

Abstract
Spontaneous regression/complete resistance (SR/CR) mice resist very high doses of cancer cells that are lethal to WT mice even at low doses. In this study, we show that this resistance is mediated by rapid infiltration of leukocytes, mostly of innate immunity, in both primary and repeated challenges. Formation of rosettes with infiltrating natural killer cells, neutrophils, and macrophages was required for the subsequent destruction of cancer cells through rapid cytolysis. Highly purified natural killer cells, macrophages, and neutrophils from the SR/CR mice independently killed cancer cells in vitro. The independent killing activity by each subset of effector cells is consistent with the observation that the resistance was abolished by depleting total infiltrating leukocytes but not by depleting only one or two subsets of leukocytes. The resistance was completely transferable to WT recipient mice through SR/CR splenocytes, bone marrow cells, or enriched peritoneal macrophages, either for prevention against subsequent cancer challenges or eradication of established malignancy at distant sites.
Keywords: cellular cancer immunity, adoptive transfer, cancer therapy, macrophages, leukocyte depletion
Resistance to challenges with aggressive mouse transplantable cancer cells, such as S180, L5178Y, or EL4, is rare for laboratory mice. Spontaneous regression/complete resistance (SR/CR) mice possess a unique autosomal dominant trait that allows them to survive challenges with aggressive mouse cancer cells at up to millions of times the lethal doses for WT mice (1, 2). In SR/CR mice, cancer cells induce rapid infiltration of leukocytes that form rosettes around cancer cells, which then rapidly undergo cytolysis. Leukocytes from SR/CR mice also kill a wide array of mouse and human cancer cell lines in vitro. The cytolytic activity is specific to cancer cells, because SR/CR mice are cancer-free and disease-free for their lifetimes, with no detectable abnormality despite repeated challenges. From the single BALB/c founder mouse discovered in 1999, this cancer-resistance trait has been bred into >2,000 descendants in >14 generations and in three additional backgrounds (C57BL/6, CAST/Ei, and C3H). The overall transmission rate remains at 30–40%. None of the WT breeding partners and control mice housed in the same cages acquired resistance to similar tumor cell challenges.
The immune system in mice, like that in humans, is composed of four major effector cell types that directly attack target cells: cytotoxic T lymphocytes, natural killer cells (NK), neutrophil and possibly eosinophil polymorphonuclear leukocytes (PMN), and macrophages (MΦ). Cytotoxic T lymphocytes belong to adaptive immunity, and their activation requires prior exposure and establishment of memory T cells. However, NK, PMN, and MΦ belong to innate immunity, and their activation requires no prior exposure but depends on a predetermined mechanism to recognize specific patterns of nonself molecules. The fact that SR/CR mice survived the initial challenge with cancer cells without any prior exposure suggests that the anticancer response is innate immunity. Alternatively, the fact that repeated challenges enhanced subsequent responses suggests a possible involvement of adaptive immunity, such as generation of memory T and B lymphocytes and antibody production. For example, older SR/CR mice challenged with cancer cells for the first time showed a delayed onset of the resistance mechanism, resulting in an SR phenotype. SR only happened once to a particular mouse, after which the mouse became completely resistant to subsequent challenges. Furthermore, frequent challenges with cancer cells in SR/CR mice shortened the response time from a few days to even <1 h (unpublished data). The roles of innate and adaptive immunity and the identity of effector cells in SR/CR resistance were not clear. Identification of effector cell type(s) involved in resistance would help clarify the resistance mechanism.
One important implication of cancer resistance in SR/CR mice is its therapeutic potential. The components of the immune system are amenable to adoptive cell transfer (AT), a practice used widely in clinical settings. AT of leukocytes has shown promise against certain types of malignancy but has limitations (reviewed in refs. 3–7) and often requires ex vivo stimulation of donor leukocytes before transfer (8–9). To evaluate the therapeutic potential of this resistance in mice, critical questions needed to be addressed before a feasible strategy could be developed: (i) Do SR/CR leukocytes function only in SR/CR mice or also in WT mice? (ii) Can SR/CR leukocytes only prevent cancer or also treat established malignancy? (iii) Can SR/CR leukocytes act against cancer only locally or also systemically? (iv) Which organs or tissues provide the most effective donor cells? (v) Which subsets of immune cells function as donor cells? (vi) How long can the transferred resistance protect the recipients? In this paper, we report findings related to these questions.
Results
Cell Type Composition of Cancer-Infiltrating SR/CR Leukocytes.
We profiled subset compositions of infiltrating leukocytes to primary and subsequent cancer challenges to determine whether there was a shift from innate to adaptive immunity. We retrieved peritoneal leukocytes after injection with S180 cells, labeled them with fluorescent cell surface markers, and analyzed them with flow cytometry. During the first response, peritoneal cells consisted of 3% CD8+, 7% CD4+, 12% CD19+ (B cells), 29% F4/80+ (MΦ), 26% Ly6G+ (PMN), 30% NK1.1+, and 15% CD11c+ (dendritic cells) (Fig. 1). Crossreactivity of markers caused the sum of percentages to be >100%. MΦ and PMN, as major infiltrates, were verified by counting based on cell morphology. During responses to the second, third, and fourth challenges, at least 2 weeks apart, the leukocyte compositions of the peritoneal cells remained largely unchanged from the first response.
Fig. 1.

Leukocyte subset compositions of the peritoneal infiltrates during responses to primary and secondary challenges with cancer cells. TG-induced peritoneal leukocytes were isolated from unchallenged pups of SR/CR lineage and analyzed for the ability to kill S180 cancer cells in vitro as described in Materials and Methods. The pups displaying the SR/CR phenotype were then challenged with i.p. injection of 106 S180 cells for the first time. The responding leukocytes were washed from the peritoneal cavities, stained with the indicated cell surface markers, and profiled with flow cytometry (dark red bars indicate the first challenge). The populations of responding immune cells were also profiled for the second (green bars), third (brown bars), and fourth (orange bars) challenges with S180 cells. Subset percentages of total infiltrates are shown.
Identification of Leukocyte Subsets in Rosettes with Cancer Cells.
Formation of rosettes between leukocytes and cancer cells is a specific property of the anticancer response in SR/CR mice and has not been observed in WT mice challenged with cancer cells. Using surface marker labeling, we profiled the leukocyte composition of rosettes that showed 38% Ly6G+, 35% NK1.1+, and 26% F4/80+ (Fig. 2A). Fig. 2 B and C shows examples of PMN, MΦ, and NK surrounding S180 in rosettes. In contrast, CD8+ and CD4+ T cells and CD11c+ and CD19+ cells were mostly absent from rosettes.
Fig. 2.

Formation of leukocyte rosettes during the killing of cancer cells in SR/CR mice. SR/CR mice were challenged i.p. with 107 S180 cells, and infiltrating peritoneal leukocytes were harvested after 24 h and analyzed to determine which leukocyte subsets were present in rosettes. (A) Composition of leukocyte subsets in the rosettes. Cytopreps were prepared and stained with cell-specific markers for each leukocyte subset. The percentage of each subset was quantified by direct counting. (B) Contact site morphology of rosettes. The contact sites between cancer cells (T) and PMN (arrowhead) and MΦ (arrow) were viewed by transmission electron microscopy. (C) Immunofluorescence microscopy of rosettes. SR/CR mice were injected i.p. with GFP-transfected S180 cells, and responding leukocytes were removed and analyzed by light microscopy. (a) Phase-contrast view of a rosette. (b) DAPI staining showing nuclear morphology. (c) GFP fluorescence of S180. (d) Rhodamine-conjugated anti-NK1.1 labeling of leukocytes in the rosette (arrows). (e) Composite picture of S180 and anti-NK1.1 staining in a rosette.
Effect of Depletion of Effector Subsets on the Resistance Phenotype.
We next determined whether leukocytes were required for cancer resistance by depleting the cancer-infiltrating leukocytes. We challenged 20 SR/CR mice i.p. with S180 cells, followed by peritoneal lavage to remove cells. When peritoneal cells were depleted repeatedly by this process every day or every other day, up to five times, 19 of 20 SR/CR mice developed palpable peritoneal tumors (ranging from 2 to 10 mm) (Fig. 7, which is published as supporting information on the PNAS web site). However, 2–3 weeks after peritoneal lavage was halted, the tumors regressed in 17 of 19 SR/CR mice, and the recovered mice remained healthy and cancer-free for their lifetimes. Two of the 19 SR/CR donor mice died of cancer due to a large tumor burden before the cessation of depletion. This finding suggests that resistance was entirely dependent on SR/CR leukocytes.
To determine whether resistance required a specific subset of leukocytes, we performed specific depletions. NK or PMN were individually depleted by injection of anti-NK1.1 or anti-Ly6G, respectively (10, 11). To verify depletion, the S180-induced peritoneal leukocytes were analyzed by surface marker-based flow cytometry. NK were absent in the NK-depleted infiltrates, and PMN were absent in the PMN-depleted infiltrates (Fig. 8, which is published as supporting information on the PNAS web site). Because no depleting antibodies were available for MΦ, they were inhibited by i.p. injection of 2-chloroadenosine (Fig. 8) (12). The depletions did not affect unrelated leukocyte populations in the infiltrates (data not shown). The effect of depletion on resistance was evaluated by both short- and long-term observations. For short-term observation, we analyzed the presence or absence of S180 cells in the peritoneum 1 day after challenge to determine whether the resistance was impaired. For long-term observation, we observed whether mice developed ascites or solid tumors. Depletion of single or double subsets had no apparent effect on resistance (Fig. 8). This finding suggests that resistance involved more than one subset independent of one another.
Identification of SR/CR Mice Without Challenge With Live Cancer Cells.
Previously, in the in vitro killing assay (1), resistant leukocytes were isolated from SR/CR mice that had been screened at least twice with cancer cells. One way to confirm the innate nature of the anticancer immunity is to demonstrate the cancer-killing activity of leukocytes isolated from SR/CR mice before challenge with cancer cells. The design of this screen was to use the in vitro killing assay to determine whether leukocytes had tumoricidal activity before the generation of adaptive immunity. From litters of SR/CR mice bred to WT mice, we isolated peritoneal MΦ from each pup by Brewer thioglycollate medium (TG) stimulation (13) and measured S180 killing in vitro (Table 1, which is published as supporting information on the PNAS web site). Then we challenged the pups i.p. with 106 S180 cells to identify resistant mice. Of 31 screened mice, of which ≈40% were expected to be resistant, all 16 mice that had in vitro killing activity survived the in vivo challenges. Thirteen of the 15 mice that had no in vitro killing activity were sensitive to the in vivo challenge. Thus, the in vitro killing assay predicted resistance in mice without challenge with live cancer cells. A second benefit of this design was that it validated the in vitro killing assay as a reliable recapitulation of in vivo activity.
Individual Subsets of SR/CR Leukocytes Have Independent Killing Activity Against Cancer Cells.
Next, we determined which leukocyte subset(s) killed cancer cells in SR/CR mice by using the in vitro killing assay. We measured the killing activity of purified leukocyte subsets. All purified subsets (NK, MΦ, or PMN) from SR/CR mice had purities >95% (Fig. 9, which is published as supporting information on the PNAS web site) and displayed killing similar to that of mixed leukocytes from SR/CR mice (Fig. 3). These results indicate that all major subsets of innate leukocytes in SR/CR mice independently killed S180. To determine whether this killing was specific to cancer cell lines or general to cultured cell lines, we similarly cultured SR/CR leukocytes either with CHO cells or NIH 3T3 fibroblasts. The SR/CR leukocytes did not kill the noncancerous cell lines (Fig. 3).
Fig. 3.

In vitro killing of cancer cells by purified leukocyte subsets from SR/CR mice. Individually purified leukocyte subsets from WT and SR/CR mice (described in Materials and Methods) were examined for their ability to kill S180 cells in a standard in vitro killing assay. The effector cells were incubated with S180 cells at a 10:1 ratio at 39°C for 24 h. Live S180 cells were identified by size, morphology, and fluorescence, and viability was determined by trypan blue exclusion. S, splenocytes; T, thioglycollate-induced peritoneal MΦ; M, MΦ; N/A, no leukocytes added. As a control, mixed populations of SR/CR leukocytes were incubated as described above with CHO or NIH 3T3 mouse fibroblast cells to determine whether S180 killing was specific to cancer cells or general to cultured cell lines.
Treatment of Established Tumors at Distant Sites by Transferred Leukocytes.
We determined whether leukocytes from SR/CR mice could treat established cancers in WT mice. Although our results showed that SR/CR mice resisted several lethal murine cancer cell lines, including L5178Y, EL4, and J774 (1), S180 was chosen as the tumor model system for this study. Some unique properties of S180 cells suitable for this study include the ability to uniformly establish aggressive, rapidly lethal malignancy in all inbred strains of WT mice and the ability to form both solid tumors and ascites fluid (14, 15). We established solid tumors ranging from 5 to 10 mm in surface diameter or 300 to 800 mg in weight on the backs of female WT BALB/c nude mice either by direct s.c. injection of S180 cells in suspension or by implantation of S180-induced tumor fragments with stroma from syngeneic donors (16). We extracted leukocytes for the transfers from the spleens and bone marrow of male BALB/c SR/CR or WT donors. Then we divided mice with day-4 tumors (verified by histology of similar tumors for development of vascular networks and invasion into normal tissues) into three groups: (i) recipients that received a single i.p. injection of ≈4 × 108 SR/CR leukocytes from spleens and bone marrow; (ii) controls that received a single i.p. injection of 4 × 108 similarly prepared WT leukocytes; or (iii) controls that received no leukocyte transfer. In both control groups, the s.c. S180 tumors grew both outwardly and inwardly, penetrating through normal tissues into major cavities until the mice became moribund and were euthanized. In the group that received the SR/CR leukocytes, tumors were noticeably larger than those of the control groups in the first 4 days after leukocyte transfer, became noticeably smaller than the control groups for another 7 days, and then regressed completely (9 of 14 tumors) or stabilized (5 of 14 tumors) 3 weeks after the transfer of SR/CR leukocytes (Fig. 4A). Solid tumors often regressed by necrosis from a scab-covered center (no open wound or bleeding), followed by regression of the residual rings and complete healing of scabs (Fig. 4B). Mice with complete regression remained healthy and tumor-free at the time of publication, 10 months after the experiment (Fig. 4C).
Fig. 4.

Transfer of resistance from SR/CR mice to WT mice against established s.c. tumors. s.c. tumors were implanted in WT nude mice and allowed to grow for 4 days. Tumor-bearing mice received AT of SR/CR leukocytes, WT leukocytes, or no leukocytes. (A) Changes in tumor volume after tumor challenge and subsequent AT of leukocytes (n = 10 for SR and n = 8 for WT). The average tumor volume for control mice and mice receiving AT from SR/CR mice was calculated for each day. Control tumor volume was arbitrarily set at 100%, and tumor volume with AT from SR/CR mice was reported as a percentage compared with control. Experiments included five mice with AT of SR/CR leukocytes (■) and four control mice (♦). (B) Images of necrosis of tumor centers and subsequent regression in a single mouse. (C) Survival of the AT recipients and controls. Experiments included seven mice with AT of SR/CR leukocytes (▴) and five control mice (■).
Samples of stable and regressing tumors in the treatment group and growing tumors in control groups were collected for histological examination. Regressing tumors showed necrotic tumor cell cores bordered closely by a layer of infiltrating MΦ. The MΦ were bordered by a layer of infiltrating PMN, which were in turn bordered by a layer of infiltrating plasma cells and other lymphocytes (Fig. 5). Effector cells of the same subsets grouped together during attack on the cancer cells. By histology, the stable tumors showed a massive infiltration of leukocytes and desmoplasia interspersed with occasional live cancer cells (data not shown). In contrast, growing tumors in control groups showed no infiltration of leukocytes at all (data not shown). Staining for the Y chromosome by fluorescent in situ hybridization showed that most infiltrating leukocytes were from the donor SR/CR male cells (Fig. 10, which is published as supporting information on the PNAS web site). Upon continuous observation, no lethality or indication of graft vs. host disease in adult recipients or newborn syngeneic recipients was seen. When surviving recipients were challenged again with i.p. or s.c. injections of S180 cells, they remained resistant to cancer cells.
Fig. 5.

Leukocyte infiltration in established S180 tumors. s.c. tumors were established in WT mice, which then received AT of SR/CR leukocytes. Regressing S180 tumors were sectioned, fixed, and stained with hematoxylin and eosin 24 days after receiving AT of SR/CR leukocytes. (A) Tumor cells are surrounded by MΦ and an outer layer of neutrophils, whereas a surprising number of plasma cells are observed in the outermost layer. (B and C) At higher power, small, scattered islands of surviving tumor cells are observed surrounded by MΦ and PMNs, with plasma cells seen in the periphery. Areas of scar tissue formation with fibroblast proliferation were also noticed. T, S180 tumor; M, MΦ; PMN, neutrophil; p, plasma cell; f, fibroblast. The brackets in C delineate the layers of infiltrating leukocytes.
Transfer of SR/CR Leukocytes Depleted of T and B Lymphocytes into Rag1−/−-Recipient Mice.
One of the greatest challenges confronting cell transfer therapy is the concern about how long the transferred cells remain effective in the recipients. Donor cells from bone marrow and spleen likely contained stem cells expected to engraft and survive indefinitely in the syngeneic recipients. One critical question was how long the transferred resistance could be maintained by terminally differentiated effector cells. To address this question, we used different methods to collect donor cells for transfer and used a simplified model of i.p. cancer cell challenge. We challenged male SR/CR donor mice with S180 cells i.p. to induce peritoneal infiltrating effector cells. The infiltrating cells were retrieved by lavage and injected i.p. into H2-compatible, immune-competent recipient mice. One week after transfer, recipients received a first challenge of 106 S180 cells. With each challenge, control mice receiving no transferred leukocytes or transferred WT leukocytes were also challenged with S180 cells. Recipient mice with SR/CR leukocytes survived multiple challenges with S180 cells when challenged multiple times for up to 12 months after leukocyte transfer, whereas all control mice developed ascites within 2 weeks of challenge. The potency of resulting resistance was proportional to the total number of donor cells regardless of whether the transfer was completed at one time or over multiple times.
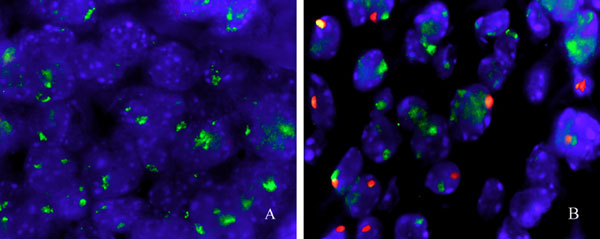
We also determined whether the transferred resistance persisted in the absence of adaptive immune components of both donors and recipients. T and B cells constituting ≈15% of isolated donor cells were removed by using anti-T and -B lymphocyte antibodies conjugated with magnetic beads. The removal of T and B cells was confirmed by immunocytochemistry. Donor cells with <1% T and B lymphocytes, but predominantly composed of MΦ, PMN, and NK, were transferred into Rag1−/−-recipient mice (17), which lacked adaptive immunity. The recipient mice were challenged with S180 cells. The majority of recipients survived multiple challenges, with the final challenge at 12 weeks after AT (Fig. 6). The ability to survive for such an extended period in the absence of adaptive immune components in the donor cells and recipients suggests that the leukocytes of innate immunity were sufficient for long-term resistance. We further validated this result by eliciting MΦ with TG from donor mice. Adaptive immune components were largely absent. The TG-induced MΦ from SR/CR donors, but not WT donors, offered long-term protection against challenge of S180 cells in Rag1−/− mice (Fig. 6). This finding suggests that the differentiated MΦ may have potential to confer long-term survival in new hosts. The mechanism of such long-term survival for innate immune cells is currently unknown. The effective protection of recipients by enriched MΦ is consistent with the results from the in vitro killing assay that showed killing ability in purified populations of SR/CR leukocytes. Male donor cells were identified by fluorescent in situ hybridization for mouse Y chromosome in the cancer infiltrates 3 months after AT into female recipients (Fig. 11, which is published as supporting information on the PNAS web site).
Fig. 6.

Donor cells devoid of adaptive immune components were transferred into recipients deficient in adaptive immune responses. Rag1−/− WT mice received total tumor-induced leukocytes, innate immune cells (T/B depleted), or TG-induced leukocytes (>90% MΦ) from SR/CR donors. The recipient WT mice were then injected i.p. with S180 cells. Survival curves are shown for WT controls and for Rag1−/− recipients after initial S180 challenge.
Discussion
The resistance to cancer cells in SR/CR mice involves the ability of host leukocytes to detect unique signals from cancer cells, to migrate to the cancer site, and to establish tight physical contact, resulting in the rupture of the cancer cells. SR/CR leukocytes apparently rely on a predetermined mechanism for recognizing some signal common to all cancer cell lines tested so far. The concept that the resistance against cancers is mediated by innate immunity is supported by several facts. First, the genetically determined anticancer response required no prior exposure. Second, innate immune cells remained the major responding effector cells during repeated challenges. Third, leukocytes from naïve SR/CR mice displayed killing activity in vitro before any in vivo exposure to cancer cells. Fourth, transfer of highly purified MΦ conferred a similar resistance in WT recipients. In contrast, involvement of transplant rejection for the anticancer response is highly unlikely, because it usually takes at least 2 weeks after the primary exposure to the target.
However, there has been an obvious enhancement of resistance in terms of speed and tumor burden during repeated responses. Despite a lack of evidence that the components of adaptive immunity contributed to the resistance, we still could not completely rule out a possible role for adaptive immunity in the enhancement of anticancer responses.
The independent involvement of MΦ, PMN, or NK in the resistance is somewhat surprising in comparison with the conventional understanding of anticancer immune responses that often implicate one subset of effector cells, such as cytotoxic T lymphocytes or NK. An obvious advantage of such an unconventional but more effective immune strategy is that cancer cells can be killed by multiple effector mechanisms, and the resistant hosts can still be protected in cases of failure in one or more immune components.
The second major finding of this study was that resistance could be entirely transferred to cancer-sensitive mice for both treatment and prevention of malignancy. Even highly aggressive forms of malignancy with large tumor burdens could be completely eradicated systemically. This efficacy in mice suggests that the anticancer strategy in SR/CR mice may point to a future therapeutic direction in which adverse side effects of treatment are minimal. The transferred resistance functioned in both immune-competent (Fig. 12, which is published as supporting information on the PNAS web site) and immune-deficient recipients. Our findings from AT also suggest that effective resistance to transplanted cancer cells in SR/CR mice is mediated entirely by transferable cellular components of the immune system. The successful long-term retention of resistance was achieved in the absence of adaptive immunity of either donor or recipient. In addition to the independent ability to kill S180 cells in vitro, purified MΦ from SR/CR mice could be conveniently transferred to a new host for somewhat unexpected long-term cancer resistance. Currently, the mechanism for this long-term retention of resistance phenotype is unknown. Nevertheless, this result raises the possibility that AT of peripheral leukocytes can be used therapeutically as a long-term intervention for cancer treatment or prevention.
Another observation in this study was that large established solid s.c. tumors in WT mice completely regressed after transfer of SR/CR leukocytes. This finding is remarkable given that reversal of this kind of malignancy has, to our knowledge, never before been reported. This regression took place with or without the presence of stroma. Together, these findings suggest that the resistance has implications in both prevention and treatment of cancers in mice. These findings suggest that the resistance mechanism may recognize a common property when malignancies are presented, locally or distantly, in different forms or different cell types.
Materials and Methods
Cell Lines and Mouse Strains.
S180, CHO, and NIH/3T3 cell lines were purchased from American Type Culture Collection and propagated in DMEM with 10% FCS. BALB/c and BALB/c nude mice were purchased from Charles River Laboratories. C57BL/6 and C57BL/6 Rag1−/− were purchased from The Jackson Laboratory. SR/CR mice (1, 2) were bred at the Animal Research Programs of Wake Forest University School of Medicine. Mice were housed in plastic cages, which were covered with individual air filter tops and contained hardwood shavings as bedding. Mice were allowed free access to water and regular chow and were exposed to a 12-h fluorescent light/dark cycle.
Cytoprep and Histology.
For immunocytochemistry of surface markers, fixed cells in cytopreps were blocked by using anti-mouse IgG Fab and then probed with anti-CD4, -CD8, -CD19, -Ly6G, -F4/80, -NK1.1, or -CD11c. These antibodies were followed by rhodamine-conjugated secondary antibodies and counted with a Zeiss Axioplan2 fluorescence microscope.
Flow Cytometry.
Cells from peritoneal washes were stained with specific antibodies to cell surface markers according to standard procedures recommended by the supplier and analyzed on a FACSCalibur (BD Biosciences, Mountain View, CA). Forward and side scatter gain settings were tuned to sort live cells from cell fragments.
Depletion of Leukocyte Subsets.
For PMN depletion, three i.p. injections of 500 μg of anti-Ly6G (10) were given to each SR/CR mouse at 2-day intervals. For NK depletion, three i.p. injections of 500 μg of anti-NK1.1 antibody (11) were administered at 2-day intervals. For MΦ depletion, 25 mg/kg 2-chloradenosine (2-CAdo) was injected i.p. into SR/CR mice (12). The treated mice were then injected i.p. with 2 × 107 S180.
Isolation of SR/CR Leukocyte Subsets.
PMN were isolated from the SR/CR peritoneal wash by layering mixed cells over Histopaque 1083 (Sigma) and centrifuging at 400 × g for 30 min according to the manufacturer’s instructions. MΦ were isolated by plating the mixed cells of peritoneal wash in culture dishes in DMEM with 10% FCS at 37°C for 1 h. The nonadherent cells were removed from adherent MΦ by rinsing with PBS. NK were isolated by anti-DX5-labeled microbeads (Miltenyi Biotec, Auburn, CA) according to the manufacturer’s protocol. The purified subsets of leukocytes were verified by microscopic examination to have ≥95% purity, according to unique morphology for MΦ, polymorphic nuclei for PMN, and NK1.1 staining and lymphocytic morphology for NK.
In Vitro Assays of Target Cell Killing by Leukocytes.
In vitro killing of S180 cells by leukocytes was performed as described in ref. 1. Briefly, total abdominal leukocytes or specific subsets were incubated with S180 cells labeled with 3,3′-dioctadecyloxacarbocyaninine at a 10:1 ratio. Equal numbers of S180 cells cultured alone in the same conditions served as no-killing controls (100% survival). After 24 h, live S180 cells were identified by size, morphology, and fluorescence. Live S180 cells were characterized by trypan blue exclusion and fluorescence retention. Dead S180 cells were characterized by trypan blue retention and fluorescence loss.
AT of Donor Leukocytes.
SR/CR mice were challenged with i.p. injection of S180 cells. The donor cells were injected i.p. into recipient mice at a donor/recipient ratio of 1:1 for a total of up to eight transfers. Rag1−/− female recipients are unable to mount an adaptive response against sex-mismatched donor cells and therefore received haplotype-compatible leukocytes from male donor mice to facilitate tracking of donor cells in the female recipient. For donor leukocytes depleted of T and B cells, S180-induced leukocytes were incubated with anti-CD90 and anti-CD45R microbeads (Miltenyi Biotec) according to the manufacturer’s instructions. The T/B-depleted leukocytes were transferred to four Rag1−/− recipients for a total of five transfers. SR/CR and WT mice were injected i.p. with 3% TG to elicit MΦ infiltration. Three days after TG elicitation, the infiltrating cells (>90% MΦ) were recovered and transferred to recipient mice for a total of 10 transfers. All recipients were then challenged with 106 S180 cells. Splenocytes were obtained from SR/CR or WT mice and transferred to recipient mice at a 1:1 ratio.
Acquired Resistance to Established Solid Tumor at a Distant Site.
Six-week-old female BALB/c nude mice were used as recipients of cancer cells and subsequent adoptive transfers of leukocytes. Solid s.c. tumors were established by injecting 2 × 107 S180 cells at two sites on the back and allowed to reach diameters of 5–10 mm. Leukocytes were extracted from the spleens and femur bone marrow of donor mice, according to standard protocols. Four days after tumor cell transplantation, isolated donor leukocytes were transferred i.p. with a ratio of two donors to one recipient. Each recipient received ≈4 × 108 leukocytes, which accounted for 80% of the estimated endogenous leukocytes in a mouse. Tumor volume (V) was calculated by using the formula V = (W× W× L)/2, where W was the shorter of the two diameters and L was the longer one. Tumor volume was reported as the mean ± SEM. Moribund mice with tumors were euthanized according to Institutional Animal Care and Use Committee guidelines. For histological evaluation of stabilized tumors, the remaining tumor was removed 28 days after tumor injection (24 days after AT) from the euthanized mouse and fixed in 3.7% formaldehyde overnight. Tumors were embedded in paraffin, sectioned, and stained with hematoxylin and eosin. Data presented are representative of results observed from four samples.
Identification of Male Donor Cells in Female Recipients by XY Fluorescent in Situ Hybridization.
Fragments of stable, regressing, and progressing tumors were embedded in paraffin, and sections were mounted on glass slides and deparaffinized. S180-induced peritoneal cells were fixed to slides. Slides were processed according to standard cytogenetic methodology and codenatured with mouse XY whole-chromosome probes (Vysis). The X chromosome probe was directly labeled with FITC (green) and was mixed 1:1 with the Y probe labeled with Cy3 (red). Finally, slides were counterstained in DAPI.
Supplementary Material
Acknowledgments
This work was supported by grants from the Cancer Research Institute, the National Cancer Institute, and the Charlotte Geyer Foundation (to Z.C.). G.R. is supported by National Cancer Institute Training Grant CA79448.
Abbreviations
- AT
adoptive cell transfer
- CR
complete resistance
- MΦ
macrophages
- NK
natural killer cell(s)
- PMN
polymorphonuclear leukocyte(s)
- SR
spontaneous regression.
Footnotes
Conflict of interest statement: No conflicts declared.
References
- 1.Cui Z., Willingham M. C., Hicks A. M., Alexander-Miller M. A., Howard T. D., Hawkins G. A., Miller M. S., Weir H. M., Du W., DeLong C. J. Proc. Natl. Acad. Sci. USA. 2003;100:6682–6687. doi: 10.1073/pnas.1031601100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cui Z. Cancer Immun. 2003;3:14. [PubMed] [Google Scholar]
- 3.Morris E., Hart D., Gao L., Tsallios A., Xue S. A., Stauss H. Blood Rev. 2006;20:61–69. doi: 10.1016/j.blre.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 4.Mocellin S., Rossi C. R., Lise M., Marincola F. M. Cancer Immunol. Immunother. 2002;51:583–595. doi: 10.1007/s00262-002-0308-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Knutson K. L., Almand B., Mankoff D. A., Schiffman K., Disis M. L. Expert Opin. Biol. Ther. 2002;2:55–66. doi: 10.1517/14712598.2.1.55. [DOI] [PubMed] [Google Scholar]
- 6.Locatelli F., Comoli P., Montagna D., Rossi F., Daudt L., Maccario R. Best Pract. Res. Clin. Haematol. 2004;17:479–492. doi: 10.1016/j.beha.2004.06.0054. [DOI] [PubMed] [Google Scholar]
- 7.Ruggeri L., Mancusi A., Capanni M., Marelli M. F., Velardi A. Curr. Opin. Immunol. 2005;17:211–217. doi: 10.1016/j.coi.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 8.Chang A. E., Shu S. Crit. Rev. Oncol. Hematol. 1996;22:213–228. doi: 10.1016/1040-8428(96)00194-1. [DOI] [PubMed] [Google Scholar]
- 9.Overwijk W. W. Curr. Opin. Immunol. 2005;17:187–194. doi: 10.1016/j.coi.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 10.Conlan J. W., North R. J. J. Exp. Med. 1994;179:259–268. doi: 10.1084/jem.179.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koo G. C., Dumont F. J., Tutt M., Hackett J., Jr., Kumar V. J. Immunol. 1986;137:3742–3747. [PubMed] [Google Scholar]
- 12.Kikkawa H., Imafuku H., Tsukada H., Oku N. FEBS Lett. 2000;146:211–216. doi: 10.1016/s0014-5793(00)01144-3. [DOI] [PubMed] [Google Scholar]
- 13.Leijh P. C. J., van Zwet T. L., ter Kuile M. N., van Furth R. Infect. Immun. 1984;46:448–452. doi: 10.1128/iai.46.2.448-452.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tarnowski G. S., Mountain I. M., Stock C. C. Cancer Res. 1973;33:1885–1888. [PubMed] [Google Scholar]
- 15.Alfaro G., Lomeli C., Ocadiz R., Ortega V., Barrera R., Ramirez M., Nava G. Vet. Immunol. Immunopathol. 1992;30:385–398. doi: 10.1016/0165-2427(92)90107-2. [DOI] [PubMed] [Google Scholar]
- 16.Singh S., Ross S. R., Acena M., Rowley D. A., Shreiber H. J. Exp. Med. 1992;175:139–146. doi: 10.1084/jem.175.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mombaerts P., Iacomini J., Johnson R. S., Herrup K., Tonegawa S., Papaioannou V. E. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}