity in either the mutant mice or in ANT1-mutant PEO patients has not been observed to increase apoptosis,26 whereas ANT1 expression due to Ant1 cDNA transfection into nonmyogenic cultured cells has been reported to induce apoptosis.27 However, our data suggest that increased apoptosis may not be a concern during the transduction of skeletal muscle with the Ant1 cDNA. This is because myoblasts, myotubes, skeletal muscle and heart all naturally express high levels of ANT1 and thus must be resistant to any proapoptotic effects that this protein might have. Other cell types, which generally express only ANT2, could well be induced to undergo apoptosis as a consequence of the nonphysiological expression of ANT1. Therefore, we feel that it should be possible to transduce the Ant1 cDNA into autosomal-dominant PEO patients harboring Ant1 missense mutations in sufficiently high levels to ameliorate symptoms without also inducing muscle cell necrosis and apoptosis.
While in vivo, injection of the AAV-ANT into the muscles of Ant1−/− mice resulted in a 5–30% increase in ANT1 protein and a restitution of between 25 and 45% of the normal ATP secretion levels, AAV-ANT1 transduction of Ant1−/− myoblasts did not restore the normal growth rate. Since myoblasts express both ANT1 and ANT2, the reduced growth rate of the Ant1−/− myoblasts may not be entirely related to deficiency in ATP export. Perhaps other physiological factors may be limiting growth rate, such as nucleotide pools28 or signal transduction pathways,29 which require constant higher levels of ANT1.
Irrespective of the inability of AAV-ANT1 transduction to restore normal myoblast growth rate, it substantially increased the level of ANT1 protein and ATP export in the skeletal muscle of the ANT1-deficient mice. Moreover, AAV-ANT1 transduction resulted in the amelioration of many of the histopathological abnormalities associated with mitochondrial myopathy, including reduction of the number of RRFs and suppression of the hyperinduction of the OXPHOS enzymes, NADH-DH, SDH and COX. Hence, these results demonstrate that mitochondrial myopathy can be locally treated by AAV-mediated transduction of the Ant1 gene.
Having achieved this success, there are several ways that this procedure could be improved. Transgene expression might be enhanced by employing other promoters such as the endogenous Ant1 promoter or the muscle mitochondrial creatine phosphokinase promoter. Muscle infectivity could be improved by changing the AAV vector capsid genes to serotypes with higher muscle affinity.30,31 Viral replication and thus transduction efficiency could be enhanced by using dimeric or self-complementary AAV vectors.32
One potential difficulty with using the current AAV-ANT1 system for treating human mitochondrial myopathy resulting from Ant1 mutations is that the human disease is inherited as a dominant, rather than a recessive as in the current mouse model. The most likely reason for this difference is that the mouse mutant is a null, while the reported human mutants are missense mutations. Since the ANTs are thought to function as homo-oligomeric proteins, one defective subunit could inactive an entire complex, thus acting as a dominant negative. If this is the case, then high-level expression of a functional transgene may be required to generate enough subunits to assure sufficient functional enzyme to ameliorate the symptoms of autosomal PEO in patients.
In conclusion, using the Ant1 knockout mouse as a model system, we have demonstrated that AAV transduction provides a robust method for reconstituting OXPHOS activity in muscle mitochondria. By introducing a functional copy of Ant1 into one of the most prominently affected tissues in this mouse model, we were able to increase Ant1 expression to a level that resulted in the amelioration of the biochemical deficiencies and histopathological features. Thus, AAV-mediated gene therapy may have real potential for treatment of the ophthalmoplegia in autosomal PEO.
Materials and methods
Vector construction and amplification
The mouse Ant1 cDNA (AF240002)8 cloned in pBlue-script SK+ (Stratagene) and AAV vector plasmids were propagated in Escherichia coli SURE®2 cells (Stratagene) and the plasmids purified using commercial kits (QIA-prep, Qiagen). The 1.2 kb Ant1 cDNA fragment was cloned into the AAV vector pTR-UF1,33 creating the vector pAAV-ANT1. Insert size was confirmed by XhoI digest (New England Biolabs). Orientation of the insert and integrity of cloning sites was sequence verified with primers located in the CMV promoter (cANT1-SDSA: AGG CCT GTA CGG AAG TGT TA) and polyA region (SV40polyA: CCC CTG AAC CTG AAA CAT AA). AAV-ANT1 vector stocks were produced by two-plasmid cotransfection and double CsCl purification.17
In vitro transfection and transduction
Primary myoblasts derived from 3-day-old S4/129 (wild-type) and W1/129 (ANT1-deficient) mouse gastrocnemius muscle were purified to >99%.34 Myoblasts grown in GM medium (Ham’s F10 (Gibco), 20% FBS (Hyclone), 5 ng/ml bFGF (Promega)/5% CO2/37°C) on dishes coated with E–C–L matrix (Upstate Biotech) were plated in 24-well tissue culture plates and plating efficiency was controlled to avoid significant variation among groups. At 48 h after plating, cells were transduced with the viral vectors, either with AAV-ANT1 plus pTR-UF1 at a 1:1 ratio or with AAV-ANT1 alone. To determine optimal titers for transduction of myocytes, AAV-ANT1 virus was administered at MOIs of 1 × 108, 1 × 1010 and 1 × 1012. Differentiation of myoblasts into myotubes was induced by substituting FBS with horse serum. Growth rates were estimated by determining the cell number 3 days after transduction/differentiation for cell viability by trypan blue exclusion. Assessment of transgene expression was carried out on clonal cultures of transduced myoblasts before and after differentiation. Quantitative analysis for morphological changes was carried out using light and fluorescent microscopy and by staining with Hoechst-33258 (Sigma).
In vivo expression of AAV-ANT1 from murine skeletal muscle
All animal experiments were approved by the Emory University and UCI IACUC committees. One group of S4/129 controls and W1/129 (ANT1−/−) mice were injected with 1:1 AAV-ANT1 and AAV-GFP to optimize the injection strategy. A second group was injected with phosphate-buffered saline (PBS) in the right hind limb and with the AAV-ANT1 transgene vector in the left hind limb (contralateral leg).
Adult mice were given standard intramuscular injections.35 Neonatal mice were collected at 3 days of age and anesthetized by hypothermia.36 A short longitudinal skin incision was made on the back lower hind limb and with the aid of a dissecting microscope a 33-gauge hypodermic needle (Hamilton; Reno, NV, USA) was inserted into the posterior group of muscles (gastrocnemius, soleus) parallel to the fiber orientation and 2 μl were injected in three separate longitudinal injections (a total of 6 μl and 1 × 109 infectious units in PBS/10% glycerol). The incision was closed with tissue adhesive (Vectabond™). At 2, 7, 14, 30 and 90 days as well as 1 year after injection, the animals were killed by cervical dislocation and tissues were collected for analysis.
Isolation and purification of mitochondria
Skeletal muscle mitochondria were isolated by tissue disruption, differential centrifugation and Iodixanol-gradient centrifugation37 (OptiPrep™, Invitrogen/AXIS-SHIELD) using a self-generating continuous gradient.38 Respiration of mitochondria was assessed in a Clark-type O2 electrode39 (Hansatech). Mitochondria from cultured cells were prepared using hypoosmotic swelling40 and Iodixanol density-gradient centrifugation. Purified mitochondria were subfractionated into outer membrane, inner membrane and matrix using digitonin (Roche) and differential centrifugation.41
DNA and RNA analysis
The Ant1 genotype of animals was determined by differential PCR.9 Nucleic acids were isolated from soleus and gastrocnemius muscle using Trizol® (Life Technologies), and RNA, DNA and protein were collected taking into account the small samples available (1 × 106 myocytes, 250 mg of muscle tissue). RNA expression of AAV-ANT1 was assayed by semi-quantitative RT-PCR (Roche). Purified RNA was reverse transcribed using a vector-specific primer (ANT1: SV40polyAR: TTC ACT GCA TTC TAG TTG AGG A). cDNA was synthesized using nested oligonucleotides (cANT1-SDSA: see above; ANT-1-E1R: AGC AGC AGT TTG ACC CTC TC).
Western blot analysis
Western blots were developed by using affinity-purified polyclonal antibodies.9 The protein content of cell and tissue lysates was determined by Lowry protein assay (Pierce). Protein (25 μg) of total tissue lysate or 15 μg of isolated mitochondrial lysate were separated using 10% TG-SDS–PAGE in an MES-buffer system (NOVEX/Invitrogen), blotted onto nitro-cellulose membranes (Biorad) and the blots stained with Ponceau S (Sigma). Blots were reacted with 2 μg/ml ANT1, 4 μg/ml ANT2, COX IV (Molecular Probes) and Hsp70 (Stressgen) antibodies. Bound antibody was detected by a peroxidase-conjugated secondary antibody (Sigma) and signals were visualized by chemiluminescence substrate (ECL, Amersham/Pharmacia).
Assays for ANT1 function
Functional ANT1 protein in the muscle homogenates was assessed with fluorescent N-ATR.42 Mitochondrial protein (100 μg) were assayed in 300 μl reaction mix in a temperature-controlled (25°C) stirred quartz cuvette on a LS50B fluorescence spectrometer (Perkin-Elmer). The mitochondria were reacted with N-ATR, the N-ATR competed off the ANT with C-ATR (Calbiochem) and the increased fluorescence monitored by fluorimetry.43 N-ATR binding was quantified as nmol C-ATR/mg mitochondrial protein, normalized to citrate synthase (CS) activity.44
The mitochondrial export of ATP by the ANT carrier too was assayed using a kinetic bioluminescence assay45 (ATP Bioluminescence Kit CLS II, Roche). Coupled mitochondrial protein (25 μg) were combined with 25 μl of bioluminescent reagent in a 200 μl reaction in a Sirius-Luminometer (Berthold Detection Systems). Pyruvate and malate were used as respiration substrate, the reaction started by adding ADP, and the ATP secreted into the reaction buffered quantified every 15 s for 10 min in duplicate (40 readings). ATP production and ATP export were inhibited with C-ATR and Oligomycin (Sigma), respectively. Bioluminescence ATP determinations were correlated with ATP standards and expressed as nm ATP/min/mg mitochondrial protein and normalized to CS activity.
Pathophysiological assessment
The posterior muscles of the lower hind limb from injected mice and controls were removed at day 7, 14, 28 and 96 after viral transduction. After removal of tendons, blood vessels and connective tissue, muscles were weighted and then either frozen or fixed with formalin.
Immunohistochemistry
Myocytes cultured on chamber slides (Tissue-Tek) and stained with 250 ng/ml Mitotracker CMX-ROS (Molecular Probes) were fixed with ice-cold acetone/methanol (3/1) and permeabilized with 0.1% Triton X-100 (Calbiochem). After blocking with 2% serum in PBS/1% BSA, cells were incubated with 2 μg/ml ANT-specific antibody (Santa Cruz Biotech.) in PBS/0.05% Triton X-100 and the bound antibody detected by an Alexa-Fluor 488-conjugated secondary antibody (Molecular Probes).
Snap-frozen tissues were embedded in OCT compound #4583 (Tissue-Tek/VWR). Serial cryosections (10 μm thick) from the midbelly of the muscle were transferred on Vectabond™-treated glass slides, air-dried (20–30 min at RT) and washed 2 × in PBS/0.1% Tween-20, blocked in 10% normal serum in PBS/1% BSA and incubated with the ANT-specific antibody (4 μg/ml). Endogenous peroxidase was quenched with 0.3% H2O2 in PBS for 5 min. After 30 min, the secondary biotinylated antibody (Vector Laboratories) was applied for 30 min and detected by a biotinylated horseradish peroxidase conjugate (Vectastain Elite ABC Kit). After DAB-substrate solution color development, sections were dehydrated, cleared and mounted (Histo-Clear™, Histomount™, Nat. Diagnostics).
Histochemistry
Transverse sections from gastrocnemius muscles were analyzed by H&E and Gomori Tri-Chrome staining46 and also stained for NADH-DH, SDH and COX.47
Images were acquired using a CCD camera (SPOT-Camera/Software) mounted on an Axioplan 2 (Carl Zeiss) and 10 different sections, defined by their position relative to the poplieatal artery, were analyzed using image processing software (Adobe® Photoshop 7) and the results averaged.
Acknowledgments
This work was supported by NIH Grants NS21328, NS41850, AG131154, HL64017 and an Ellison Foundation Senior Investigator Grant awarded to DCW. Support for RJS was provided by an NIH/NHLBI Grant 5P01HL051818-06-10 of the Cystic Fibrosis Foundation.
References
- 1.Morgan-Hughes JA. Mitochondrial diseases of muscle. Curr Opin Neurol. 1994;7:457–462. doi: 10.1097/00019052-199410000-00014. [DOI] [PubMed] [Google Scholar]
- 2.Wallace DC. Mitochondrial diseases in man and mouse. Science. 1999;283:1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- 3.Zeviani M, et al. An autosomal dominant disorder with multiple deletions of mitochondrial DNA starting at the D-loop region. Nature. 1989;339:309–311. doi: 10.1038/339309a0. [DOI] [PubMed] [Google Scholar]
- 4.Kaukonen J, et al. Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science. 2000;289:782–785. doi: 10.1126/science.289.5480.782. [DOI] [PubMed] [Google Scholar]
- 5.Pebay-Peyroula E, et al. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature. 2003;426:39–44. doi: 10.1038/nature02056. [DOI] [PubMed] [Google Scholar]
- 6.Gropp T, et al. Kinetics of electrogenic transport by the ADP/ATP carrier. Biophys J. 1999;77:714–726. doi: 10.1016/S0006-3495(99)76926-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim JS, He L, Lemasters JJ. Mitochondrial permeability transition: a common pathway to necrosis and apoptosis. Biochem Biophys Res Commun. 2003;304:463–470. doi: 10.1016/s0006-291x(03)00618-1. [DOI] [PubMed] [Google Scholar]
- 8.Levy SE, Chen YS, Graham BH, Wallace DC. Expression and sequence analysis of the mouse adenine nucleotide translocase 1 and 2 genes. Gene. 2000;254:57–66. doi: 10.1016/s0378-1119(00)00252-3. [DOI] [PubMed] [Google Scholar]
- 9.Graham BH, et al. A mouse model for mitochondrial myopathy and cardiomyopathy resulting from a deficiency in the heart/muscle isoform of the adenine nucleotide translocator. Nat Genet. 1997;16:226–234. doi: 10.1038/ng0797-226. [DOI] [PubMed] [Google Scholar]
- 10.Smythe GM, Hodgetts SI, Grounds MD. Immunobiology and the future of myoblast transfer therapy. Mol Ther. 2000;1:304–313. doi: 10.1006/mthe.2000.0049. [DOI] [PubMed] [Google Scholar]
- 11.Li S, Benninger M. Applications of muscle electroporation gene therapy. Curr Gene Ther. 2002;2:101–105. doi: 10.2174/1566523023348183. [DOI] [PubMed] [Google Scholar]
- 12.Watchko J, et al. Adeno-associated virus vector-mediated mini-dystrophin gene therapy improves dystrophic muscle contractile function in mdx mice. Hum Gene Ther. 2002;13:1451–1460. doi: 10.1089/10430340260185085. [DOI] [PubMed] [Google Scholar]
- 13.Pruchnic R, et al. The use of adeno-associated virus to circumvent the maturation-dependent viral transduction of muscle fibers. Hum Gene Ther. 2000;11:521–536. doi: 10.1089/10430340050015716. [DOI] [PubMed] [Google Scholar]
- 14.Hamilton H, Gomos J, Berns KI, Falck-Pedersen E. Adeno-associated virus site-specific integration and AAVS1 disruption. J Virol. 2004;78:7874–7882. doi: 10.1128/JVI.78.15.7874-7882.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Owen RI, et al. Recombinant adeno-associated virus vector-based gene transfer for defects in oxidative metabolism. Hum Gene Ther. 2000;11:2067–2078. doi: 10.1089/104303400750001381. [DOI] [PubMed] [Google Scholar]
- 16.Seo BB, et al. Use of the NADH-quinone oxidoreductase (NDI1) gene of Saccharomyces cerevisiae as a possible cure for complex I defects in human cells. J Biol Chem. 2000;275:37774–37778. doi: 10.1074/jbc.M007033200. [DOI] [PubMed] [Google Scholar]
- 17.Xiao X, Li J, Samulski RJ. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol. 1998;72:2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Snyder RO, et al. Efficient and stable adeno-associated virus-mediated transduction in the skeletal muscle of adult immunocompetent mice. Hum Gene Ther. 1997;8:1891–1900. doi: 10.1089/hum.1997.8.16-1891. [DOI] [PubMed] [Google Scholar]
- 19.Snow MH. The effects of aging on satellite cells in skeletal muscles of mice and rats. Cell Tissue Res. 1977;185:399–408. doi: 10.1007/BF00220299. [DOI] [PubMed] [Google Scholar]
- 20.Pette D, Staron RS. Myosin isoforms, muscle fiber types, and transitions. Microsc Res Technol. 2000;50:500–509. doi: 10.1002/1097-0029(20000915)50:6<500::AID-JEMT7>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 21.Black JT, Judge D, Demers L, Gordon S. Ragged-red fibers. A biochemical and morphological study. J Neurol Sci. 1975;26:479–488. doi: 10.1016/0022-510x(75)90048-9. [DOI] [PubMed] [Google Scholar]
- 22.Hamalainen N, Pette D. Patterns of myosin isoforms in mammalian skeletal muscle fibres. Microsc Res Technol. 1995;30:381–389. doi: 10.1002/jemt.1070300505. [DOI] [PubMed] [Google Scholar]
- 23.Abadie J, et al. Recombinant adeno-associated virus type 2 mediates highly efficient gene transfer in regenerating rat skeletal muscle. Gene Therapy. 2002;9:1037–1043. doi: 10.1038/sj.gt.3301773. [DOI] [PubMed] [Google Scholar]
- 24.McFarland DC, et al. Variation in fibroblast growth factor response and heparan sulfate proteoglycan production in satellite cell populations. Comp Biochem Physiol C. 2003;134:341–351. doi: 10.1016/s1532-0456(02)00272-7. [DOI] [PubMed] [Google Scholar]
- 25.Kokoszka JE, et al. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature. 2004;427:461–465. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fagiolari G, et al. Lack of apoptosis in patients with progressive external ophthalmoplegia and mutated adenine nucleotide translocator-1 gene. Muscle Nerve. 2002;26:265–269. doi: 10.1002/mus.10172. [DOI] [PubMed] [Google Scholar]
- 27.Bauer MK, Schubert A, Rocks O, Grimm S. Adenine nucleotide translocase-1, a component of the permeability transition pore, can dominantly induce apoptosis. J Cell Biol. 1999;147:1493–1502. doi: 10.1083/jcb.147.7.1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marti R, Nishigaki Y, Vila MR, Hirano M. Alteration of nucleotide metabolism: a new mechanism for mitochondrial disorders. Clin Chem Lab Med. 2003;41:845–851. doi: 10.1515/CCLM.2003.128. [DOI] [PubMed] [Google Scholar]
- 29.Luciakova K, et al. Repression of the human adenine nucleotide translocase-2 gene in growth-arrested human diploid cells: the role of nuclear factor-1. J Biol Chem. 2003;278:30624–30633. doi: 10.1074/jbc.M303530200. [DOI] [PubMed] [Google Scholar]
- 30.Rabinowitz JE, et al. Cross-packaging of a single adeno-associated virus (AAV) type 2 vector genome into multiple AAV serotypes enables transduction with broad specificity. J Virol. 2002;76:791–801. doi: 10.1128/JVI.76.2.791-801.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi W, Arnold GS, Bartlett JS. Insertional mutagenesis of the adeno-associated virus type 2 (AAV2) capsid gene and generation of AAV2 vectors targeted to alternative cell-surface receptors. Hum Gene Ther. 2001;12:1697–1711. doi: 10.1089/104303401750476212. [DOI] [PubMed] [Google Scholar]
- 32.McCarty DM, Monahan PE, Samulski RJ. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Therapy. 2001;8:1248–1254. doi: 10.1038/sj.gt.3301514. [DOI] [PubMed] [Google Scholar]
- 33.Zolotukhin S, et al. A "humanized" green fluorescent protein cDNA adapted for high-level expression in mammalian cells. J Virol. 1996;70:4646–4654. doi: 10.1128/jvi.70.7.4646-4654.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rando TA, Blau HM. Primary mouse myoblast purification, characterization, and transplantation for cell-mediated gene therapy. J Cell Biol. 1994;125:1275–1287. doi: 10.1083/jcb.125.6.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kessler PD, et al. Gene delivery to skeletal muscle results in sustained expression and systemic delivery of a therapeutic protein. Proc Natl Acad Sci USA. 1996;93:14082–14087. doi: 10.1073/pnas.93.24.14082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Daly TM, et al. Neonatal gene transfer leads to widespread correction of pathology in a murine model of lysosomal storage disease. Proc Natl Acad Sci USA. 1999;96:2296–2300. doi: 10.1073/pnas.96.5.2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ford T, Graham J, Rickwood D. Iodixanol: a nonionic isoosmotic centrifugation medium for the formation of self-generated gradients. Anal Biochem. 1994;220:360–366. doi: 10.1006/abio.1994.1350. [DOI] [PubMed] [Google Scholar]
- 38.Graham J, Ford T, Rickwood D. The preparation of subcellular organelles from mouse liver in self- generated gradients of iodixanol. Anal Biochem. 1994;220:367–373. doi: 10.1006/abio.1994.1351. [DOI] [PubMed] [Google Scholar]
- 39.Chance B, Williams GR. The respiratory chain and oxidative phosphorylation. Adv Enzymol Relat Subj Biochem. 1956;17:65–134. doi: 10.1002/9780470122624.ch2. [DOI] [PubMed] [Google Scholar]
- 40.Marsh M, et al. Rapid analytical and preparative isolation of functional endosomes by free flow electrophoresis. J Cell Biol. 1987;104:875–886. doi: 10.1083/jcb.104.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pedersen PL, et al. Preparation and characterization of mitochondria and submitochondrial particles of rat liver and liver-derived tissues. Methods Cell Biol. 1978;20:411–481. doi: 10.1016/s0091-679x(08)62030-0. [DOI] [PubMed] [Google Scholar]
- 42.Boulay F, Brandolin G, Lauquin GJ, Vignais PV. Synthesis and properties of fluorescent derivatives of atractyloside as potential probes of the mitochondrial ADP/ATP carrier protein. Anal Biochem. 1983;128:323–330. doi: 10.1016/0003-2697(83)90382-2. [DOI] [PubMed] [Google Scholar]
- 43.Fiore C, et al. Fluorometric detection of ADP/ATP carrier deficiency in human muscle. Clin Chim Acta. 2001;311:125–135. doi: 10.1016/s0009-8981(01)00581-2. [DOI] [PubMed] [Google Scholar]
- 44.Moriyama T, Srere PA. Purification of rat heart and rat liver citrate synthases. Physical, kinetic, and immunological studies. J Biol Chem. 1971;246:3217–3223. [PubMed] [Google Scholar]
- 45.Manfredi G, Yang L, Gajewski CD, Mattiazzi M. Measurements of ATP in mammalian cells. Methods. 2002;26:317–326. doi: 10.1016/S1046-2023(02)00037-3. [DOI] [PubMed] [Google Scholar]
- 46.Bancroft JDG, Marylin. Theory and Practice of Histological Techniques WB Saunders Co: Philadelphia, 2002.
- 47.Zheng XX, Shoffner JM, Voljavec AS, Wallace DC. Evaluation of procedures for assaying oxidative phosphorylation enzyme activities in mitochondrial myopathy muscle biopsies. Biochim Biophys Acta. 1990;1019:1–10. doi: 10.1016/0005-2728(90)90118-n. [DOI] [PubMed] [Google Scholar]