

Abstract
Presequence protease PreP is a novel protease that degrades targeting peptides as well as other unstructured peptides in both mitochondria and chloroplasts. The first structure of PreP from Arabidopsis thaliana refined at 2.1 Å resolution shows how the 995-residue polypeptide forms a unique proteolytic chamber of more than 10 000 Å3 in which the active site resides. Although there is no visible opening to the chamber, a peptide is bound to the active site. The closed conformation places previously unidentified residues from the C-terminal domain at the active site, separated by almost 800 residues in sequence to active site residues located in the N-terminal domain. Based on the structure, a novel mechanism for proteolysis is proposed involving hinge-bending motions that cause the protease to open and close in response to substrate binding. In support of this model, cysteine double mutants designed to keep the chamber covalently locked show no activity under oxidizing conditions. The manner in which substrates are processed inside the chamber is reminiscent of the proteasome; therefore, we refer to this protein as a peptidasome.
Keywords: peptidase, peptide degradation, PreP protease, protein import, targeting peptide
Introduction
Degradation of small peptides in subcellular compartments is an essential function that prevents the accumulation of potentially toxic species in the cell. Presequence protease PreP was recently identified in plants as a novel 110 kDa protease responsible for the degradation of targeting peptides in both mitochondria and chloroplasts (Ståhl et al, 2002; Bhushan et al, 2003, 2005; Moberg et al, 2003). Mitochondrial and chloroplastic targeting peptides are about 30–50 amino acids long and show very little sequence similarity except that they are generally enriched in positively charged, hydroxylated and hydrophobic amino-acid residues (Pfanner and Geissler, 2001; Zhang and Glaser, 2002). Targeting peptides are cleaved off following import by either the mitochondrial processing peptidase (MPP) or the stromal processing peptidase (SPP), respectively (Richter and Lamppa, 1998; Glaser and Dessi, 1999; Neupert and Brunner, 2002). PreP from Arabidopsis thaliana displays very low substrate specificity and degrades targeting peptides as well as a number of nonrelated peptides such as the insulin B-chain and galanin (Moberg et al, 2003; Ståhl et al, 2005). Human PreP, known as human metalloprotease 1 (Mzhavia et al, 1999), is approximately 30% identical to AtPreP and has a similar function to the plant enzyme (Ståhl et al, submitted). Notably, both human and plant PreP can degrade the degenerative amyloid-β peptide associated with Alzheimer's disease (Ståhl et al, in preparation). PreP's ability to degrade amyloid-β and other potentially harmful peptides in mitochondria is particularly interesting in the light of recent findings, which link amyloid-β to the mitochondrial toxicity associated with Alzheimer's disease (Lustbader et al, 2004; Caspersen et al, 2005). Therefore, a better understanding of PreP is highly desirable from a medical perspective.
PreP belongs to the pitrilysin family of proteases, which includes several distantly related metalloendoproteases with the ability to cleave or degrade peptides (peptidase family M16 in the MEROPS database, Rawlings et al, 2004). They are also called inverzincins since they have an inverted version of the common zinc-binding motif, HXXEH (Becker and Roth, 1992). The family can be divided into subgroups, where M16A and M16C comprise proteases of about 1000 residues while M16B consists of heterodimers formed by two monomers of approximately 500 residues each. Subfamily M16A includes well-known human proteases such as insulin-degrading enzyme (insulysin, IDE) and nardilysin (di-basic convertase, NRD), in addition to the Escherichia coli periplasmic protease pitrilysin (protease III, PTRA). Both IDE and pitrilysin are known to cleave amyloid-β peptide (Kurochkin, 2001; Cornista et al, 2004). Subfamily M16B includes matrix MPP and membrane bound MPP integrated as Core proteins 1 and 2 of the cytochrome bc1 complex (Glaser and Dessi, 1999). The role of MPP is to cleave off presequences from imported proteins once they reach the mitochondrial matrix. This makes MPP different from other pitrilysin proteases, which only degrade peptides and do not act on larger substrates. PreP belongs to subfamily M16C that also includes falcilysin, an essential protease responsible for hemoglobin degradation in the malaria parasite Plasmodium falciparum (Eggleson et al, 1999).
Here, we report the 2.1 Å resolution structure of presequence protease PreP from A. thaliana, which presents the first closed, substrate-bound conformation of an M16 protease. It reveals how the two enzyme halves come together to form a large peptidasome chamber to hold the substrate. The closed conformation places previously unidentified amino acids at the active site, and these residues ensure that the chamber is kept closed during proteolysis to avoid unintentional degradation. This study also reveals the importance of magnesium or other cations for the proper function of AtPreP1. A mechanism is proposed wherein access to the active site involves a hinge-bending motion that causes the peptidasome to open and close in response to substrate binding. In support of this model, proteolytic studies of cysteine double mutants in which disulfide bonds keep the two halves covalently locked show that there is no activity when these bonds are intact while proteolysis is normal when they are reduced.
Results and discussion
Description of the structure
The crystal structure of the catalytically inactive mutant AtPreP1 E80Q was refined at 2.1 Å resolution. The asymmetric unit comprises two protein molecules, each with a zinc atom and a six-residue peptide bound at the active site (Figure 1A). Magnesium ions facilitate crystallization of AtPreP1 and two hydrated ions are found in each protein molecule coordinated by acidic residues (Figure 1B). The 995-residue polypeptide folds into four topologically similar domains, which form two bowl-shaped halves. Each of the four domains comprise a five to seven-stranded β-sheet (XABFCED) with two or three α-helices (2, 3 and 8) packed against one face of the sheet and five additional α-helices (1, 4, 5, 6 and 7) situated at one end of the sheet (Figure 1C). Comparison of the four domains shows that the sequence identity is only 6–11% and the root-mean-square deviation (r.m.s.d.) after superimposing the main chains is 2 Å or higher despite a similar topology. Within each half of the protein, the two domains are attached through extensive contacts between helix 5 from one domain and the β-sheet of the other domain. Additional interactions are also formed between the respective DE-loops. A surface exposed linker of 22 residues connects the first and second domains and an analogous linker comprising 20 residues connects the third and fourth domains.
Figure 1.
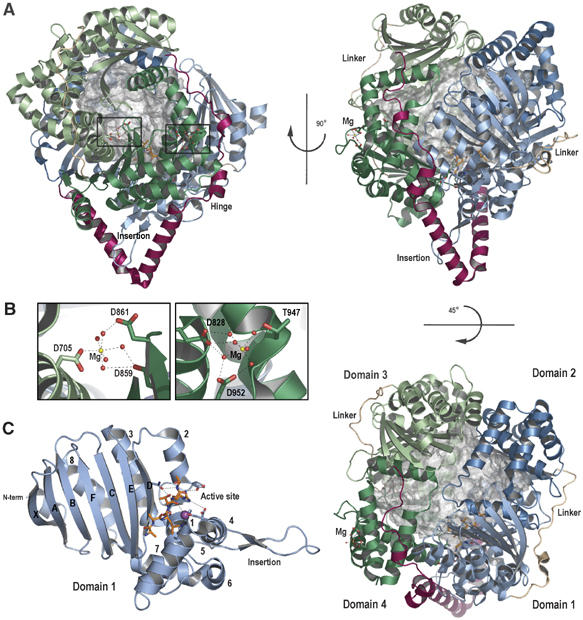
Structure of the presequence protease PreP. (A) Ribbon presentation of AtPreP E80Q with the four domains, hinge, and connecting linkers distinguished by color. The 10 000 Å3 proteolytic chamber is shown as a gray surface representation with the zinc displayed as a purple sphere and the two magnesium binding sites, active site and substrate peptide (orange) drawn as sticks. (B) Detailed view of the magnesium atoms coordinated by acidic residues. (C) The isolated first domain indicating the topology of the secondary structure and the position of the active site and the large insertion in helix 4.
A unique hinge region of 82 residues joins the two enzyme halves. Following β-strand F in the second domain, the hinge forms two long α-helices, which protrude from the protease in the shape of a V. They are followed by a long linker consisting of two additional small helices that spans the surface of the first and fourth domains before it connects to β-strand A in the third domain. Consequently, the hinge almost exclusively interacts with the first and fourth domains in the vicinity of the catalytic site. In addition, there is a large insertion in helix 4 of the first domain that rests right between the two V-shaped helices and forms a bridge that connects the active site and the hinge.
Unpredicted residues complete the active site
The arrangement of the active site is that of a typical metalloprotease, albeit that the zinc-binding motif is inverted (Figure 2A). The first domain forms the major part of the active site; the inverted zinc-binding motif (HXXEH) is part of helix 1 where His77 and His81 coordinate the zinc and Glu80 (substituted for Gln in the crystallized mutant) acts as a base catalyst. The inactive mutants H77L, E80Q and H81L generated in a previous study confirm the importance of these residues in proteolysis (Moberg et al, 2003). However, the third zinc ligand Glu177 situated in helix 4 was previously unknown. It is also essential for catalysis as demonstrated by the inactive E177Q mutant (Figures 2D and E, lane 11). Despite the fact that the protein was crystallized in the absence of a substrate, the electron density reveals a peptide of six residues bound in the active site (Figure 2C). The peptide forms an antiparallel β-strand to strand D located at the inside edge of the first β-sheet, with three main-chain hydrogen bonds to Ala110 and Thr112. The side chain of Asn109 forms additional hydrogen bonds to the main chain of P1′ and P2′ in the substrate, supporting the sharp turn immediately following the scissile bond. The N109A mutant would be unable to form these hydrogen bonds and its proteolytic activity is reduced (Figures 2D and E, lane 10).
Figure 2.
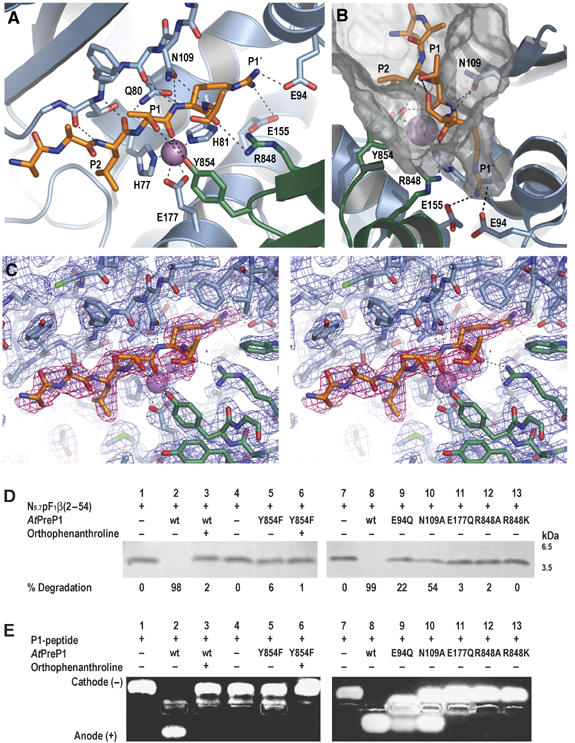
The active site. (A) Details of the substrate binding to the D strand (drawn as sticks) and active site residues from domains 1 (blue) and 4 (green). Selected hydrogen bonds are shown as black dashes. (B) The S1′ pocket with the two acidic residues Glu94 and Glu155 forming salt bridges to the basic P1′ Arg in the substrate. The inner surface of the chamber is shown in gray. (C) Stereo representation of an early 2Fo–Fc electron density map calculated before the peptide was built into the model. The whole map is contoured at 1σ (blue) and the density for the peptide is also shown for 0.5σ (red). (D–E) Proteolytic activity of native (wt) AtPreP1 and active site mutants measured as the degradation of (D) the presequence N5.7pF1β(2–54) and (E) the P1 peptide. Substrate in the absence of protease is shown in lanes 1, 4 and 7. Degradation by native AtPreP1 is shown in lanes 2, 3 and 8 and by different active site mutants in lanes 5–6 and 9–13. For comparison, the inhibitory effect on proteolysis by ortho-phenanthroline is shown in lanes 3 and 6.
The most astonishing discovery is the presence of C-terminal residues at the active site, namely Arg848 and Tyr854 from the loop preceding strand D in the fourth domain. Consequently, residues separated by almost 800 residues in sequence form the active site. This explains why a C-terminal deletion of SPP results in a protease that can bind substrate but not cleave it (Richter and Lamppa, 2003) and why a similar C-terminal deletion of nardilysin produces an inactive but soluble protease (Ma et al, 2002). Arg848 forms a hydrogen bond to the main chain oxygen of P2′, while Tyr854 binds to the main chain oxygen of the scissile bond. Substituting Arg848 for Ala or even Lys abolishes proteolytic activity of AtPreP1 (Figures 2D and E, lanes 12 and 13). Similarly, mutating Tyr854 to Phe also results in inactivation (Figures 2D and E, lane 5) compatible with the reaction mechanism for thermolysin in which this residue stabilizes the transition state (Tronrud et al, 1992). This particular combination of side chains is similar to that of the botulinum neurotoxins (BoNTs) as replacing Arg362 and Tyr365 in BoNT/A results in a severe effect on catalysis (Binz et al, 2002). Substitution of tyrosine for phenylalanine in other metalloproteases has also resulted in inactive mutants (Vazeux et al, 1997; Marie-Claire et al, 1998; Thompson et al, 2003). Tyr854 is conserved in other members of the pitrilysin subfamilies M16A and M16C, and can be identified by sequence alignments based on the structures or by threading (Jones, 1998) (Supplementary Figure 1).
Substrate binding supports specificity towards basic residues
With the exception of the S1′ site, the specificity pockets that hold the substrate side chains are large and open to the proteolytic chamber. Hence, they would be expected to accommodate most side chains in agreement with the observed low substrate specificity. However, the S1′ pocket following the scissile bond is long and narrow with two acidic residues at the bottom: Glu94 and Glu155 (Figure 2B). The electron density for the P1′ side chain in the substrate is consistent with an arginine, which forms favorable salt bridges to the glutamates. In a previous study, the E155Q mutation showed decreased activity towards one of two substrates tested (Moberg et al, 2003). A similar effect was also observed from the E94Q mutation generated for this study (Figures 2D and E, lane 9). The decrease in activity can be explained by the contribution of Glu94 and Glu155 to the negative charge of the S1′ pocket and perhaps their proximity to the hinge region.
The electron density at the N-terminal side of the scissile bond suggests a small P1 side chain and was built as a threonine that can form hydrogen bonds to both P2′ and Tyr854. The density for the P2 residue agrees with leucine, and there are several hydrophobic residues lining the S2 pocket. Based on the electron density, shape and charge of the specificity pockets the optimal sequence for binding is similar to LTR. In agreement with these conclusions, recent proteolytic studies of AtPreP1 show a preference for cleavage after small polar residues and before basic residues (Ståhl et al, 2005).
The peptidasome chamber controls proteolysis
The two enzyme halves completely enclose a large proteolytic chamber that shields the active site (shown as a gray surface representation in Figure 1A). The chamber has a volume of more than 10 000 Å3 as calculated using the program VOIDOO (Kleywegt and Jones, 1994). The inner surface is primarily polar comprising a large number of hydroxylated and acidic residues with local patches of aromatic side chains. The chamber appears spacious enough to hold peptide substrates such as the presequences processed by PreP, but is sufficiently small to exclude larger, folded proteins. Since the active site includes residues from both the N- and C-terminal part of the protein, proteolysis can occur only when the chamber is closed. Thus, the proteolytic chamber protects against unwarranted degradation by preventing folded proteins from entering the active protease and limiting the size of the substrate. In fact, substrate specificity studies show that AtPreP1 can degrade peptides between 10 and 65 residues in length (Ståhl et al, 2005). Similar results have also been reported for IDE (Kurochkin, 2001).
Divalent cations are essential for AtPreP1 function
Magnesium or calcium ions were required for crystallization of AtPreP1. In the crystal structure, two hydrated Mg2+ ions are found in each protein molecule, coordinated by acidic residues (Figure 1B). One of the [Mg(H2O)6]2+-binding sites is located on the inside of the proteolytic chamber between the third and fourth domains (Figure 1B, left panel). Asp705 Oδ2 from helix 5 in domain 3 binds directly to Mg2+ and replaces one of the six water molecules. Asp859 and Asp861 from strand D in the fourth domain also coordinate this ion. The other [Mg(H2O)6]2+-site is located on the surface of the fourth domain and is coordinated by Asp828 from helix 1, Thr947 from helix 6 and Asp952 from helix 7 (Figure 1B, right panel). In molecule A, this magnesium ion also forms crystal contacts to Asp510 located in the hinge of another molecule A.
The position of the [Mg(H2O)6]2+-sites suggests that binding of magnesium ions may have an affect on the conformation of the fourth domain. The position of this domain is crucial for proteolytic activity, since it holds the active site residues Arg848 and Tyr854. Both [Mg(H2O)6]2+ sites are in the vicinity of these residues; the first site is at the other end of helix 1 with respect to Arg848 and connects helix 1 to helices 6 and 7, while the second site is at the opposite end of strand D compared to Tyr854 and forms a link between the third and fourth domains. In order to determine the impact of cations for the proper function of AtPreP1, the proteolytic activity was studied using various concentrations of MgCl2 and CaCl2 (Figure 3). AtPreP1 was inactive in the absence of MgCl2 and CaCl2. Addition of MgCl2 or CaCl2 resulted in gaining of the proteolytic activity with full activation at 10 mM concentrations of MgCl2 and CaCl2. These results suggest the requirement of additional metal ions beside Zn for the activity of AtPreP1.
Figure 3.
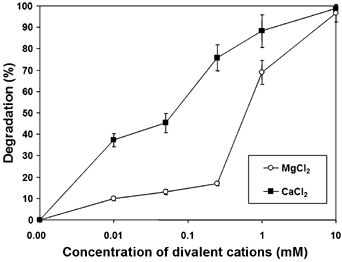
Effect of divalent cations on the proteolytic activity of AtPreP1. Degradation of presequence N5.7pF1β(2–54) by native AtPreP1 in absence or presence of different concentrations of MgCl2 and CaCl2. The results are derived from three independent experiments (standard deviation is indicated).
The other pitrilysin structures display an open conformation
The very recently determined structure of the pitrilysin protease from E. coli (PDB code 1Q2L, Maskos K, Jozic D, Fernandez-Catalan C, Crystal Structure of Pitrilysin, the Prototype of Insulin-Degrading Enzymes, unpublished) and the structure of yeast MPP (PDB code 1HR6, Taylor et al, 2001) display four topologically similar domains organized in two halves as we observe in AtPreP1. However, in contrast to PreP, the pitrilysin and MPP structures are in an open conformation (Figures 4A and B). The sequence identity and main chain r.m.s.d. are very low between the different M16 family members, but they display the same fold. The highest sequence identity (about 20%) is observed in the first domain, which binds the substrate and holds the majority of the active site residues. Overall, the secondary structure elements are similar and the organization within each enzyme half is preserved such as the contacts formed between helix 5 from one domain and the β-sheet of the other domain and the long linkers connecting the two domains. However, the relative position and the interactions formed between the two halves are very different. In pitrilysin, the two halves are separated by a rotation of more than 50° compared to AtPreP1 and in MPP this rotation is approximately 38°. The distance between the catalytic zinc and the Cα atom in Tyr854 is 8 Å in AtPreP1, while the corresponding distance is 38 Å in pitrilysin and 22 Å in MPP.
Figure 4.
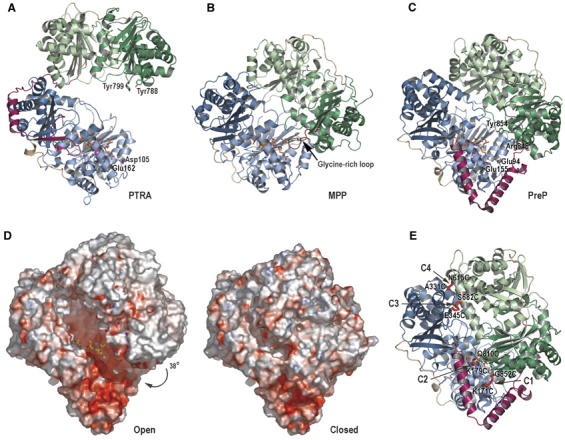
Structure of different pitrilysin family members. (A) E. coli pitrilysin (PTRA) from subfamily M16A (Maskos K, Jozic D, Fernandez-Catalan C, unpublished) and (B) Yeast MPP from subfamily M16B (Taylor et al, 2001). The position of the substrate peptide from the AtPreP1 structure is shown for clarity. (C) Model of the open conformation of AtPreP1 from subfamily M16C created by superimposing the two enzyme halves onto the subunits of MPP. (D) Surface representation of the closed and proposed open form of AtPreP1 colored according to the electrostatic potential (negative red/positive blue). (E) Positions of the introduced cysteine pairs designed to form disulfide bonds that lock AtPreP1 in a closed conformation: K171C-G852C (C1), K179C-Q810C (C2), E345C-S682C (C3) and A331C-N615C (C4).
The two enzyme halves of AtPreP1 are connected by the hinge region, which forms extensive interactions with the first and fourth domains. There are also numerous contacts between the second and third domains. In pitrilysin, a linker that spans the surface of the second and third domains joins the two halves, and there are no contacts between the first and fourth domains (Figure 4A). Thus, the pitrilysin structure appears to be more flexible than AtPreP1. In MPP, there is no covalent link between the two halves, but there is a large network of hydrogen bonds and hydrophobic contacts holding the four domains together (Figure 4B). Furthermore, MPP has a long, flexible, glycine-rich loop located in the fourth domain that closes the gap between the first and fourth domains in the vicinity of the active site. The function of this loop is unclear, but MPP containing a deletion in this loop displays a lower affinity for substrate as well as a reduction in catalytic activity (Nagao et al, 2000).
Modeling a closed conformation of pitrilysin by superimposing its four domains individually onto AtPreP1 suggests that it must also be closed during proteolysis. Tyr788 and Tyr799 are easily identified as the residues equivalent to Arg848 and Tyr854, and thus complete the active site. Interestingly, the shape and charge of the S1′ pocket is also similar to AtPreP1 with the two acidic residues Asp105 and Glu162 corresponding to Glu94 and Glu155. In the MPP structure, it is difficult to establish a side chain that stabilizes the transition state since the glycine-rich loop prevents the two halves from closing completely (Figure 4B). However, an open conformation of MPP is more compatible with its function to cleave off presequences from folded proteins that are too large to fit inside the enzyme.
Proposed mechanism for substrate binding and release
The closed conformation of the peptidasome suggests an underlying mechanism to allow substrate access to the active site. The peptidasome will need to open for the substrate to bind and then close in order to bring Arg848 and Tyr854 from the C-terminus into contact with the bound substrate. Given that the structures are related, it seems likely that the open conformation of PreP is similar to those observed in pitrilysin (Maskos K, Jozic D, Fernandez-Catalan C, unpublished) and MPP (Taylor et al, 2001). Modeling an open conformation of AtPreP1 using the structure of MPP suggests that the closed and open states may differ by as much as a 38° rotation of the two halves around the hinge (Figure 4C). It is possible that the opening and closing of the peptidasome is simply the result of thermal motion, but the proximity to the active site and the S1′ pocket suggests that the hinge may move in response to substrate binding. We propose that the unbound state is open and that substrate binding triggers a movement that brings the two halves of the enzyme together so that Arg848 and Tyr854 can complete the active site and stabilize the transition state (Figure 5). The proximity to the active site and the S1′ pocket would allow the hinge to move in response to substrate binding. In fact, smaller hinge-bending motions associated with inhibitor and substrate binding have already been described for thermolysin and other related neutral proteases (Holland et al, 1992; Hausrath and Matthews, 2002).
Figure 5.
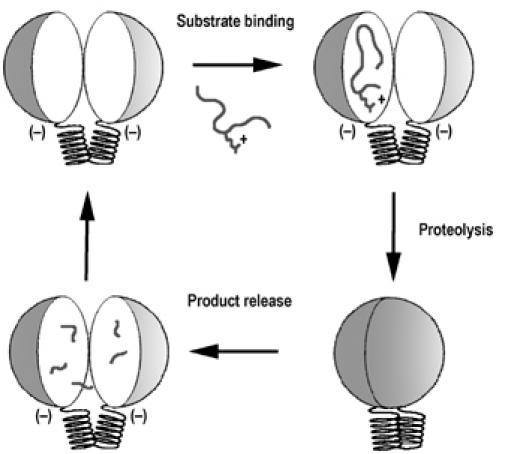
Proposed mechanism for the PreP peptidasome substrate binding and release.
While the opening and closing of the peptidasome revolves around the hinge, it appears electrostatic forces could be driving these movements. The electrostatic surface potential for AtPreP1 E80Q was calculated using APBS tools (Baker et al, 2001) created by Michael Lerner for the program PyMOL (DeLano Scientific LLC, USA). The protease is very acidic, especially around the active site (Figure 4D). As the two negatively charged halves repel each other, the proteasome stays open. However, when a substrate containing basic residues is bound, part of that negative charge is neutralized allowing the chamber to close. It is possible that cations such as magnesium reduce negative charge and further favoring the closed conformation. After proteolysis, the cleaved products are released from the active site and the interactions to Arg848 and Tyr854 are broken, signaling the unbound state to the hinge region. The two negatively charged inner surfaces then contribute to push the two halves open by repulsion.
AtPreP1 is inactive if locked in a closed formation
The mechanism involving the opening and closing of the enzyme was investigated by introducing disulfide bonds between the two enzyme halves, thus preventing PreP from opening. Disulfide bonds are expected to form under oxidizing conditions, thus locking the enzyme closed, whereas it would be able to open and close normally under reducing conditions due to the absence of these disulfide bonds (Figure 6A). Consequently, mutants with appropriately positioned cysteine pairs would be inactive under oxidizing conditions and fully active under reducing conditions. Residues in a position to form such disulfide bonds upon substitution to cysteine were predicted using the program Disulfide by Design (Dombkowski, 2003). Four double mutants were created by site directed mutagenesis: AtPreP1 K171C/G852C (C1), AtPreP1 K179C/Q810C (C2), AtPreP1 E345C/S682C (C3) and AtPreP1 A331C/N615C (C4) (Figure 4D).
Figure 6.
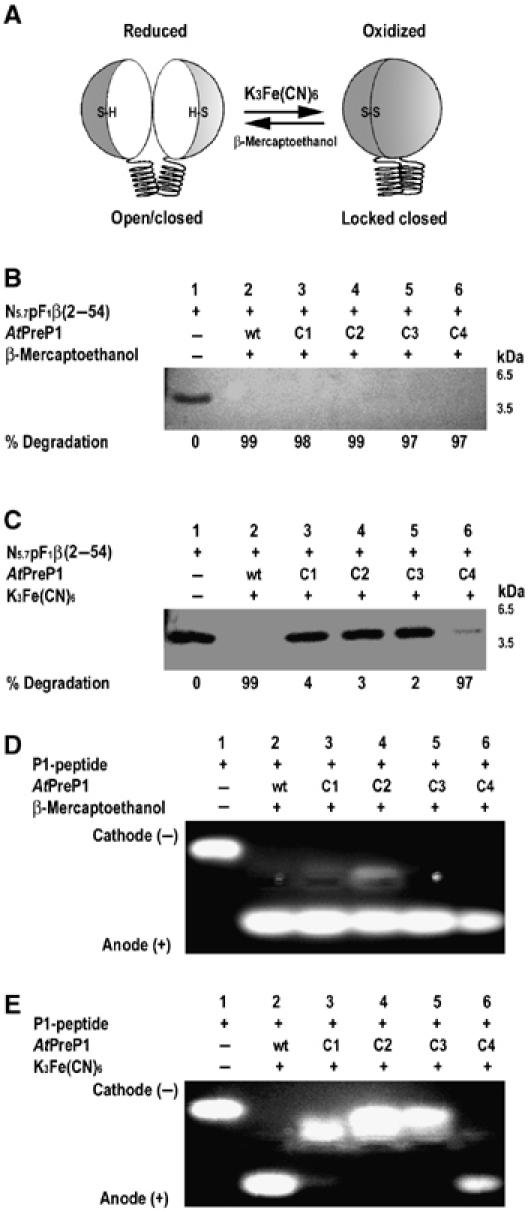
AtPreP1 is inactive if locked in a closed conformation. (A) Schematic representation of the AtPreP1 cysteine double mutants K171C-G852C (C1), K179C-Q810C (C2), E345C-S682C (C3) and A331C-N615C (C4) under reducing and oxidizing conditions. Proteolytic activity of native (wt) AtPreP1 and the cysteine double mutants measured as the degradation of N5.7pF1β(2–54) under reducing (B) and oxidizing conditions (C) and degradation of the P1 peptide under reducing (D) and oxidizing conditions (E).
Oxidizing and reducing conditions were established using potassium ferricyanide (K3Fe(CN)6)) and β-mercaptoethanol (β-ME) as oxidizing and reducing agents, respectively. Catalytic activity was tested at both conditions using the Nicotiana plumbaginifolia presequence N5.7pF1β(2–54) and the P1 peptide as substrates. The proteolytic activity of the wild type was not affected under any of the given conditions (Figures 6B–E, lane 2). The activity of all four cysteine double mutants was normal under reducing conditions using both N5.7pF1β(2–54) and the P1 peptide as substrates (Figures 6B and D, lanes 3–6). Interestingly, under oxidizing conditions, the C1, C2 and C3 mutants were catalytically inactive (Figures 6C and E, lanes 3–5) whereas the C4 mutant had almost normal activity (Figures 6C and E, lane 6). The introduced disulfide bonds are located in various positions between the two halves of AtPreP1 (Figure 4E). The disulfide bond in the C4 mutant is farthest away from the active site, in a position where the conformational change upon opening and closing would be expected to be small. This can explain why this mutant displayed little effect on catalytic activity under oxidizing conditions. The remaining three disulfide bonds are located in regions where larger conformational changes would be necessary.
Free SH groups in native AtPreP1 and the cysteine double mutants were quantitatively estimated using Ellman's Reagent (5,5′-dithio-bis-(2-nitrobenzoic acid), DTNB) and protein purified under both oxidizing and reducing conditions (Table I). There are 10 estimated free SH groups in native AtPreP1 under both oxidizing and reducing conditions while the cysteine double mutants have 10 (10.33–10.59) free SH groups under oxidizing conditions and 12 (12.07–12.33) free SH groups under reducing conditions, suggesting that the introduced cysteines form a disulfide bond under oxidizing conditions. In order to prove that the loss of activity was due to the formation of disulfide bonds, the free SH groups of all cysteine residues were irreversibly modified using N-ethylmaleimide (NEM). Binding of NEM prevents cysteines from forming disulfide bonds even under oxidizing conditions (Figure 7A). Thus, all four cysteine double mutants as well as native AtPreP1 were proteolytically active under oxidizing conditions after being treated with NEM prior to oxidation (Figures 7B and C, lanes 3–6). The result confirms that the loss of activity observed in the cysteine double mutants under oxidizing conditions is due to the formation of disulfide bonds covalently locking the two halves of the protease.
Table 1.
Number of free SH groups (mol/mol of AtPreP1)
| AtPreP1 variant | Reducing conditions | Oxidizing conditions | Reduced–Oxidized |
|---|---|---|---|
| Native (wt) | 10.30 | 10.07 | 0.23 |
| C1 | 12.30 | 10.55 | 1.80 |
| C2 | 12.19 | 10.33 | 1.86 |
| C3 | 12.33 | 10.61 | 1.72 |
| C4 | 12.07 | 10.59 | 1.48 |
Figure 7.
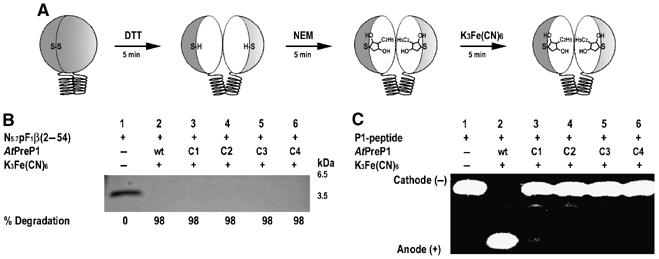
Blocking of free SH-groups restores the activity of the AtPreP1 cysteine double mutants. (A) Covalent modification of free SH-groups by NEM. Degradation of the presequence N5.7pF1β(2–54) (B) and the P1 peptide (C) by native (wt) AtPreP1 and the cysteine double mutants K171C-G852C (C1), K179C-Q810C (C2), E345C-S682C (C3) and A331C-N615C (C4) under oxidizing conditions after modification by NEM.
The suppression of proteolytic activity by the introduction of disulfide bonds supports the mechanism involving the opening and closing of the two enzyme halves. Under oxidizing conditions, the disulfide bonds restrict the opening of the protease so that the substrate is unable to reach the active site. The proteolytic activity of the cysteine double mutants was still intact under reducing conditions and native AtPreP1 showed normal function in both oxidizing and reducing conditions. Modification by NEM shows that the change in activity is directly linked to the formation of disulfide bonds. Taken together, these results verify that neither the cysteine substitutions per se nor the addition of oxidizing or reducing agents have a negative impact on proteolysis. Overall, the results support the proposed mechanism involving opening and closing of the enzyme in response to substrate binding.
Materials and methods
Generation of the AtPreP1 mutants
The single mutations E94Q, N109A, E177Q, R848K, R848A, Y854F and the double mutations K171C/G852C, K179C/Q810C, A331C/N615C, E345C/S682C were introduced to a pGEX-6P-2 plasmid (Amersham Biosciences) containing wild-type AtPreP1 using the Quick-Change Site Directed Mutagenesis Kit (Stratagene). The constructs were verified by DNA sequencing utilizing the DYEnamic Sequencing Kit (Amersham Biosciences). All primers are listed in Supplementary Table I.
Expression and purification of the AtPreP1 mutants
The pGEX-6P-2 vector containing different AtPreP1 mutants with an N-terminal fusion of the GST was used to transform E. coli BL21(DE3). Colonies were grown at 37°C in 100 ml LB medium containing 100 μg/ml ampicillin, induced with 1 mM IPTG at OD600 of 0.5 and incubated at 30°C to OD600 of 2. The cells were harvested by centrifugation, resuspended in phosphate-buffered saline (PBS) buffer (140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4, pH 7.3) and stored at −80°C. Lysis was induced by the addition of 0.5 mg/ml lysozyme followed by sonication. The lysate was centrifuged for 10 min at 75 000 g to remove cell debris and the supernatant filtered through a 0.45 μm membrane. The filtered lysate was applied to Glutathione Sepharose® (Amersham Biosciences) equilibrated with PBS buffer and incubated at 4°C for 4 h with light agitation. Unbound proteins were washed away with PBS buffer and then the bound AtPreP1 was cleaved off with PreScission™ Protease in PreScission cleavage buffer (50 mM Tris–HCl, 150 mM NaCl, pH 7.5) at 4°C for 4 h. AtPreP1 was collected from the supernatant and purity was analyzed on 12% SDS gels in the presence of 4% urea and stained with Coomassie Brilliant Blue protein. The protein concentration was measured using BioRad Protein Assay (BioRad). In total, 50% glycerol was added prior to storage at −20°C to preserve protease activity.
Selenomethionine-labeled AtPreP1 E80Q was produced using the metabolic inhibition method (Van Duyne et al, 1993) with initial growth at 37°C followed by expression at 20°C for 16 h. The GST-tagged protein was bound to Glutathione Sepharose® (Amersham Biosciences) and eluted with 10 mM GSH in PBS supplemented with 10 mM KCl and 2 mM β-ME. After desalting into the appropriate cleavage buffer, the GST-tag was removed by incubation at 4°C for 16 h with PreScission™ Protease (Amersham Biosciences). β-Octylglucoside (0.1% (w/v)) was added to the cleavage reaction before concentration by ultrafiltration and separation on a Superdex200 gel filtration column (Amersham) (equilibrated with 10 mM HEPES–KOH, 50 mM NaCl and 1 mM β-ME).
Proteolytic activity assay
The proteolytic activity of AtPreP1 was analyzed by the degradation of two different substrates, the N. plumbaginifolia presequence N5.7pF1β(2–54) and the fluorescent PepTag®C1 peptide, P1 (Moberg et al, 2003), in a degradation buffer (20 mM HEPES–KOH and 10 mM MgCl2, pH 8.0) at 30°C for 30 min. Each reaction contained 2 μg N5.7pF1β(2–54) peptide and 0.5 μg purified protease or 1 μl P1 peptide and 0.5 μg protease. For the inhibition of proteolytic activity, 10 mM ortho-phenanthroline was added to the reaction mixture prior to the addition of substrate. The reactions containing the N5.7pF1β(2–54) peptide were stopped by addition of 2 × Laemmli sample buffer and analyzed on 10–20% Tris–Tricine gels (BioRad) followed by staining with Coomassie Brilliant Blue. Glycerol (10%) was added to the reactions containing P1 peptide and the samples were analyzed on a 0.8% agarose gel. The fluorescent peptide was visualized by UV light. Proteolytic activity of the cysteine double mutants was investigated under oxidizing conditions (1 mM K3Fe(CN)6) and reducing conditions (1 mM β-ME). The protease was incubated under the respective conditions at room temperature for 5 min prior to the addition of substrates.
Blocking of free SH groups
For native and the cysteine double mutants, 0.5 μg AtPreP1 was incubated with 1 mM DTT in a 4 μl volume reaction at room temperature for 5 min. The reduced protease was then treated with 1 mM NEM at room temperature for another 5 min, and diluted to a volume of 40 μl with degradation buffer before 1 mM K3Fe(CN)6 was added to the reaction. The reaction mixture was further incubated at room temperature for 5 min before addition of substrates. The proteolytic activity of NEM treated AtPreP1 mutants was studied as degradation of the N5.7pF1β(2–54) peptide and the P1 peptide compared with the native protein as described previously.
Estimation of free SH groups
Free SH groups (Table I) were quantitatively estimated using Ellman's Reagent (DTNB) as described by the supplier (Pierce). Native and cysteine double mutants of AtPreP1 were purified under both oxidizing and reducing conditions using either 1 mM K3Fe(CN)6 and 1 mM β-ME as oxidizing and reducing agent, respectively.
Crystallization and data collection
Crystals were grown by the hanging-drop vapor diffusion method using 2 μl drops consisting of 1 μl AtPreP1 E80Q at 5 mg/ml in 10 mM HEPES pH 7.0 with 50 mM NaCl and 1 mM β-ME, plus 1 μl crystallization solution with 22% (w/v) PEG 6 K, 0.1 M HEPES pH 6.5, 25 mM MgCl2 and 0.1% β-ME suspended over 1 ml of the same solution. The best crystals appeared after approximately 1 week as thin plates with the maximum dimensions of 0.6 × 0.4 × 0.04 mm3. They belong to the primitive orthorhombic space group P212121 with two molecules in the asymmetric unit and cell dimensions a=110.8 Å, b=114.3 Å and c=163.0 Å. The crystals were flash-frozen in liquid nitrogen after brief soaks in crystallization solution with 20% PEG 400 as a cryo protectant. Two data sets were collected at the beamline ID14 at the European Synchrotron Radiation Facility (Table I). The first, phasing data to 2.7 Å were processed at the beamline using the programs IPMOSFLM (Leslie, 2006) for indexing and integration, SCALA (Evans, 1997) for scaling and XPREP (Schneider and Sheldrick, 2002) for analyzing anomalous differences (19 719 pairs). All 40 selenium atoms in the asymmetric unit were located by SHELXD (Schneider and Sheldrick, 2002) and phases were calculated and improved first with MLPHARE (Otwinowski, 1991) to confirm the solution and then with SOLVE (Terwilliger and Berendzen, 1999) and RESOLVE (Terwilliger, 2000) to create the two-fold averaged electron density map at 3.0 Å resolution used to build the model. The second 2.1 Å resolution data set was processed and scaled using the program package DENZO/SCALEPACK (Otwinowski and Minor, 1997). Details of the data collection are shown in Table I.
Structure building and refinement
The first molecule (A) was built as a polyalanine model and copied to create the second molecule (B) by rigid body fitting into the initial 3 Å resolution electron density map in XtalView (McRee, 1999). The coordinates were transferred to the second data set that extends to 2.1 Å resolution by rigid body refinement using REFMAC5 (Murshudov et al, 1997). The majority of side chains were then built automatically using Arp/Warp (Perrakis et al, 1999), followed by subsequent runs of manual fitting using XtalView (McRee, 1999) and O (Jones et al, 1991) and refinement by REFMAC5 (Murshudov et al, 1997). Simulated annealing using CNS (Brunger et al, 1998) was performed to help with the positioning of less ordered loops. The final coordinates comprise residues 15 to 993 from each molecule, a peptide of six residues in each active site, two zinc ions, four magnesium ions, one chloride ion and 948 water molecules. The structures display good stereochemistry with 93.0% of the main chain angles in the most favored regions of the Ramachandran plot, 6.9% in the allowed regions and 0.1% in the additionally allowed regions. Rwork and Rfree of the final structure is 20.6 and 25.6%. Refinement statistics are included in Table II.
Table 2.
Data collection, phasing and refinement statistics
| Crystal 1 | Crystal 2 | |
|---|---|---|
| Data collection | ||
| Space group | P212121 | P212121 |
| Cell dimensions | ||
| a, b, c (Å) | 111.1, 115.7, 162.6 | 110.8, 114.3, 163.0 |
| α, β, γ (deg) | 90, 90, 90 | 90, 90, 90 |
| Wavelength | 0.979 | 0.979 |
| Resolution (Å) | 79–2.7 | 40–2.10 (2.14–2.10)a |
| Rsym or Rmerge | 10.7 | 11.9 (41.7) |
| I/σI | 17.0 | 10.4 (2.26) |
| Completeness (%) | 99.96 | 96.5 (86.6) |
| Redundancy | 7.17 | 4.7 (2.3) |
| Refinement | ||
| Resolution (Å) | 40–2.10 | |
| No. reflections | 116 872 | |
| Rwork/Rfree | 20.6/25.6 | |
| No. of atoms | ||
| Protein | 15 462 | |
| Ligands/ions | 82/8 | |
| Water | 948 | |
| Average B-factor | 38.44 | |
| r.m.s.d. | ||
| Bond lengths (Å) | 0.011 | |
| Bond angles (deg) | 1.311 | |
| Two crystals were used for AtPreP1 structure determination: Crystal 1 for phasing and Crystal 2 for refinement. | ||
| aHighest resolution shell is shown in parenthesis. | ||
Modeling the open conformation of PreP
The open conformation of PreP was modeled by superimposing the two halves of AtPreP1 onto the subunits of yeast MPP (Taylor et al, 2001) using the program Sequoia (Bruns et al, 1999). The second and third domains were merged at the point where the hinge from each half came together at Ala533 in the small helix following the long V-shaped helices.
Accession numbers
The coordinates and structure factors have been deposited in the Protein Data Bank under the accession code 2FGE.
Supplementary Material
Supplementary Figure 1
Supplementary Table 1
Acknowledgments
We thank P Nordlund for his support and use of his laboratory, which is financed by the Swedish Research Council and the Swedish Cancer Society, and K-M Larsson and P Moberg for their participation in the early stages of the project. This research was supported by grants from the Knut and Alice Wallenberg Foundation, the Carl Trygger Foundation and the Magn Bergwall Foundation to TE and from the Swedish Research Council to EG.
References
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA 98: 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker AB, Roth RA (1992) An unusual active site identified in a family of zinc metalloendopeptidases. Proc Natl Acad Sci USA 89: 3835–3839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhushan S, Lefebvre B, Ståhl A, Wright SJ, Bruce BD, Boutry M, Glaser E (2003) Dual targeting and function of a protease in mitochondria and chloroplasts. EMBO Rep 4: 1073–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhushan S, Ståhl A, Nilsson S, Lefebvre B, Seki M, Roth C, McWilliam D, Wright SJ, Liberles DA, Shinozaki K, Bruce BD, Boutry M, Glaser E (2005) Catalysis, subcellular localization, expression and evolution of the targeting peptides degrading protease, AtPreP2. Plant Cell Physiol 46: 985–996 [DOI] [PubMed] [Google Scholar]
- Binz T, Bade S, Rummel A, Kollewe A, Alves J (2002) Arg(362) and Tyr(365) of the botulinum neurotoxin type a light chain are involved in transition state stabilization. Biochemistry 41: 1717–1723 [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Bruns CM, Hubatsch I, Ridderström M, Mannervik B, Tainer JA (1999) Human glutathione transferase A4–4 crystal structures and mutagenesis reveal the basis of high catalytic efficiency with toxic lipid peroxidation products. J Mol Biol 288: 427–439 [DOI] [PubMed] [Google Scholar]
- Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, Wei Xu H, Stern D, McKhann G, Yan SD (2005) Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer's disease. FASEB J 19: 2040–2041 [DOI] [PubMed] [Google Scholar]
- Cornista J, Ikeuchi S, Haruki M, Kohara A, Takano K, Morikawa M, Kanaya S (2004) Cleavage of various peptides with pitrilysin from Escherichia coli: kinetic analyses using beta-endorphin and its derivatives. Biosci Biotechnol Biochem 68: 2128–2137 [DOI] [PubMed] [Google Scholar]
- Dombkowski AA (2003) Disulfide by design: a computational method for the rational design of disulfide bonds in proteins. Bioinformatics 19: 1852–1853 [DOI] [PubMed] [Google Scholar]
- Eggleson KK, Duffin KL, Goldberg DE (1999) Identification and characterization of falcilysin, a metallopeptidase involved in hemoglobin catabolism within the malaria parasite Plasmodium falciparum. J Biol Chem 274: 32411–32417 [DOI] [PubMed] [Google Scholar]
- Evans PR (1997) Scala. Joint CCP4 and ESF-EACMB Newsletter Protein Crystallogr 33: 22–24 [Google Scholar]
- Glaser E, Dessi P (1999) Integration of the mitochondrial-processing peptidase into the cytochrome bc1 complex in plants. J Bioenerg Biomembr 31: 259–274 [DOI] [PubMed] [Google Scholar]
- Hausrath AC, Matthews BW (2002) Thermolysin in the absence of substrate has an open conformation. Acta Crystallogr D 58: 1002–1007 [DOI] [PubMed] [Google Scholar]
- Holland DR, Tronrud DE, Pley HW, Flaherty KM, Stark W, Jansonius JN, McKay DB, Matthews BW (1992) Structural comparison suggests that thermolysin and related neutral proteases undergo hinge-bending motion during catalysis. Biochemistry 31: 11310–11316 [DOI] [PubMed] [Google Scholar]
- Jones DT (1998) THREADER: protein sequence threading by double dynamic programming. In Computational Methods in Molecular Biology, Salzberg S, Searls D, Kasif S (eds), Chapter 13 Amsterdam: Elsevier Science [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Kleywegt GJ, Jones TA (1994) Detection, delineation, measurement and display of cavities in macromolecular structures. Acta Crystallogr D 50: 178–185 [DOI] [PubMed] [Google Scholar]
- Kurochkin IV (2001) Insulin-degrading enzyme: embarking on amyloid destruction. Trends Biochem Sci 26: 421–425 [DOI] [PubMed] [Google Scholar]
- Leslie AG (2006) The integration of macromolecular diffraction data. Acta Crystallogr D Biol Crystallogr 62: 48–57 [DOI] [PubMed] [Google Scholar]
- Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, Lue LF, Walker DG, Kuppusamy P, Zewier ZL, Arancio O, Stern D, Yan SS, Wu H (2004) ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science 304: 448–452 [DOI] [PubMed] [Google Scholar]
- Ma Z, Chow KM, Csuhai E, Hersh LB (2002) The use of proteolysis to study the structure of nardilysin. Arch Biochem Biophys 401: 198–204 [DOI] [PubMed] [Google Scholar]
- Marie-Claire C, Ruffet E, Tiraboschi G, Fournie-Zaluski MC (1998) Differences in transition state stabilization between thermolysin (EC 3.4.24.27) and neprilysin (EC 3.4.24.11). FEBS Lett 438: 215–219 [DOI] [PubMed] [Google Scholar]
- McRee DE (1999) XtalView/Xfit—a versatile program for manipulating atomic coordinates and electron density. J Struct Biol 125: 156–165 [DOI] [PubMed] [Google Scholar]
- Moberg P, Ståhl A, Bhushan S, Wright SJ, Eriksson A, Bruce BD, Glaser E (2003) Characterization of a novel zinc metalloprotease involved in degrading targeting peptides in mitochondria and chloroplasts. Plant J 36: 616–628 [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Mzhavia N, Berman YL, Qian Y, Yan L, Devi LA (1999) Cloning, expression, and characterization of human metalloprotease 1: a novel member of the pitrilysin family of metalloendoproteases. DNA Cell Biol 18: 369–380 [DOI] [PubMed] [Google Scholar]
- Nagao Y, Kitada S, Kojima K, Toh H, Kuhara S, Ogishima T, Ito A (2000) Glycine-rich region of mitochondrial processing peptidase alpha-subunit is essential for binding and cleavage of the precursor proteins. J Biol Chem 275: 34552–34556 [DOI] [PubMed] [Google Scholar]
- Neupert W, Brunner M (2002) The protein import motor of mitochondria. Nat Rev Mol Cell Biol 3: 555–565 [DOI] [PubMed] [Google Scholar]
- Otwinowski S, Minor W (1997) Processing of X-ray diffraction data collected in oscillation method. Methods Enzymol 276: 307–325 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z (1991) MLPHARE. Daresbury Study Weekend, proceedings [Google Scholar]
- Perrakis A, Morris R, Lamzin VS (1999) Automated protein model building combined with iterative structure refinement. Nat Struct Biol 6: 458–463 [DOI] [PubMed] [Google Scholar]
- Pfanner N, Geissler A (2001) Versatility of the mitochondrial protein import machinery. Nat Rev Mol Cell Biol 2: 339–349 [DOI] [PubMed] [Google Scholar]
- Rawlings ND, Tolle DP, Barrett AJ (2004) MEROPS: the peptidase database. Nucleic Acids Res 32: D160–D164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter S, Lamppa GK (1998) A chloroplast processing enzyme functions as the general stromal processing peptidase. Proc Natl Acad Sci USA 95: 7463–7468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter S, Lamppa GK (2003) Structural properties of the chloroplast stromal processing peptidase required for its function in transit peptide removal. J Biol Chem 278: 39497–39502 [DOI] [PubMed] [Google Scholar]
- Schneider TR, Sheldrick GM (2002) Substructure solution with SHELXD. Acta Crystallogr D 58: 1772–1779 [DOI] [PubMed] [Google Scholar]
- Ståhl A, Moberg P, Ytterberg J, Panfilov O, Brockenhuus Von Löwenhielm H, Nilsson F, Glaser E (2002) Isolation and identification of a novel mitochondrial metalloprotease (PreP) that degrades targeting presequences in plants. J Biol Chem 277: 41931–41939 [DOI] [PubMed] [Google Scholar]
- Ståhl A, Nilsson S, Lundberg P, Bhushan S, Biverståhl H, Moberg P, Morisset M, Vener A, Mäler L, Langel U, Glaser E (2005) Two novel targeting peptide degrading proteases, PrePs, in mitochondria and chloroplasts, so similar and still different. J Mol Biol 349: 847–860 [DOI] [PubMed] [Google Scholar]
- Taylor AB, Smith BS, Kitada S, Kojima K, Miyaura H, Otwinowski Z, Ito A, Deisenhofer J (2001) Crystal structures of mitochondrial processing peptidase reveal the mode for specific cleavage of import signal sequences. Structure 9: 615–625 [DOI] [PubMed] [Google Scholar]
- Terwilliger TC (2000) Maximum-likelihood density modification. Acta Crystallogr D 56: 965–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC, Berendzen J (1999) Automated MAD and MIR structure solution. Acta Crystallogr D 55: 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson MW, Govindaswami M, Hersh LB (2003) Mutation of active site residues of the puromycin-sensitive aminopeptidase: conversion of the enzyme into a catalytically inactive binding protein. Arch Biochem Biophys 413: 236–242 [DOI] [PubMed] [Google Scholar]
- Tronrud DE, Roderick SL, Matthews BW (1992) Structural basis for the action of thermolysin. Matrix Suppl 1: 107–111 [PubMed] [Google Scholar]
- Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J (1993) Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J Mol Biol 229: 105–124 [DOI] [PubMed] [Google Scholar]
- Vazeux G, Iturrioz X, Corvol P, Llorens-Cortes C (1997) A tyrosine residue essential for catalytic activity in aminopeptidase A. Biochem J 327: 883–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XP, Glaser E (2002) Interaction of plant mitochondrial and chloroplast signal peptides with the Hsp70 molecular chaperone. Trends Plant Sci 7: 14–21 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Table 1