

Abstract
To investigate whether genetic alteration of the STK11/LKB1 tumor-suppressor gene is involved in the carcinogenesis of head and neck squamous cell carcinoma (HNSCC), the entire encoding exons and flanking intronic sequences of the STK11/LKB1 gene were analyzed with direct genomic sequencing of fifteen HNSCC specimens. A novel missense mutation with presumed loss of heterozygosity (LOH) and ten polymorphisms were identified in these samples. The novel mutation of STK11/LKB1 at nucleotide position 613 G → A, which causes the amino acid substitution from alanine to threonine at residue 205 within the catalytic kinase domain, was identified in cell line RPMI2650. To further determine whether this point mutation affects the gene function, constructs of the wild-type and A205T mutant of the STK11/LKB1 gene expression vectors were created and transfected into RPMI2650 cells. Our results showed that the reintroduction of the wild-type but not the mutant STK11/LKB1 construct into RPMI2650 cells induced suppression of the cell growth. The mutation also affected the kinase activity of the Stk11/Lkb1 protein. This led us to conclude that the A205T point mutation of the STK11/LKB1 gene produces functionally inactive proteins. This is the first described mutation of the STK11/LKB1 gene in HNSCC. While the mutation frequency of the STK11/LKB1 gene in HNSCC remains to be determined in future studies, our data strongly suggests that STK11/LKB1 is involved in the carcinogenesis of HNSCC.
Keywords: HNSCC, PJS, STK11, LKB1, genetic mutation, tumor-suppressor gene
The abbreviations used are: HNSCC, Head and neck squamous cell carcinoma; PJS, Peutz-Jeghers syndrome; LOH, loss of heterozygosity; HA, hemagglutinin; HRP, horseradish peroxidase
INTRODUCTION/RESULTS/DISCUSSION
STK11 (serine/threonine kinase 11), also named LKB1, is located on chromosome 19p13.3 (Hemminki et al., 1997). STK11/LKB1 was identified because germline mutations of the gene are responsible for Peutz–Jeghers syndrome (PJS) (Hemminki et al., 1998; Jenne et al., 1998). PJS is an autosomal dominant syndrome characterized by germline mutations in STK11/LKB1, the development of multiple PJS polyps in the gastrointestinal tract and a markedly increased risk of developing malignant tumors derived mostly from epithelial origin (Giardiello et al., 2000; Giardiello et al., 1987).
The human Stk11/Lkb1 protein comprises 433 residues with a kinase domain at residues 44–319. The Stk11/Lkb1protein is ubiquitously expressed in all human fetal and adult tissues (Rowan et al., 2000). Homozygous deletion of STK11/LKB1 in mice leads to embryonic lethality at midgestation (E11.0), indicating that STK11/LKB1 plays an important role in embryogenesis (Jishage et al., 2002; Ylikorkala et al., 2001). Interestingly, most of the STK11/LKB1+/− mice developed intestinal polyps by the age of 45 weeks, identical to those observed in patients with PJS (Bardeesy et al., 2002; Jishage et al., 2002; Nakau et al., 2002). These results suggested that haploinsufficiency in the STK11/LKB1+/− animals is sufficient to induce polyposis as in humans (Rossi et al., 2002; Su et al., 1999). Tumors only arise in PJS patients when the remaining wild-type allele of STK11/LKB1 is also inactivated (Su et al., 1999).
Biallelic inactivation of the STK11/LKB1 gene is also observed in a small percentage of sporadic cancers (Avizienyte et al., 1999; Avizienyte et al., 1998; Chen et al., 1999; Guldberg et al., 1999). For example, we previously reported germline and somatic mutations of the STK11/LKB1 gene in 4–6% of pancreatic and biliary cancers (Su et al., 1999). Mutation frequency of the STK11/LKB1 gene in sporadic lung adenocarcinomas has been reported at 8.3–28% (Avizienyte et al., 1999; Ghaffar et al., 2003; Sanchez-Cespedes et al., 2002).
A novel mutation of STK11/LKB1 gene was identified in HNSCC
To investigate whether the STK11/LKB1 gene is mutated in head and neck cancer, the entire encoding exons and flanking intronic sequences of STK11/LKB1 gene were analyzed by PCR amplification and direct sequencing from the genomic DNA of fifteen HNSCC specimens including eight cell lines (RPMI 2650, FaDu, SW579, SCC-15, CAL 27, SCC-25, A-253, Detroit 562). A missense mutation of STK11/LKB1 at nucleotide 613 from G to A, leading to an amino acid change at codon 205 in exon 5 (alanine (GCG) substituted by threonine (ACG)), was identified in a HNSCC cell line RPMI 2650 (Fig. 1A. and Table 1). LOH was presumed because the mutation was observed in the absence of a wild-type allele. To further confirm this point mutation and the LOH status, an independent PCR product from the original genomic template was cloned into pGEM-T easy vector and transformed into bacteria. Six bacterial colonies with DNA inserts were randomly chosen for sequencing. Only sequences with the A205T point mutation were obtained in all six clones. Therefore 100% (6/6) of the DNA sequences were mutant; the wild-type sequences were completely absent. A search of the mutational databases for STK11/LKB1 revealed that this mutation was novel and had not been described before. This is also the first report demonstrating that STK11/LKB1 gene mutations arise in HNSCC.
Fig. 1. Genetic alterations identified in STK11LKB1 exons in 15 HNSCC specimens.
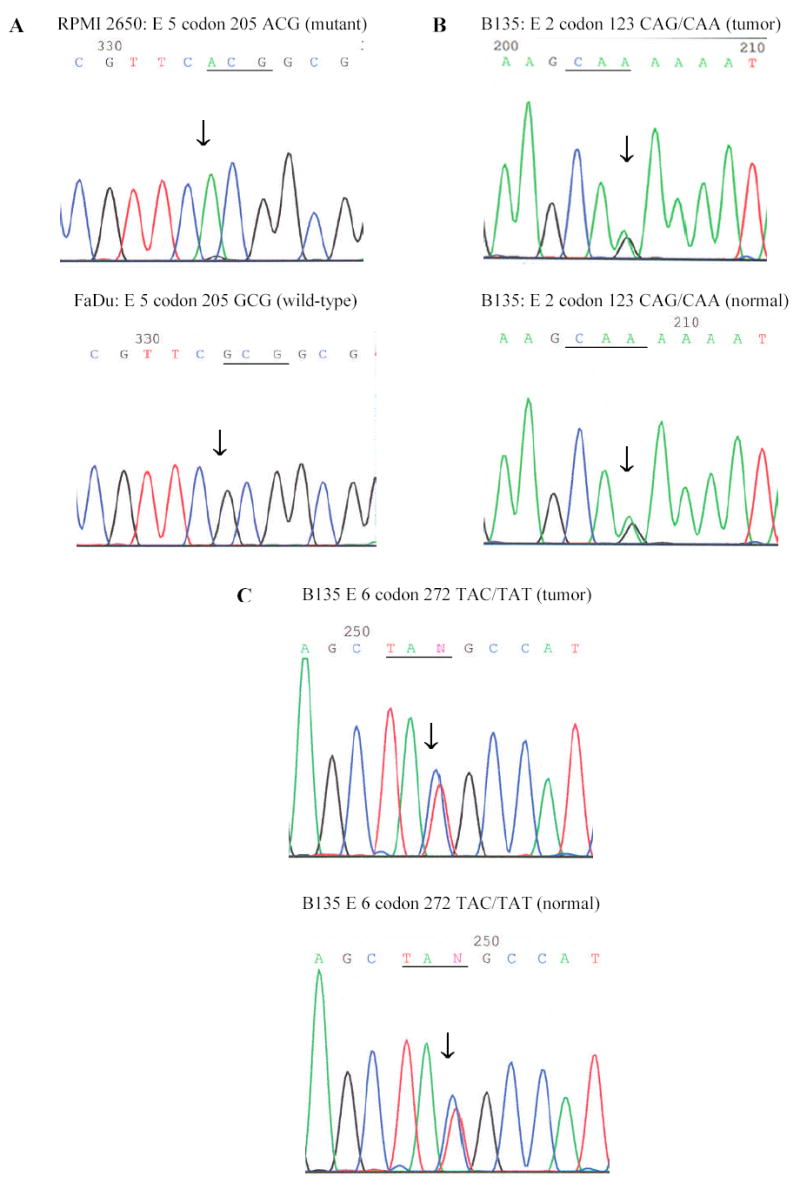
Genomic sequences are presented in the 5’ to 3’ direction and the location of the mutation is marked by an arrow. Seven primary HNSCC tumors and matching normal tissues were collected from the Tumor Bank facility of the Herbert Irving Comprehensive Cancer Center at Columbia University. Fresh-frozen tumor samples were meticulously dissected to ensure that the specimen contained at least 75% cancer cells. The eight HNSCC cancer cell lines were obtained from ATCC. Genomic DNAs from the fifteen HNSCC specimens were amplified for the entire encoding sequences and flanking intronic sequences of STK11/LKB1 with eight sets of primers and then subject to direct sequencing. All samples found to have a mutation were sequenced in the reverse direction as the initial confirmation of the mutation. Subsequent verification of the mutation was accomplished by sequencing of a second PCR product derived independently from the original template. A, RPMI 2650 cell line: a missense mutation in codon 205 GCG (Ala) → ACG (Thr); FaDu cell line: wild-type. B, Patient B135: Tumor tissue, a heterozygous silent mutation in Exon 2 code 123 CAG (Gln) → CAA (Gln); Normal tissue: the same variant. C, Patient B135: Tumor tissue, a heterozygous silent mutation in exon 6 codon 272 TAC (Tyr) → TAT (Tyr); Normal tissue: the same variant.
Table 1.
Genetic variants identified in the exons of STK11/LKB1
| Sample | Tissue | Codon* | Exon | Nucleotide change | Amino acid change | Presence of wildtype allele | Type of alteration |
|---|---|---|---|---|---|---|---|
| RPMI 2650 | nose | 205 | E5 | GCG → ACG | Ala → Thr | no | N/A |
| B135 | larynx | 272 | E6 | TAC → TAT | silent | yes | germline |
| 123 | E2 | CAG → CAA | silent | yes | germline | ||
| B182 | tongue | 88 | E1 | ATC → ATA | silent | yes | germline |
| 2221 | oral | 88 | E1 | ATC → ATA | silent | yes | germline |
| 3017 | nose | 88 | E1 | ATC → ATA | silent | yes | germline |
Numbering according to GenBank accession number: AF035625
Reintroduction of the wild-type STK11/LKB1 but not mutant STK11/LKB1 into RPMI 2650 cells induced cell growth suppression
Because the variant was identified in a cell line, the possibility of the variant being a rare polymorphism could not be excluded. A comprehensive search of the NCBI SNP database showed that our mutation is not a known polymorphism. Although this still does not completely rule out the possibility, together with the fact that the A205T alteration is localized to the catalytic kinase domain of the Stk11/Lkb1 protein, it is unlikely that the alteration is a polymorphism. We chose the colony formation assay to examine whether the A205T mutation of STK11/LKB1 affects its tumor-suppressor function. RPMI 2650 cells were transfected with pcDNA3 empty vector alone, pcDNA3 wild-type STK11/LKB1-hemagglutinin (HA) vector, or with pcDNA3 A205T-mutant STK11/LKB1-HA vector, respectively. First, to evaluate the amounts of wild-type and mutant Stk11/Lkb1 proteins expressed in the transfected RPMI 2650 cells, we performed western blotting with anti-HA antibody two days after the initial transfection (Fig. 2A). The amount of exogenous protein expressed in the cells transfected with the wild-type STK11/LKB1 vector was similar to that transfected with the mutant STK11/LKB1. The anti-β-actin western blotting assay showed the amount of proteins was equally loaded in each lane (Fig. 2A).
Fig. 2. Mutation at the codon 205 of the Stk11/Lkb1 protein led to the loss of its cell growth inhibition function.
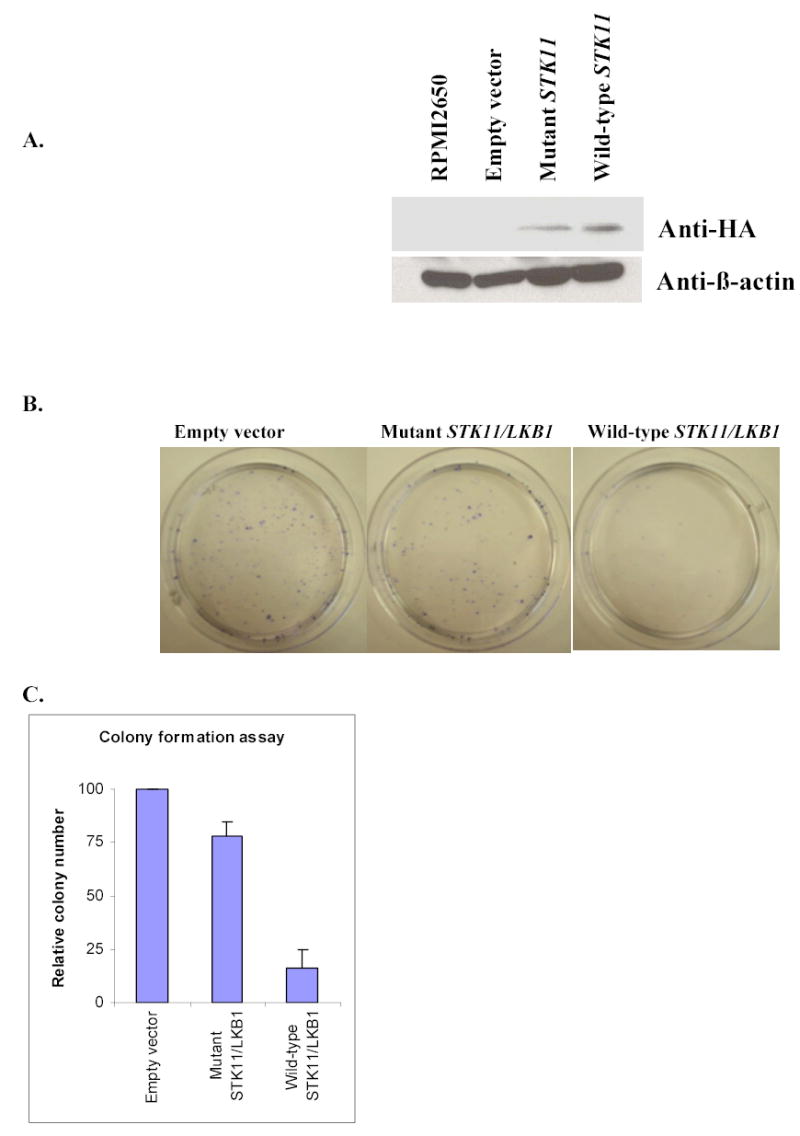
A, Western blotting analysis of RPMI 2650 cells transiently transfected with expression vectors carrying STK11/LKB1 wild-type-HA, A205T mutant-HA, or empty vector respectively. Twenty-five μg of total protein was resolved on 10%SDS-PAGE gel and transferred onto nitrocellulose membrane. Detection was performed using primary antibody mouse monoclonal anti-β-actin (Sigma) or mouse monoclonal anti-HA antibody (Roche) at 1:5000 and 1:500 respectively. The secondary antibody goat anti-mouse IgG-HRP (Sigma) was used at a final dilution of 1:5000. RPMI 2650 without transfection was loaded in first lane as an endogenous Stk11/Lkb1 protein control. Anti-β-actin bands showed that the proteins of four samples were loaded equally. Anti-HA bands showed that the ectopic protein expression level from the mutant STK11/LKB1 vector was equal to that of the wild-type in transfected RPMI 2650 cells. B, Giemsa-stained G418-resistant colonies of the RPMI 2650 cells transfected with indicated expression vectors. RPMI 2650 cells were cultured to 60–70% confluence in 10-cm dishes and transfected respectively with 1 mg of the wild-type Stk11/Lkb1, A205T mutant, or pcDNA3 empty vector using Fugene-6 transfection reagent (Roche). After 48 h transfection, G418 was added to the medium to give a final concentration of 2 mg/ml. After 18–20 day’s selection, the cells were stained with Giemsa, and the average number of the colonies present in each dish was photographed and counted. C, Relative numbers of G418-resistant colonies after transfection with indicated vectors, represented as the percentage of colonies with respect to the control (pcDNA3 empty vector). Standard deviations are derived from four independent experiments.
Eighteen days after the initial transfection and selection with G418, a significant reduction in the number of colonies expressing wild-type Stk11/Lkb1 was observed in comparison with those expressing mutant Stk11/Lkb1 (p<0.05) and those carrying the empty vector (p<0.05) (Fig. 2B). This shows that ectopic overexpression of the wild-type Stk11/Lkb1 strongly inhibited growth of RPMI 2650 cells. Thus, reintroduction of the wild-type but not mutant Stk11/Lkb1 into RPMI2650 cells suppresses cell growth. The results indicate that the A205T point mutation abrogates Stk11/Lkb1 growth suppression function and indicates that STK11/LKB1 gene is a tumor suppressor gene in head and neck cells.
A205T mutation of the Stk11/Lkb1 protein led to its reduced kinase activity
Stk11/Lkb1 is an upstream regulator of AMP-activated protein kinase (AMPK) (Hawley et al., 2003; Hong et al., 2003). AMPK is a sensor of cellular energy that is conserved throughout eukaryotes. AMPK is activated by stimuli that increase the cellular AMP/ATP ratio, including H2O2, AICAR, and sorbitol. Stk11/Lkb1 has been shown to directly phosphorylate Thr-172 of AMPKalpha in vitro and activates its kinase activity (Hawley et al., 2003; Shaw et al., 2004b). To examine how the A205T mutation may affect the kinase activity of Stk11/Lkb1, AMPK phosphorylation was evaluated in RPMI 2650 cells transfected with pcDNA3 wild-type STK11/LKB1-HA vector and with pcDNA3 A205T-mutant STK11/LKB1-HA vector. Diminished AMPK phosphorylation at the Thr-172 was observed in H2O2-treated cells overexpressing mutant Stk11/Lkb1 (Figure 3). The expression level of non-phosphorylated AMPK remained unchanged. The expression level of wild-type and mutant Stk11/Lkb1 were comparable. The Thr-172 phosphorylation of AMPK was not completely abolished in the presence of mutant Stk11/Lkb1 because Stk11/Lkb1 is the dominant but not the only upstream regulator of AMPK (Shaw et al., 2004b; Suzuki et al., 2004). This data demonstrated that the A205T mutation also disrupted the kinase activity of the Stk11/Lkb1 protein.
Fig 3. Mutant Stk11/Lkb1 displayed reduced kinase activity in activating its down stream target, AMPK, in RPMI 2650 cells.
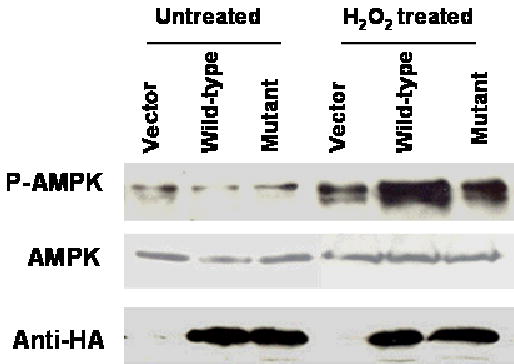
RPMI 2650 cell transfected with empty vector, wild-type or mutant Stk11/Lkb1 were treated with or without 0.1 mM H2O2 for 20 min. Western blotting was performed as previously described. Total cell extracts were immunoblotted with antibodies for phosphor-Thr-172 AMPK (p-AMPK) (Cell Signaling, 1:1,000 dilution), total AMPK (Cell Signaling, 1:1,000 dilution) and Stk11 (Cell Signaling, 1:1,000 dilution).
Multiple exonic and intronic polymorphisms were observed
Three exonic and seven intronic polymorphisms were also identified in our study (Figure 1, B, C, Table 1 and 2). The three exonic polymorphisms are silent mutations and heterozygous in the normal and tumor samples. These three polymorphic variants were identified respectively in the exon 1 codon 88 (Ile), exon 2 codon 123 (Gln), and exon 6 codon 272 (Tyr). They represent single nucleotide polymorphisms (Fig. 1, B, C, and Table 1). The exon 1 ATC88ATA and exon 6 TAC272TAT are previously described SNPs (Sanchez-Cespedes et al., 2002), while exon 2 CAG123CAA is a novel SNP. Five of the seven intronic polymorphic variants observed in the study, occurred in the intron 1 (+36 G → T), intron 2 (+24 G → C, −49 G → A), intron 3 (−51 T → C) and intron 7 (+7 G → C), have been reported previously (Avizienyte et al., 1998; Rowan et al., 1999; Su et al., 1999; Westerman et al., 1999). The other 2 polymorphic changes in the intron 1 (−32 C → T), and intron 4 (−16 C → G) are not noted in previous studies (Table 2).
Table 2.
Polymorphic changes of STK11/LKB1 found in the introns in 15 HNSCCs
| Intron | Nucleotide position♣ | Polymorphic change | Genotype (number of samples) |
|---|---|---|---|
| 1 | +36 | G → T | Homozygous (2) Heterozygous (2) |
| 1 | −32 | C → T | Heterozygous (1) |
| 2 | +24 | G → T | Homozygous (2) Heterozygous (5) |
| 2 | −49 | G → A | Homozygous (5) |
| 3 | −51 | T → C | Homozygous (2) Heterozygous (2) |
| 4 | −16 | C → G | Heterozygous (1) |
| 7 | +7 | G → C | Homozygous (1) Heterozygous (1) |
On contrast to our findings, a previous report failed to identify STK11/LKB1 mutations in 16 laryngeal cancer samples analyzed by single strand conformation polymorphism (SSCP) analysis (Chen et al., 1999). The reasons for the differences in our result and this prior study may be partly due to the relatively low mutation rate in head and neck tumors. In addition, the previous study only focused on laryngeal squamous cell carcinoma. Third, the sensitivity of the SSCP as a mutational screening method is only at 70% to 80% (Jordanova et al., 1997).
Interestingly the STK11/LKB1 gene abnormalities were detected only in adenocarcinomas of the lung, not in squamous cell carcinomas, in a panel of lung cancers (Sanchez-Cespedes et al., 2002). Some previous studies also show that the malignant tumors from PJS are commonly adenocarcinoma but not squamous cell carcinoma (Giardiello et al., 1987). Our finding appears to demonstrate that the STK11/LKB1 gene mutation is also implicated in the development of squamous cell carcinoma.
To date, 145 distinct mutations have been described in PJS patients and 21% of them (30/145) are missense mutations (Launonen, 2005). Interestingly, all except one of these missense mutations are located in the kinase domain (Launonen, 2005). A total of 40 different somatic STK11/LKB1 mutations have been published to date and 45% (18/40) of that are missense mutations (Launonen, 2005). Similar to the germline cases, the majority (78% or 14/18) of the missense mutations observed in sporadic cancers are localized to the kinase domain of the STK11/LKB1 gene (Launonen, 2005). Taken together, missense alterations detected in the kinase domain must be functionally significant to the Stk11/Lkb1 protein. Three of the germline mutants found in the catalytic kinase domain have been further examined for the Stk11/Lkb1 protein function (Boudeau et al., 2003; Tiainen et al., 1999). A missense mutation G163D of the STK11/LKB1 gene was shown to cause the loss of its cell growth suppression function in a previous study (Tiainen et al., 1999). And another study also showed two other missense mutations I177N and R304W of the STK11/LKB1 gene also led to the loss of its cell growth inhibition function (Boudeau et al., 2003). All these three missense mutations occur in the catalytic kinase domain of Stk11/Lkb1. The novel missense mutation A205T of STK11/LKB1 identified in the current study is also located at the catalytic kinase region of Stk11/Lkb1. Here we showed that the mutation affected the growth suppression function and disrupted the kinase activity of the Stk11/Lkb1 protein. It is consistent that this novel mutation also leads to the loss of STK11/LKB1 gene function.
The detailed mechanism of STK11/LKB1 gene function as a tumor suppressor is still not fully understood. In additional to its role in AMPK-regulated energy metabolism, the STK11/LKB1 gene is implicated in a variety of cellular processes, including the control of cell cycle arrest (Tiainen et al., 1999), p53-mediated apoptosis (Karuman et al., 2001), Wnt signaling (Ossipova et al., 2003; Spicer et al., 2003), TGF-β signaling (Bardeesy et al., 2002), ras induced cell transformation (Smith et al., 2001), and cell polarity (Lizcano et al., 2004). Our findings suggest that the cell growth inhibition function and AMPK-regulation of the Stk11/Lkb1 protein are important in head and neck cancers.
Acknowledgments
This work was supported by the NCI Temin Award CA95434 and the NCI R01 CA109525.
References
- Avizienyte E, Loukola A, Roth S, Hemminki A, Tarkkanen M, Salovaara R, Arola J, Butzow R, Husgafvel-Pursiainen K, Kokkola A, Jarvinen H, Aaltonen LA. Am J Pathol. 1999;154:677–81. doi: 10.1016/S0002-9440(10)65314-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avizienyte E, Roth S, Loukola A, Hemminki A, Lothe RA, Stenwig AE, Fossa SD, Salovaara R, Aaltonen LA. Cancer Res. 1998;58:2087–90. [PubMed] [Google Scholar]
- Bardeesy N, Sinha M, Hezel AF, Signoretti S, Hathaway NA, Sharpless NE, Loda M, Carrasco DR, DePinho RA. Nature. 2002;419:162–7. doi: 10.1038/nature01045. [DOI] [PubMed] [Google Scholar]
- Boudeau J, Kieloch A, Alessi DR, Stella A, Guanti G, Resta N. Hum Mutat. 2003;21:172. doi: 10.1002/humu.9112. [DOI] [PubMed] [Google Scholar]
- Chen RW, Avizienyte E, Roth S, Elivo I, Makitie AA, Aaltonen LM, Aaltonen LA. J Med Genet. 1999;36:943–4. [PMC free article] [PubMed] [Google Scholar]
- Ghaffar H, Sahin F, Sanchez-Cepedes M, Su GH, Zahurak M, Sidransky D, Westra WH. Clin Cancer Res. 2003;9:2998–3003. [PubMed] [Google Scholar]
- Giardiello FM, Brensinger JD, Tersmette AC, Goodman SN, Petersen GM, Booker SV, Cruz-Correa M, Offerhaus JA. Gastroenterology. 2000;119:1447–53. doi: 10.1053/gast.2000.20228. [DOI] [PubMed] [Google Scholar]
- Giardiello FM, Welsh SB, Hamilton SR, Offerhaus GJA, Gittelsohn AM, Booker SV, Krush AJ, Yardley JH, Luk GD. N England J Med. 1987;316:1511–4. doi: 10.1056/NEJM198706113162404. [DOI] [PubMed] [Google Scholar]
- Guldberg P, thor Straten P, Ahrenkiel V, Seremet T, Kirkin AF, Zeuthen J. Oncogene. 1999;18:1777–80. doi: 10.1038/sj.onc.1202486. [DOI] [PubMed] [Google Scholar]
- Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, Bignell G, Warren W, Aminoff M, Hoglund P, Jarvinen H, Kristo P, Pelin K, Ridanpaa M, Salovaara R, Toro T, Bodmer W, Olschwang S, Olsen AS, Stratton MR, de la Chapelle A, Aaltonen LA. Nature. 1998;391:184–7. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- Hemminki A, Tomlinson I, Markie D, Jarvinen H, Sistonen P, Bjorkqvist AM, Knuutila S, Salovaara R, Bodmer W, Shibata D, de la Chapelle A, Aaltonen LA. Nature Genetics. 1997;15:87–90. doi: 10.1038/ng0197-87. [DOI] [PubMed] [Google Scholar]
- Hong SP, Leiper FC, Woods A, Carling D, Carlson M. Proc Natl Acad Sci U S A. 2003;100:8839–43. doi: 10.1073/pnas.1533136100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenne DE, Reimann H, Nezu J, Friedel W, Loff S, Jeschke R, Muller O, Back W, Zimmer M. Nature Genetics. 1998;18:38–43. doi: 10.1038/ng0198-38. [DOI] [PubMed] [Google Scholar]
- Jishage K, Nezu J, Kawase Y, Iwata T, Watanabe M, Miyoshi A, Ose A, Habu K, Kake T, Kamada N, Ueda O, Kinoshita M, Jenne DE, Shimane M, Suzuki H. Proc Natl Acad Sci U S A. 2002;99:8903–8. doi: 10.1073/pnas.122254599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordanova A, Kalaydjieva L, Savov A, Claustres M, Schwarz M, Estivill X, Angelicheva D, Haworth A, Casals T, Kremensky I. Hum Mutat. 1997;10:65–70. doi: 10.1002/(SICI)1098-1004(1997)10:1<65::AID-HUMU9>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Karuman P, Gozani O, Odze RD, Zhou XC, Zhu H, Shaw R, Brien TP, Bozzuto CD, Ooi D, Cantley LC, Yuan J. Mol Cell. 2001;7:1307–19. doi: 10.1016/s1097-2765(01)00258-1. [DOI] [PubMed] [Google Scholar]
- Launonen V. Hum Mutat. 2005;26:291–7. doi: 10.1002/humu.20222. [DOI] [PubMed] [Google Scholar]
- Lizcano JM, Goransson O, Toth R, Deak M, Morrice NA, Boudeau J, Hawley SA, Udd L, Makela TP, Hardie DG, Alessi DR. Embo J. 2004;23:833–43. doi: 10.1038/sj.emboj.7600110. Epub 2004 Feb 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakau M, Miyoshi H, Seldin MF, Imamura M, Oshima M, Taketo MM. Cancer Res. 2002;62:4549–53. [PubMed] [Google Scholar]
- Ossipova O, Bardeesy N, DePinho RA, Green JB. Nat Cell Biol. 2003;5:889–94. doi: 10.1038/ncb1048. [DOI] [PubMed] [Google Scholar]
- Rossi DJ, Ylikorkala A, Korsisaari N, Salovaara R, Luukko K, Launonen V, Henkemeyer M, Ristimaki A, Aaltonen LA, Makela TP. Proc Natl Acad Sci U S A. 2002;99:12327–32. doi: 10.1073/pnas.192301399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan A, Bataille V, MacKie R, Healy E, Bicknell D, Bodmer W, Tomlinson I. J Invest Dermatol. 1999;112:509–11. doi: 10.1046/j.1523-1747.1999.00551.x. [DOI] [PubMed] [Google Scholar]
- Rowan A, Churchman M, Jefferey R, Hanby A, Poulsom R, Tomlinson I. J Pathol. 2000;192:203–6. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH686>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Sanchez-Cespedes M, Parrella P, Esteller M, Nomoto S, Trink B, Engles JM, Westra WH, Herman JG, Sidransky D. Cancer Res. 2002;62:3659–62. [PubMed] [Google Scholar]
- Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, Cantley LC. Cancer Cell. 2004a;6:91–9. doi: 10.1016/j.ccr.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. Proc Natl Acad Sci U S A. 2004b;101:3329–35. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DP, Rayter SI, Niederlander C, Spicer J, Jones CM, Ashworth A. Hum Mol Genet. 2001;10:2869–77. doi: 10.1093/hmg/10.25.2869. [DOI] [PubMed] [Google Scholar]
- Spicer J, Rayter S, Young N, Elliott R, Ashworth A, Smith D. Oncogene. 2003;22:4752–6. doi: 10.1038/sj.onc.1206669. [DOI] [PubMed] [Google Scholar]
- Su GH, Hruban RH, Bansal RK, Bova GS, Tang DJ, Shekher MC, Westerman AM, Entius MM, Goggins M, Yeo CJ, Kern SE. Am J Pathol. 1999;154:1835–40. doi: 10.1016/S0002-9440(10)65440-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Kusakai G, Kishimoto A, Shimojo Y, Ogura T, Lavin MF, Esumi H. Biochem Biophys Res Commun. 2004;324:986–92. doi: 10.1016/j.bbrc.2004.09.145. [DOI] [PubMed] [Google Scholar]
- Tiainen M, Ylikorkala A, Makela TP. Proc Natl Acad Sci U S A. 1999;96:9248–51. doi: 10.1073/pnas.96.16.9248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerman AM, Entius MM, Boor PP, Koole R, de Baar E, Offerhaus GJ, Lubinski J, Lindhout D, Halley DJ, de Rooij FW, Wilson JH. Hum Mutat. 1999;13:476–81. doi: 10.1002/(SICI)1098-1004(1999)13:6<476::AID-HUMU7>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Ylikorkala A, Rossi DJ, Korsisaari N, Luukko K, Alitalo K, Henkemeyer M, Makela TP. Science. 2001;293:1323–6. doi: 10.1126/science.1062074. [DOI] [PubMed] [Google Scholar]