

Abstract
The phenotype of the human genetic disorder Cockayne syndrome (CS) is not only due to DNA repair defect but also (and perhaps essentially) to a severe transcription initiation defect. After UV irradiation, even undamaged genes are not transcribed in CSB cells. Indeed, neither RNA pol II nor the associated basal transcription factors are recruited to the promoters of the housekeeping genes, around of which histone H4 acetylation is also deficient. Transfection of CSB restores the recruitment process of RNA pol II. On the contrary, the p53-responsive genes do not require CSB and are transcribed in both wild-type and CSB cells upon DNA damage. Altogether, our data highlight the pivotal role of CSB in initiating the transcriptional program of certain genes after UV irradiation, and also may explain some of the complex traits of CS patients.
Keywords: CSB, p53, RNA polymerase II, transcription initiation
Introduction
Cockayne syndrome (CS) is an autosomal recessive multisystem disorder also characterized as a premature aging disease. CS patients also display an array of clinical symptoms that include mental and physical growth retardation, gait defects, ocular anomalies such as progressive pigmentary retinopathy, cataracts and optic disk atrophy, sensorineural hearing loss, dental caries and mild hypersensitivity to sunlight (Nance and Berry, 1992). Two complementation groups (A and B) have been identified among CS patients (Troelstra et al, 1992; Henning et al, 1995). One of the hallmarks of CS cells is their inability to resume transcription after UV irradiation (Mayne and Lehmann, 1982), which has been commonly ascribed to the defect that these cells display in transcription coupled repair (TCR), a subpathway of nucleotide excision repair (NER). TCR rapidly and preferentially removes lesions from the transcribed strand of active genes (de Laat et al, 1999). The other NER subpathway, known as global genome repair (GGR), removes lesions located any where in the genome. The inability of CS cells to resume transcription after UV irradiation, however, appears not to be only due to a defect in removing the damage induced by UV light. In fact, it is difficult to reconcile the clinical symptoms observed in CS patients with solely an impairment of TCR. This is especially the case when CS patients are compared to xeroderma pigmentosum (XP) patients, which can carry mutations that inactivate both GGR and TCR pathways and do not display most of the symptoms of CS patients (Lehmann, 2003). Indeed, except for the sun sensitivity, the clinical features of CS cannot be obviously attributed to defects in DNA repair, which is the case for XP. It has therefore been suggested that CS proteins may have additional functions beyond their role in DNA repair, possibly an involvement in transcription following genotoxic attack (Friedberg, 1996).
CSB is a 168-kDa protein that belongs to the SWI2/SNF2 family of chromatin remodelling proteins (Matson et al, 1994), exhibits ATPase activity (Selby and Sancar, 1997), and has conserved helicase motifs (Troelstra et al, 1992). This protein has been shown to play a role in both remodelling the chromatin structure and disrupting protein–DNA interactions (Citterio et al, 2000; Beerens et al, 2005). CSB is also part of, and able to stimulate the enzymatic activity of, complexes containing RNA polymerase I and RNA polymerase II (RNA pol II). These complexes also contain TFIIH, a basal transcription factor (Selby and Sancar, 1997; Bradsher et al, 2002). Additionally, CSB cells were shown to have a defect in transcription, both in vivo and in vitro, in the absence of stress such as UV light (Balajee et al, 1997). Significantly, an adequate model describing at which stage of transcription, CSB is operational, beyond its involvement in TCR, is missing. To address some of these issues, we have investigated whether the CSB mutation would affect specific transcriptional events, such as initiation and elongation.
In this study, we unveiled the crucial role played by CSB in the transcription initiation of a certain set of protein coding genes after UV irradiation. We further demonstrated that under these conditions, p53-responsive genes do not require CSB for transcription, explaining their unaltered and constant availability to be transcribed in case of a genotoxic insult.
Results
In CSB cells, UV irradiation inhibits transcription of an undamaged DNA template
We investigated the RNA synthesis recovery in wild-type (WT) cells, as well as in two CSB cell lines, CS1PV and CS8PV, expressing truncated and nonfunctional forms of the CSB protein. RNA synthesis was measured by [3H]uridine incorporation during a 30-min pulse performed 2, 4, 8 and 24 h after exposure of cells to 10 J/m2 UV-C light. While WT cells showed a rapid resumption of the initial UV-induced RNA synthesis inhibition, both CSB cell lines were unable to recover transcription throughout the entire time course (Figure 1A), as observed earlier (Mayne and Lehmann, 1982).
Figure 1.
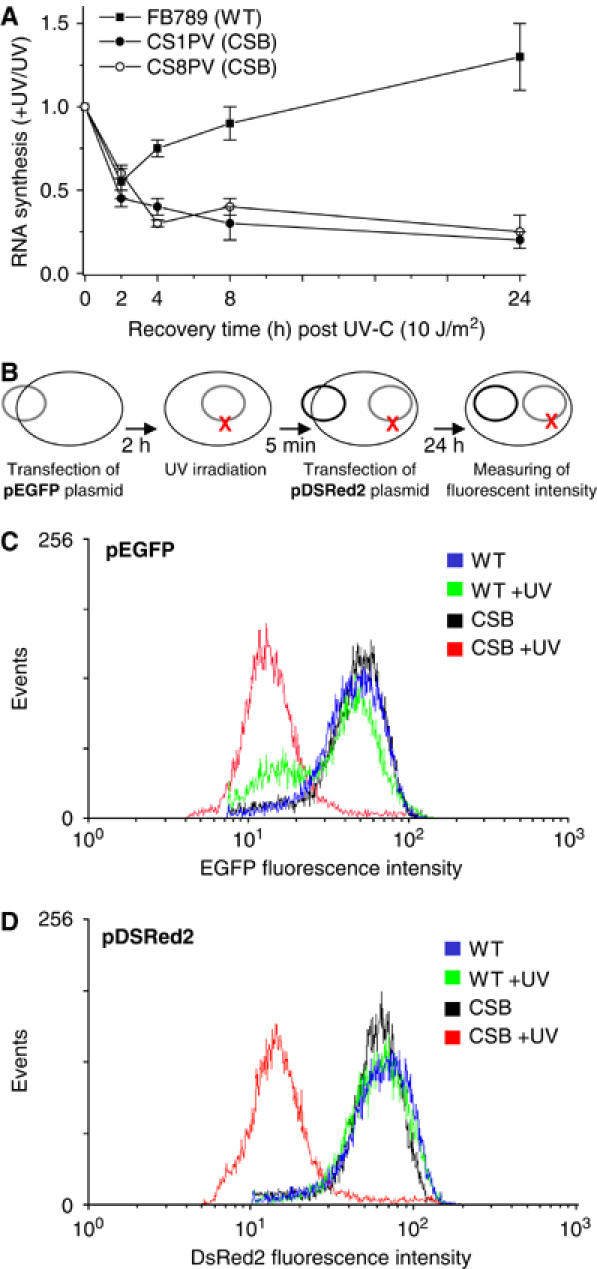
(A) RNA synthesis recovery of WT (FB789) and CSB (CS1PV and CS8PV) primary fibroblasts after UV irradiation. After prelabelling with [14C]thymidine (0.02 μCi/ml) for 2 days, unirradiated or UV (10 J/m2) irradiated cells were pulse labelled for 30 min with [3H]uridine at different incubation times after irradiation, and the acid-insoluble radioactivity was determined. Schematic representation (B) of the temporal order of transfection and irradiation. Histograms generated from flow cytometry analysis of WT and CS8PV cells 24 h after transfection with pEGFP (C) and pDSRed2 plasmid (D). Cells were either unirradiated or UV irradiated after the pEGFP transfection while pDSRed2 was transfected after UV irradiation. Relative EGFP and DSRed2 fluorescence are represented on the X-axis, and number of events (cells) is represented on the Y-axis. Similar patterns of fluorescence were observed in at least three independent experiments.
To further understand why RNA synthesis was not recovered in CSB cells following UV irradiation, we monitored the expression of pEGFP and pDsRed2, two reporter plasmids expressing proteins that emit fluorescence in response to excitation at different wavelengths. At 2 h after pEGFP transfection, WT and CSB cells were UV irradiated and then transfected with pDsRed2 (Figure 1B). Under such conditions, pEGFP was exposed to UV irradiation. After 24 h, protein expression was measured by flow cytometry. WT and CSB unirradiated cells showed a comparable mean fluorescent relative intensity of EGFP (Figure 1C, blue and black curves). Most of the irradiated WT cells were able to express a fluorescent relative intensity of EGFP similar to that observed in the unirradiated cells (green curve). However, even at such low UV doses, one cannot exclude that pEGFP was partially damaged and therefore not totally repaired when measuring EGFP expression. This would justify the presence of a little population expressing lower fluorescence. On the contrary, the majority of the UV-irradiated CSB cells showed a drastic reduction of the EGFP protein expression as indicated by the clear leftward shift of the EGFP fluorescent peak (red curve). In this later case, at this low UV dose, it is very unlikely that all the copies of EGFP, introduced into the cells, were damaged. It is therefore far more likely, and not surprising given what is known about transcription in CSB cells after UV, that transcription from both the damaged and the undamaged plasmids is simply reduced because transcription initiation in these cells is reduced.
Then we investigated the expression of pDsRed2, which was not exposed to the UV irradiation. While in unirradiated and UV-irradiated WT cells as well as in unirradiated CSB cells, DsRed2 protein expression occurs normally (Figure 1D, blue, green and black curves, respectively), DsRed2 protein expression was dramatically inhibited in UV-irradiated CSB cells according to the leftward shift of the DSRed2 fluorescent peak (Figure 1D, red curve). Southern blot analysis reveals that transfection efficiency was not affected by UV irradiation. In addition, RT–PCR analysis also revealed that mRNA synthesis from either UV-irradiated pEGFP or unirradiated pDsRed2 was similarly inhibited in CSB irradiated cells (data not shown).
Altogether, these results suggest that the inhibition of RNA synthesis observed in CSB cells after UV irradiation does not seem to be exclusively a result of a defect in DNA repair, but also and more likely, could derive from a defect in the transcription process. Such in vivo data are in agreement with what was previously observed in vitro (Balajee et al, 1997; Dianov et al, 1997; Rockx et al, 2000).
RNA pol II recruitment at the DHFR promoter is deficient in UV irradiated CSB cells
We next investigated whether the gene transcription machinery was somehow affected, after UV irradiation. Therefore, using chromatin immunoprecipitations (ChIPs), we examined the kinetics of the occupancy of RNA pol II and its associated partners to the promoter (initiation) and to a distal DNA region (elongation) of a set of genes after UV irradiation (Figure 2A). Antibodies, directed against the various components of the transcription machinery, were used to precipitate genomic DNA fragments that were further analyzed by quantitative PCR. We demonstrated that in UV-irradiated WT cells, RNA pol II is present at the promoter and at exon 3 of the dihydrofolate reductase housekeeping gene (DHFR), which is known to be constitutively active. We also observed a significant increase of RNA pol II around the promoter, likely as a result of an acceleration of RNA synthesis as soon as the repair of some damaged DNA is done.
Figure 2.
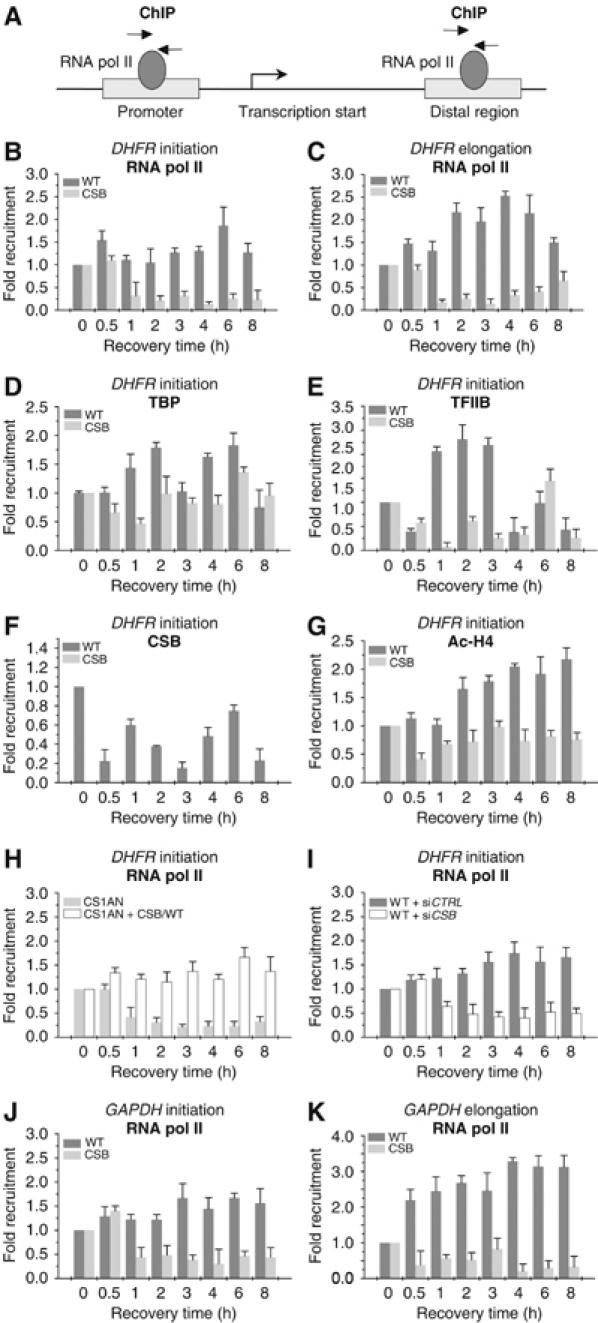
Kinetics of RNA pol II occupancy at the promoter or distal exon of DHFR and GAPDH genes (A) after UV irradiation (10 J/m2). Soluble chromatin was prepared from WT and CS8PV cells at indicated times after UV treatment and subjected to ChIP assay using the indicated antibodies. Real-time PCR using specific primers was performed to test the relative enrichment for either the proximal promoter (initiation) or the distal regions (elongation) of the gene as compared to the unirradiated samples. Kinetics of RNA pol II occupancy either at the promoter (B) or at a distal region (C) of DHFR gene after UV irradiation, in WT and CSB deficient cells. Kinetics of TBP (D), TFIIB (E), CSB (F) and acetylated H4 (G) at the promoter of DHFR gene. RNA pol II occupancy after UV irradiation, at the promoter of DHFR, either in CSB (CS1AN) or in CS1AN cells (CS1AN+CSB/WT) transfected with WT CSB gene (H). RNA pol II occupancy at the promoter of DHFR gene in WT cells previously transfected with a pool of oligonucleotides either of control (siCTRL) or silencing CSB gene (siCSB) (I). RNA pol II occupancy either at the promoter (J) or at a distal region (K) of GAPDH gene in WT and CSB cells. The results are expressed as fold enrichment relative to the untreated cells. Data are from three independent experiments. The values shown are means±s.d.
The presence of RNA pol II drastically decreased 1 h post-UV irradiation in CSB cells, without any evident recovery throughout the entire time course (Figures 2B and C). We also observed a decrease of TBP (the TATA-box recognizing factor), as well as that of TFIIB its stabilizing partner, in the UV-irradiated CSB cells (Figures 2D and E). It should be noticed that in irradiated CSB cells, a certain amount of TBP was still present on the DHFR promoter (as compared to that of either RNA pol II or TFIIB), suggesting a partial/constitutive positioning of some components of the transcriptional machinery. ChIP also reveals that in WT cells, the CSB protein is present at the DHFR promoter; its level sharply decreased after UV stress and appears to be cyclic (Figure 2F). ChIP performed in CSB cells using the antibody against CSB protein failed to immunoprecipitate any genomic DNA, likely due to the absence of the truncated CSB protein at the promoter.
To gain insight into the chromatin modification generated upon gene activation, we investigated the level of nucleosomal histone acetylation around the promoter that would condition transcription. Using antibody directed towards the acetylated histone H4, we found a reduced acetylation of histone H4 at the DHFR promoter of the UV irradiated CSB cells when compared to the WT (Figure 2G).
The inability of DHFR to be transcribed in UV-irradiated CSB cells was further examined by the following experiments. We first showed that transfection of wtCSB gene in CSB cell line (CS1AN) restored the recruitment of RNA pol II at the DHFR promoter (Figure 2H). Along this line, converse experiments showed that silencing of the CSB gene, achieved by RNA interference, in WT primary cells, resulted in a failure in the recruitment of RNA pol II to the DHFR promoter (Figure 2I).
Altogether these data demonstrate that, in UV-irradiated cells, CSB mutation (or CSB silencing) prevents the recruitment of RNA pol II at the promoter of both the DHFR and the glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Figures 2J and K) genes, a key step in transcriptional initiation.
CSB is not recruited to the promoter of p53-responsive genes
Knowing that p53 responsive genes are activated upon UV irradiation, lead us to determine whether CSB mutations would also be required for the recruitment of the transcription machinery to the promoters of the p53-targeted genes MDM2 and GADD45. ChIP performed at different times after UV irradiation showed a significant increase in the recruitment of RNA pol II (Figures 3A and C), TBP, and TFIIB basal transcription factors (data not shown), at MDM2 and GADD45 promoters in CSB cells almost identical to what is observed in WT cells. ChIP also showed an increase in the occupancy of elongating RNA pol II at distal region located at exon 4 of both genes (Figures 3B and D). This likely results from the progressive increase in the recruitment of p53 (as shown for GADD45 gene), independently of the nature of the cells, WT or CSB (Figure 3E). Under these conditions, we observed a similar increase in the GADD45 and MDM2 mRNA synthesis also reflected by the accumulation of their corresponding proteins as detected by Western blot (Figures 3G and H). Indeed, the increase of GADD45 and MDM2 proteins paralleled the rapid induction of the p53 protein in CSB cells, after UV irradiation. Surprisingly, in contrast to what we observed at the DHFR promoter, we noticed that the CSB protein does not seem to be recruited to the promoter of GADD45, either before or after UV irradiation (Figure 3F), in spite of its expression (WT versus CSB cells).
Figure 3.
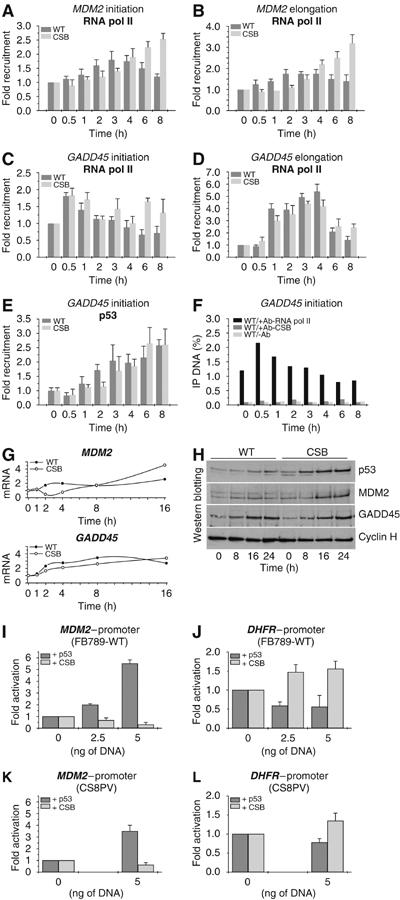
Kinetics of RNA pol II occupancy at the promoter and distal exon of MDM2 (A, B) and GADD45 (C, D) genes after UV irradiation (10 J/m2). Kinetics of p53 (E) and CSB (F) protein occupancy at the promoter of GADD45 gene after UV irradiation. (G) RT–PCR of MDM2 and GADD45 genes. RNA was extracted at the time indicated after UV irradiation. (H) Western blot analysis of p53, MDM2, GADD45 and cyclin H protein amount from nuclear extracts of either WT or CS8PV cells. Nuclear extracts were collected, after irradiation, at the time indicated. WT and CSB cells were cotransfected with either pMDM2-Luc (I, K) or pDHFR-Luc (J, L) in combination with either empty vectors or constructs expressing p53 or CSB proteins at the indicated concentrations. The results are expressed as fold activation relative to the cells transfected with the empty vector. In (F), results are expressed as the percentage of the immunoprecipitated DNA as compared to the input. Values obtained using CSB antibody do not show any enrichment as compared to the ones obtained without any antibody (negative control), which evidences the absence of CSB at the promoter.
To determine the respective role of CSB and p53 in promoting the transcription of certain genes, we designed the following experiment. Either p53 or CSB expression vectors were transfected in unirradiated cells, together with luciferase reporter vectors under the control of either MDM2 (pMDM2-Luc) or DHFR (pDHFR-Luc) promoters. We found that increasing overexpression of CSB selectively upregulates the activity of pDHFR-Luc either in WT or CSB cells (Figures 3J and L). In contrast, it has no effect on the regulation of pMDM2-Luc, as illustrated by the luciferase values (Figures 3I–K). Interestingly, increasing overexpression of p53, selectively and exclusively, upregulates expression of p53-responsive MDM2 promoter (Figures 3I and K). It should be however noticed that there is a slight inhibition of DHFR gene expression following overexpression of p53 (Figures 3J and L).
Altogether these data highlight the role of CSB in the regulation of the expression of a specific set of genes, and clearly demonstrate that p53-responsive genes are CSB independent.
VDR responsive gene expression is affected in CSB cells
We next asked whether other inducible activators could at least partially counteract the detrimental transcriptional defect due to CSB mutations. Therefore, we analyzed the stimulation operated by vitamin D (vit-D) on the promoter of CYP24, a vit-D receptor (VDR) target gene. Following vit-D addition, we observed that RNA pol II, as well as the VDR and the CSB proteins, are recruited to the promoter of CYP24 in WT cells, independent of the UV irradiation (Figures 4A–F). The activation of the CYP24 gene, in WT cells, was also paralleled by the progressive increase in acetylation of histone H4. Noteworthy is the fact that vit-D response is slightly attenuated when the cells have been irradiated with UV. On the contrary, in UV-irradiated CSB cells, the binding of VDR and RNA pol II to the CYP24 promoter as well as the acetylation of histone H4 are significantly impaired (Figures 4A, C and G). However, the pattern of vit-D mediated VDR accumulation, as visualized by Western blot, is similar in WT and CSB cells, whether the cells had been UV irradiated or not (Figures 4J and I). In nonirradiated CSB cells, however, addition of vit-D does not allow optimal recruitment of RNA pol II, VDR, or histone H4 acetylation around the CYP24 promoter as compared to WT cells (Figures 4B, D and H). In conclusion, it seems that mutations in CSB inhibit one of the first steps of the transcription reaction of VDR-responsive genes, irrespective of the CSB cells are UV irradiated or not. Activators such as the VDR nuclear receptor are unable to counteract the detrimental effect of CSB mutation, the protein p53 being the exception.
Figure 4.
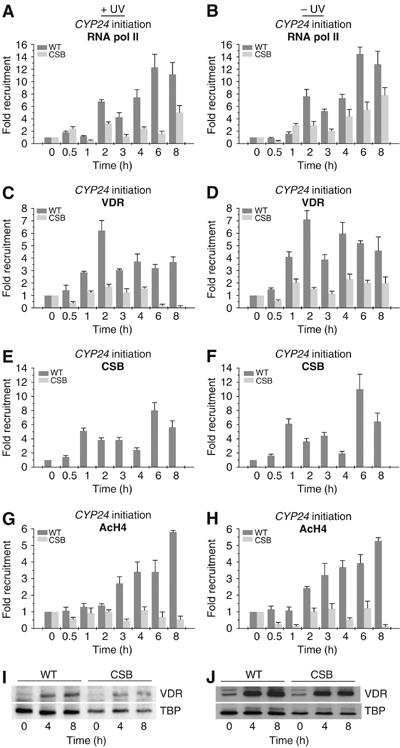
ChIP analysis on the CYP24 promoter in WT and CS8PV cells. Soluble chromatin was prepared from either UV irradiated (left panels) or unirradiated (right panels) cells co-treated with vit-D (10−7 M) for the indicated time and immunoprecipitated with antibodies against RNA pol II (A, B), VDR (C, D), CSB (E, F) or the acetylated histone H4 (G, H). The genomic DNAs were analyzed by quantitative PCR. Western blot analyzes (I, J) of VDR protein from nuclear extracts of WT and CSB cells, either UV irradiated or unirradiated, at different time points after vit-D treatment.
Defect in ongoing RNA pol II in UV irradiated CSB cells
First, having observed a deficiency in RNA pol II recruitment to certain promoters upon UV irradiation and second knowing that RNA pol II activity might have been modified upon UV irradiation (Rockx et al, 2000), lead us to investigate the status of RNA pol II in CSB cells. Indeed, during transcription, RNA pol II undergoes a series of posttranslational modifications at the C-terminal domain (CTD) of its largest subunit. First, unphosphorylated-CTD RNA pol II (RNA pol IIA) binds the promoter region. Second, CTD is phosphorylated (RNA pol IIO) by TFIIH at serine 5, which allow the RNA pol II to leave the promoter. Finally CTD is phosphorylated by pTEFb at serine 2, during full elongation (Komarnitsky et al, 2000). Additionally, RNA pol II can be dephosphorylated for another round of transcription (Lehman and Dahmus, 2000), and/or ubiquitylated and degraded following transcriptional arrest (Bregman et al, 1996; Woudstra et al, 2002; Somesh et al, 2005).
The concentration of RNA pol II was thus examined by comparing WT and CSB cells, labelled by latex green fluorescent beads (smaller for WT cells and larger for CSB) and spotted on the same slide, at different times post-UV irradiation. Using monoclonal antibodies directed towards the largest RNA pol II subunit (RPB1), confocal microscopy did not reveal any significant reduction of overall RNA pol II concentration in either WT or CSB cells 2, 4 and 8 h after the UV irradiation (Figure 5A, panels a–d). In contrast, monoclonal antibodies which specifically recognize the phosphorylated CTD, at either serine 5 (S5P-CTD) or serine 2 (S2P-CTD), showed a decrease of both phosphorylated forms of RNA pol IIO in CSB cells, after UV irradiation (panels e–h and i–l respectively). This decrease was not observed on irradiated WT cells. We also noticed that in irradiated CSB cells, the S5P-CTD RNA pol IIO concentration decreases more rapidly than the S2P-CTD RNA pol IIO (at 2 h after UV irradiation, 30 versus 60%; Figure 5B).
Figure 5.
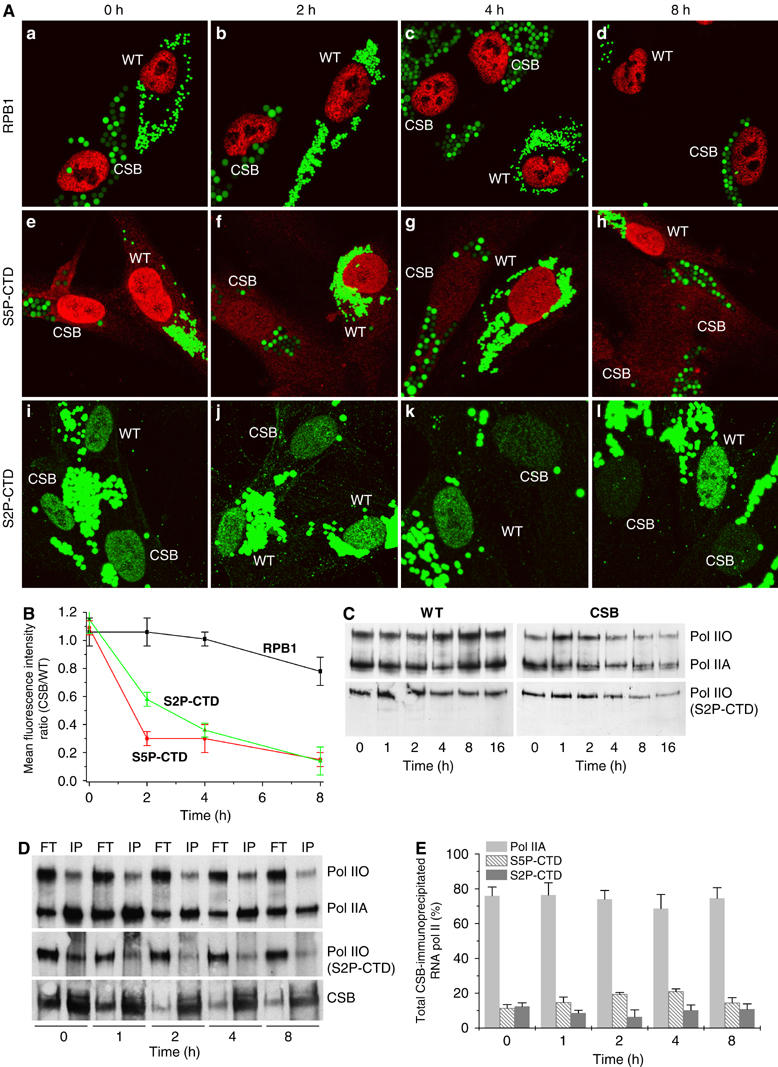
(A) Confocal microscopy analysis of RNA pol II concentration in WT or CS8PV cells using antibodies that recognize either RPB1, the largest subunit of RNA pol II (panels a–d), or the hyperphosphorylated CTD domain, at either serine 5 (S5P-CTD, panels e–h) or serine 2 (S2P-CTD, panels i–l). Cells were prelabelled with green latex fluorescent beads of different size and spotted on the same slide to further analysis of the RNA pol II concentration at the time indicated after UV irradiation (10 J/m2). (B) Graph represents mean fluorescent intensity (protein concentration) ratios calculated by dividing the mean fluorescence, relative to the protein staining, of CSB cells by the mean fluorescence of the WT cells as obtained by confocal scanner fluorescence, measuring of up to 100 cells. (C) Western blot analysis of RNA pol II amount from chromatin-bound fraction of either WT or CSB cells using antibodies that recognize RPB1, regardless its phosphorylation state (upper panel), or the phosphorylated S2P-CTD (lower panel). Pol IIO and Pol IIA indicate the hyper- and hypophosphorylated form of the CTD-RNA pol II, respectively. (D) Nuclear extracts from either unirradiated or UV-irradiated cells were immunoprecipitated (IP) using an antibody against CSB. The IP and the flowthrough (FT) fractions were analyzed by Western blot using antibody thaat recognize RPB1 (upper panel), the phosphorylated S2P-CTD RNA pol II (middle panel) or CSB (lower panel). (E) Graph represents the percentage of RNA pol IIA, and the phosphorylated S5P-CTD and S2P-CTD RNA pol II, which were CSB-IP at different time after UV irradiation.
Next, we investigated the distribution of RNA pol II in the chromatin-bound fractions following UV irradiation. Western blot analysis of chromatin-bound proteins showed that similar amounts of both RNA pol IIO and RNA pol IIA, were associated with the chromatin in unirradiated WT and CSB cells (Figure 5C). Indeed, we observe a slight but significant increase in the amounts of both RNA pol IIA and RNA pol IIO starting 4 h post-UV irradiation in WT cells (Figure 5C, upper left panel). This likely reflects an acceleration of RNA synthesis once UV-induced damage was repaired. In contrast, in CSB cells, both forms of RNA pol II progressively decreased starting 2–4 h post-UV irradiation, suggesting a decline of both initiating and elongating RNA pol II activity (Figure 5C, upper right panel). Indeed, 4 h after UV irradiation, phosphorylated S2P-CTD RNA pol IIO concentration decreases significantly in the chromatin-bound fraction (Figure 5C, lower right panel), reflecting a decrease in ongoing transcription.
The CSB-immunoprecipitated fractions from nuclear extracts performed either before or at different times after having UV irradiated WT cells, mostly contained RNA pol IIA, while RNA pol IIO, being not associated to CSB, was found in the supernatant fraction (Figure 5D). After quantification, we observed that the CSB immunoprecipitated fractions contained similar amounts of RNA pol IIA, but varied in the RNA pol IIO content (Figure 5E). Of the RNA pol IIO forms, we observed a significant decrease in the presence of the S2-CTD RNA pol IIO that represents ongoing elongation while the S5-CTD RNA pol IIO form increased until 4 h post-UV irradiation. The fact that CSB immunoprecipitates mostly (up to ∼90%) the ‘initiating' RNA pol II (RNA pol IIA+S5-CTD RNA pol IIO) forms strongly point to its role in the earlier steps of the transcription.
Discussion
CSB is required for transcription initiation after UV irradiation
We revealed that UV irradiation of CSB cells does not only prevent the recovery of transcription because genes are not repaired (defined as a TCR defect), but rather, because there is a defect in ‘reinitiating' (or maintaining) the transcription process. Here, we first showed that CSB cells cannot transcribe nondamaged genes if they were previously UV irradiated. We next discovered that the transcription of some housekeeping genes, such as DHFR or GAPDH, was inhibited in CSB cells after UV irradiation. Indeed, the formation of the transcription initiation complex at the promoters of these genes was impaired. The recruitment of TBP, which is supposed to initiate transcription, was severely decreased at these promoters; also, the recruitment of TFIIB was almost absent. Consequently, it was not surprising to observe that RNA pol II could not join the initiation complexes of these promoters in UV-irradiated cells in which CSB is either mutated or silenced.
This finding also correlated with the overall disappearance of RNA pol II from the chromatin-bound fraction. Since transcription initiation is arrested, it is not surprising to observe a progressive decrease of the elongation RNA pol II in UV irradiated CSB cells. Furthermore, histone H4 acetylation does not occur properly, highlighting a defect in one of the earlier events of the transcriptional process. The fact that CSB associates mainly with the unphosphorylated RNA pol IIA and the serine 5 phosphorylated RNA pol IIO, strongly supports a role for CSB during the first phases of the transcription reaction.
CSB does not participate in p53-responsive gene expression
In view of the above observations, we could have concluded that in UV-irradiated CSB cells, there was a defect in one of the components required to organize the transcription machinery, a prerequisite to initiate RNA synthesis. This is unlikely since, in CSB cells, p53-responsive genes are transcribed, which means that their promoters are able to recruit all the basal components of the transcription machinery. These genes can be transcribed in the absence of CSB, which is not found in the region of their promoters. These results suggest that the role of CSB in RNA pol II transcription is crucial. However, CSB is needed for the expression of genes such as DHFR, GAPDH and CYP24. Transfection of CSB, in both WT and CSB cells, stimulates and restores the expression of the above-mentioned genes, probably as a result of its recruitment to their respective promoters.
The inability to transcribe genes in CSB cells, following genotoxic attack, raises questions concerning the role of the CSB protein in general, and its relationship with p53. From our results, it seems that the recovery of RNA synthesis after genotoxic attack occurs following, at least, two distinct transcriptional pathways. The first one, which is directed by p53, concerns only p53-responsive genes and does not require CSB. The second one is mediated by CSB. In UV-irradiated CSB cells p53-responsive genes are activated, but the recovery of the transcription of housekeeping genes is affected. Moreover, we also found that in CSB cells, the vit-D dependent response is diminished, independently of UV irradiation. At this stage of our discussion, we might question why p53-transactived genes behave differently from housekeeping genes and why the VDR activator cannot bypass the CSB deficiency as the p53 activator does? The simplest answer would be that p53 activator possess in addition to its DNA binding property, some additional functions exhibited by CSB. CSB was found to play a role in remodelling the chromatin structure (Citterio et al, 2000), which could facilitate the subsequent access of the general transcriptional machinery to certain genes, such as housekeeping or VDR-targeted genes. Consequently, one can propose that p53 recruits specific chromatin remodelling factors to allow the initiation of the transcription of its own targeted genes. This observation would explain why p53-targeted genes are devoid of CSB, assuming the fact that CSB could function to convert the conformation of the promoter into an accessible structure.
Taken together, our results unveiled an essential role of CSB in transcription of specific gene programs after genotoxic attack and/or ligand stimulation.
How to reconcile the role of CSB in repair with the one in transcription?
The already established role of CSB protein in TCR and now our evidence concerning its involvement during transcription initiation allows us to speculate about the shared functions of CSB in both processes. In both cases, CSB has to deal with RNA pol II either when joining the promoter or when stalled in front of a DNA lesion. Whether or not this involves a direct interaction between these two components (Tantin et al, 1997), or induces some chromatin remodelling (Smerdon and Lieberman, 1978; Tlsty and Lieberman, 1978) is unkown. By ChIP and immunoprecipitation assays, we demonstrated that CSB operates at the transcription initiation step to allow recruitment of RNA pol IIA and associated transcription factors on a subset of genes, after UV irradiation. In WT cells, the increase of transcription machinery recruitment is related to the increased acetylation of histone H4, as observed after UV irradiation. However, a leading question remains: how to link the activity of the CSB protein and chromatin modifications, observed after UV irradiation, with the individual processes of transcription and DNA repair? CSB exhibits ATPase activity and possesses helicase motifs. Moreover, CSB belongs to the SWI2/SNF2 family of ATP-dependent chromatin remodelling factors. Accordingly, it is tempting to speculate that there is a chromatin remodelling activity (Citterio et al, 2000) mediated by CSB through the recruitment of an acetyl-transferase, such as p300 (unpublished data), in transcription and in DNA repair. Therefore, the presence of CSB at the promoter level could initiate a chromatin remodelling event, a prerequisite that would not only facilitate the recruitment of the transcription machinery but also NER factors, explaining the preferential repair around the promoter surrounding sequences (Tornaletti and Pfeifer, 1995; Frit et al, 2002).
Indeed, enhanced histone H3 and H4 acetylation and chromatin remodelling has been shown to occur at nucleotide excision and double-strand break repair (Ramanathan and Smerdon, 1989; Bird et al, 2002; Yu et al, 2005). Following such a hypothesis, it is also likely that by interacting with the RNA pol II stalled in front of a lesion, CSB would locally favor chromatin remodelling, a prerequisite for the arrival of the DNA repair factors (Sarker et al, 2005; Laine and Egly, 2006). Whether UV-induced signalling would trigger the activity of CSB following some posttranslational modifications (Christiansen et al, 2003), in order to relocate RNA pol II around the DNA damage or to release it from the template for recycling, is unknown.
We thus propose a scenario in which CSB, after UV irradiation, would act as a pivotal and nonredundant transcription factor. This transcriptional activity of CSB allows RNA pol II to initiate RNA synthesis of specific gene programs and in the case of damage on the transcribed strand, CSB would help RNA pol II to achieve transcription by allowing repair. When CSB is mutated, both of its functions in transcription and repair would not be met satisfactorily. Any genotoxic attack occurring in CSB cells during the development of the individual would thus have detrimental effects on gene expression (Kyng et al, 2003), explaining the clinical features exhibited by the CS patients.
Materials and methods
Cell lines
The normal (FB789) and CSB (CS1PV and CS8PV) primary human fibroblasts, kindly provided by M Stefanini (Pavia), were grown in minimal essential medium containing 15% fetal calf serum (FCS) and 40 mg/ml gentamycin. CSB SV40-transformed human fibroblasts (CS1AN) and a derivative expressing CSB (CS1AN/CSB) were grown in Dulbecco/Ham-F10 medium containing 10% FCS and 40 mg/ml gentamycin.
Transfections and luciferase reporter assays
FB789 and CS8PV cells were transfected using the Jet-Pei transfection reagent (Polyplus transfection). Cells (2.5 × 105) were plated in six-well plates and transfected with either DHFR- or MDM2-promoter constructs (1 μg), pCH110 (plasmid from Invitrogen coding for β-galactosidase; 1 μg) and either p53 or CSB expressing plasmid at different concentration. After 48 h, the cells were harvested and screened for galactosidase and luciferase activity. Each transfection was repeated four times.
Immunofluorescence studies
Primary fibroblasts were grown for 2 days with fluorescent latex beads of different size (Fluoresbrite Carboxylate Microspheres, Polysciences): FB789 (WT control; green, 0.75 μm), CS8PV (CSB; green, 2 μm), and seeded (in a 1:1 ratio) on microscopy slides and extracultured 2 days. Cells were fixed in 3% paraformaldehyde and permeabilized by PBS/0.5% Triton (immunodetection and confocal microscopy analysis was performed as previously described (Dubaele et al, 2003).
Flow cytometry
Cells were fixed with 70% ethanol at 24 h after the UV irradiation and processed for flow cytometry. Samples were resuspended in 20 mg/ml propidium iodide prior to analysis. EGFP and DSRed2 fluorescence (Clontech) was analyzed on a Becton Dickinson FACScan and histograms were generated using CellQuest software. Experiments were repeated at least two times and 10 000 events were recorded for each sample.
Antibodies
The monoclonal antibodies against RPB1-RNA pol II (7C2), Cyclin H, MDM2, TBP, Ac-H4 and CSB (3H8 and 1A11) were produced by the IGBMC facility. The monoclonal antibodies against the CTD-phosphorylated domain of RNA pol II (serine 5 and serine 2) were purchased from Covance. The monoclonal antibody against p53 (DO-1) and polyclonal antibodies against XPB (S19) and TFIIB (C18) were purchased from Santa Cruz.
Determination of RNA synthesis after UV irradiation
Cells in log phase were grown in the presence of [14C]thymidine (0.02 mCi/ml) for 2 days to uniformly label the DNA. The irradiated cells (10 J/m2 UV) were pulse labelled with 5 μCi/ml of [3H]uridine for 30 min at different times. The cells were collected and washed once with ice cold PBS and lysed in buffer containing 0.5% SDS and 100 μg/ml Proteinase K for 2 h at 37°C. After trichloroacetic acid (10% TCA) precipitation, the samples were spotted onto glass fiber discs (Whatmann); the filters were next sequentially washed in 5% TCA, 70% ethanol/acetone, and counted for their radioactivity.
Retrotranscription and real-time quantitative PCR
cDNA synthesis was performed by using random hexanucleotides and AMV reverse transcriptase (Sigma). Real-time quantitative PCR was performed with the QuantiTect SYBR Green PCR kit (Quiagen) and the MyIQ Real-Time PCR Detection System (Biorad). Results were normalized to 18S. Primer sequences are available upon request.
Chromatin immunoprecipitation
Cells were crosslinked with a 1% formaldehyde solution for 10 min at RT. Crosslinking was stopped by addition of glycine to 125 mM final concentration. Samples were sonicated to generate DNA fragments below 500 bp. For immunoprecipitations, 1 mg of protein extract was precleared for 2 h with 50 ml of a 50% slurry of 50:50 protein A/G-sepharose before addition of the indicated antibodies. Then, 2 mg of each antibody was added to the reactions and incubated over night at 4°C in the presence of 50 ml of protein A/G beads. After serial washings, the immunocomplexes were eluted twice for 10 min at 65°C and crosslinking was reversed by adjusting to 200 mM NaCl and incubating 5 h at 65°C. Further proteinase-K digestion was performed for 2 h at 42°C. DNA was purified by using Quiagen columns (QIAquick PCR Purification Kit). Immunoprecipitated DNA was quantified by real-time quantitative PCR. Primers sequences are available upon request.
RNA interference
A pool of four RNA oligonucleotides (Dharmacon) forming a 19 base-duplex core, specifically designed to target CSB mRNA was transfected in WT cells at the concentration of 50 nM. A pool of RNA oligonucleotides, without any target mRNA, was used as control. RNA transfection was performed by using Lipofectamine 2000 reagent (Invitrogen), according to the manufacturers instructions. Specific target reduction was analyzed 72 h posttransfection by monitoring CSB protein concentration.
Western blot analysis of the subcellular fractions
Cells were suspended in hypotonic buffer supplemented with detergent (20 mM HEPES, pH 7.5, 20 mM NaCl, 0.5% Nonidet P-40, 5 mM MgCl2, 1 mM ATP) and homogenized with a Dounce homogenizer five times before incubation on ice for 15 min. Cytosolic and nuclear supernatant was separated from the chromatin-bound fraction by centrifugation for 10 min at 10 000 r.p.m. Bound proteins were eluted from the chromatin pellet with high concentrations of salt (buffer containing 20 mM HEPES, pH 7.5, 0.5% Nonidet P-40, 0.5 mM MgCl2, 1 mM ATP plus 500 mM NaCl) to yield the ‘chromatin-bound fraction'. For Western blot, 10 mg of protein extract were loaded onto 6% SDS–PAGE and next blotted onto PVDF membranes.
Acknowledgments
We thank F Coin, M Frontini and M Stefanini for fruitful discussions. We thank N Clarke and R Velez-Cruz for critical reading of the paper. We thank T Nardo and C Braun for technical advices. This study has been supported by funds from ‘La Ligue contre le Cancer' (Contract No. EL2004), the ‘Ministere de l'Education National et de la Recherche', the ACI grants (BCMS No. 03 2 535), the Commissariat à l'Energie atomique (laboratoire correspondant) and EEC grants (MRTN-CT 2003-503618). LPDS has been supported, as research associate, by the Institut National de la Santé et de la Recherche Médicale (INSERM) and also granted by ‘Fondation pour La Recherche Médicale' (FRM). PD was granted by an EEC Grant (QLRT-1999-2002) and l'Institut de Recherche sur le Cancer de l'Appareil Digestif IRCAD Strasbourg, France.
References
- Balajee AS, May A, Dianov GL, Friedberg EC, Bohr VA (1997) Reduced RNA polymerase II transcription in intact and permeabilized Cockayne syndrome group B cells. Proc Natl Acad Sci USA 94: 4306–4311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerens N, Hoeijmakers JH, Kanaar R, Vermeulen W, Wyman C (2005) The CSB protein actively wraps DNA. J Biol Chem 280: 4722–4729 [DOI] [PubMed] [Google Scholar]
- Bird AW, Yu DY, Pray-Grant MG, Qiu Q, Harmon KE, Megee PC, Grant PA, Smith MM, Christman MF (2002) Acetylation of histone H4 by Esa1 is required for DNA double-strand break repair. Nature 419: 411–415 [DOI] [PubMed] [Google Scholar]
- Bradsher J, Auriol J, Proietti de Santis L, Iben S, Vonesch JL, Grummt I, Egly JM (2002) CSB is a component of RNA pol I transcription. Mol Cell 10: 819–829 [DOI] [PubMed] [Google Scholar]
- Bregman DB, Halaban R, van Gool AJ, Henning KA, Friedberg EC, Warren SL (1996) UV-induced ubiquitination of RNA polymerase II: a novel modification deficient in Cockayne syndrome cells. Proc Natl Acad Sci USA 93: 11586–11590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen M, Stevnsner T, Modin C, Martensen PM, Brosh RM Jr, Bohr VA (2003) Functional consequences of mutations in the conserved SF2 motifs and post-translational phosphorylation of the CSB protein. Nucleic Acids Res 31: 963–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citterio E, Van Den Boom V, Schnitzler G, Kanaar R, Bonte E, Kingston RE, Hoeijmakers JH, Vermeulen W (2000) ATP-dependent chromatin remodeling by the Cockayne syndrome B DNA repair–transcription–coupling factor. Mol Cell Biol 20: 7643–7653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dianov GL, Houle JF, Iyer N, Bohr VA, Friedberg EC (1997) Reduced RNA polymerase II transcription in extracts of cockayne syndrome and xeroderma pigmentosum/Cockayne syndrome cells. Nucleic Acids Res 25: 3636–3642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubaele S, Proietti De Santis L, Bienstock RJ, Keriel A, Stefanini M, Van Houten B, Egly JM (2003) Basal transcription defect discriminates between xeroderma pigmentosum and trichothiodystrophy in XPD patients. Mol Cell 11: 1635–1646 [DOI] [PubMed] [Google Scholar]
- Friedberg EC (1996) Cockayne syndrome—a primary defect in DNA repair, transcription, both or neither? Bioessays 18: 731–738 [DOI] [PubMed] [Google Scholar]
- Frit P, Kwon K, Coin F, Auriol J, Dubaele S, Salles B, Egly JM (2002) Transcriptional activators stimulate DNA repair. Mol Cell 10: 1391–1401 [DOI] [PubMed] [Google Scholar]
- Henning KA, Li L, Iyer N, McDaniel LD, Reagan MS, Legerski R, Schultz RA, Stefanini M, Lehmann AR, Mayne LV, Friedberg EC (1995) The Cockayne syndrome group A gene encodes a WD repeat protein that interacts with CSB protein and a subunit of RNA polymerase II TFIIH. Cell 82: 555–564 [DOI] [PubMed] [Google Scholar]
- Komarnitsky P, Cho EJ, Buratowski S (2000) Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev 14: 2452–2460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyng KJ, May A, Brosh RM Jr, Cheng WH, Chen C, Becker KG, Bohr VA (2003) The transcriptional response after oxidative stress is defective in Cockayne syndrome group B cells. Oncogene 22: 1135–1149 [DOI] [PubMed] [Google Scholar]
- de Laat WL, Jaspers NG, Hoeijmakers JH (1999) Molecular mechanism of nucleotide excision repair. Genes Dev 13: 768–785 [DOI] [PubMed] [Google Scholar]
- Laine JP, Egly JM (2006) Initiation of DNA repair mediated by a stalled RNA polymerase IIO. EMBO J 25: 387–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman AL, Dahmus ME (2000) The sensitivity of RNA polymerase II in elongation complexes to C-terminal domain phosphatase. J Biol Chem 275: 14923–14932 [DOI] [PubMed] [Google Scholar]
- Lehmann AR (2003) DNA repair-deficient diseases, xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. Biochimie 85: 1101–1111 [DOI] [PubMed] [Google Scholar]
- Matson SW, Bean DW, George JW (1994) DNA helicases: enzymes with essential roles in all aspects of DNA metabolism. Bioessays 16: 13–22 [DOI] [PubMed] [Google Scholar]
- Mayne LV, Lehmann AR (1982) Failure of RNA synthesis to recover after UV irradiation: an early defect in cells from individuals with Cockayne's syndrome and xeroderma pigmentosum. Cancer Res 42: 1473–1478 [PubMed] [Google Scholar]
- Nance MA, Berry SA (1992) Cockayne syndrome: review of 140 cases. Am J Med Genet 42: 68–84 [DOI] [PubMed] [Google Scholar]
- Ramanathan B, Smerdon MJ (1989) Enhanced DNA repair synthesis in hyperacetylated nucleosomes. J Biol Chem 264: 11026–11034 [PubMed] [Google Scholar]
- Rockx DA, Mason R, van Hoffen A, Barton MC, Citterio E, Bregman DB, van Zeeland AA, Vrieling H, Mullenders LH (2000) UV-induced inhibition of transcription involves repression of transcription initiation and phosphorylation of RNA polymerase II. Proc Natl Acad Sci USA 97: 10503–10508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarker AH, Tsutakawa SE, Kostek S, Ng C, Shin DS, Peris M, Campeau E, Tainer JA, Nogales E, Cooper PK (2005) Recognition of RNA polymerase II and transcription bubbles by XPG, CSB, and TFIIH: insights for transcription-coupled repair and Cockayne syndrome. Mol Cell 20: 187–198 [DOI] [PubMed] [Google Scholar]
- Selby CP, Sancar A (1997) Human transcription-repair coupling factor CSB/ERCC6 is a DNA-stimulated ATPase but is not a helicase and does not disrupt the ternary transcription complex of stalled RNA polymerase II. J Biol Chem 272: 1885–1890 [DOI] [PubMed] [Google Scholar]
- Smerdon MJ, Lieberman MW (1978) Nucleosome rearrangement in human chromatin during UV-induced DNA-repair synthesis. Proc Natl Acad Sci USA 75: 4238–4241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somesh BP, Reid J, Liu WF, Sogaard TM, Erdjument-Bromage H, Tempst P, Svejstrup JQ (2005) Multiple mechanisms confining RNA polymerase II ubiquitylation to polymerases undergoing transcriptional arrest. Cell 121: 913–923 [DOI] [PubMed] [Google Scholar]
- Tantin D, Kansal A, Carey M (1997) Recruitment of the putative transcription–repair coupling factor CSB/ERCC6 to RNA polymerase II elongation complexes. Mol Cell Biol 17: 6803–6814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tlsty TD, Lieberman MW (1978) The distribution of DNA repair synthesis in chromatin and its rearrangement following damage with N-acetoxy-2-acetylaminofluorene. Nucleic Acids Res 5: 3261–3273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornaletti S, Pfeifer GP (1995) UV light as a footprinting agent: modulation of UV-induced DNA damage by transcription factors bound at the promoters of three human genes. J Mol Biol 249: 714–728 [DOI] [PubMed] [Google Scholar]
- Troelstra C, van Gool A, de Wit J, Vermeulen W, Bootsma D, Hoeijmakers JH (1992) ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne's syndrome and preferential repair of active genes. Cell 71: 939–953 [DOI] [PubMed] [Google Scholar]
- Woudstra EC, Gilbert C, Fellows J, Jansen L, Brouwer J, Erdjument-Bromage H, Tempst P, Svejstrup JQ (2002) A Rad26–Def1 complex coordinates repair and RNA pol II proteolysis in response to DNA damage. Nature 415: 929–933 [DOI] [PubMed] [Google Scholar]
- Yu Y, Teng Y, Liu H, Reed SH, Waters R (2005) UV irradiation stimulates histone acetylation and chromatin remodeling at a repressed yeast locus. Proc Natl Acad Sci USA 102: 8650–8655 [DOI] [PMC free article] [PubMed] [Google Scholar]