

Abstract
Water oxidation at photosystem II Mn-cluster is mediated by the redox-active tyrosine YZ. We calculated the redox potential (Em) of YZ and its symmetrical counterpart YD, by solving the linearized Poisson-Boltzmann equation. The calculated Em(Y˙/Y−) were +926 mV/+694 mV for YZ/YD with the Mn-cluster in S2 state. Together with the asymmetric position of the Mn-cluster relative to YZ/D, differences in H-bond network between YZ (YZ/D1-His190/D1-Asn298) and YD (YD/D2-His189/D2-Arg294/CP47-Glu364) are crucial for Em(YZ/D). When D1-His190 is protonated, corresponding to a thermally activated state, the calculated Em(YZ) was +1216 mV, which is as high as the Em for PD1/D2. We observed deprotonation at CP43-Arg357 upon S-state transition, which may suggest its involvement in the proton exit pathway. Em(YD) was affected by formation of 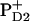 (but not
(but not 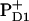 ) and sensitive to the protonation state of D2-Arg180. This points to an electrostatic link between YD and PD2.
) and sensitive to the protonation state of D2-Arg180. This points to an electrostatic link between YD and PD2.
INTRODUCTION
O2 on earth is generated by water oxidation at the Mn-cluster of the photosynthetic protein-pigment complex, photosystem II (PSII). The photosynthetic reaction in PSII is initialized by light absorption, resulting in electronic excitation that is ultimately converted to chemical potential by a charge separation process at the P680 chlorophyll a (Chla) of the PSII reaction center (RC). Charge separation leads to formation of an oxidized positively charged radical P680+˙ while the released electron travels along the electron transfer (ET) chain. The ET chain is located in the D1/D2-chain harboring a quasi-dimer Chla (PD1/D2), and pairs of accessory Chla (ChlD1/D2), of pheophytina (PheoD1/D2) and of plastoquinone (QA/B). In intact PSII, P680+˙ is reduced by the redox-active tyrosine D1-Tyr161 (YZ), which is subsequently reduced by an electron from the Mn-cluster. An overview of the location of these cofactors in the complete PSII protein complex is shown in Fig. 1.
FIGURE 1.

Location of the Mn-cluster (manganese, dark-shaded spheres; calcium, light-shaded sphere), YZ, YD (side chains only, light-shaded spheres for oxygen), and four Chla PD1/D2 and ChlD1/D2 in the reaction center (dark-shaded) of PSII. Polypeptide chains are shown by ribbon bands.
Sequential excitations of P680 drive the redox state of the Mn-cluster from the lowest S0 to the highest oxidized state S4. Release of O2 as a product of water oxidation is associated with transition from S4 to S0 that completes the redox cycle. YZ possesses D1-His190 while the symmetrical counterpart D2-Tyr160 (YD) possesses D2-His189 as H-bond partner (Fig. 2). The apparent proximity of YZ to the Mn-cluster (edge-to-edge distance between Mn-cluster and YZ is 5 Å (1)) and its redox-activity supports its significant role in water oxidation at the Mn-cluster. It is a matter of debate whether the role of YZ in water oxidation is to function as hydrogen abstractor (2) or electrostatic promoter (3). The exit pathway of protons released upon water oxidation (4) as supported by the recent crystal structure of PSII (1,5,6) is a new aspect to be considered. The other tyrosine YD is 28 Å from the Mn-cluster (1) and can therefore not directly be involved in water oxidation occurring at the Mn-cluster. Nevertheless, it plays an important role in tuning the energy of the charge state of P680 and of the S-states of the Mn-cluster before ET from YZ to P680, as has been discussed (7–9).
FIGURE 2.

H-bond network around YZ/D. Energetically optimized atomic coordinates for (a) deprotonated YZ, (b) protonated YZ, (c) deprotonated YD, and (d) protonated YD. Only for D1-His190/D2His189 and H2O, polar hydrogen atoms involved in H-bonds are displayed explicitly by small-sized dark-shaded spheres. Nitrogen atoms are displayed by medium-sized light-shaded spheres, oxygen atoms by medium-sized dark-shaded spheres, and Mn and Ca ions are shown by large spheres (manganese, dark-shaded; calcium, light-shaded). Original positions of YZ/D and D1-His190/D2-His189 from the PSII crystal structure are shown by dark-shaded sticks; energetically optimized positions are shown in light shading. Orientations of H-atoms and some H-bonds are indicated by thin lines.
There are, in principle, three possible mechanistic patterns for the involvement of tyrosine in the redox reaction between P680 and Mn-cluster. These are the following redox reactions with corresponding redox pairs (notation: YH, protonated neutral species; Y−, deprotonated anion; YH˙+, protonated cationic radical; Y˙, deprotonated neutral radical):
Y− → Y˙ + e− involving Y˙/Y− (Em(Y˙/Y−) = +680 mV, measured for N-acetyl-L-tyrosinamide).
YH → Y˙ + H+ + e− involving Y˙/YH with pKared of 9.6 referring to YH (Em(Y˙/YH) = +970 mV, measured for N-acetyl-L-tyrosinamide in aqueous solution at pH 5.0).
YH → YH˙+ + e− involving YH˙+/YH with pKaox of –2 referring to YH˙+ (Em(YH˙+/YH) = +1380 mV, measured for N-acetyl-L-tyrosinamide; reviewed in (10)).
In the following, the pKa for tyrosine is generally used as pKared, if not otherwise specified. In aqueous solution, the availability of these three possible redox reactions depends predominantly on pH. In a protein, these reactions can also be controlled by its environment as, for instance, H-bond network, presence of charged residues, and cofactors.
For efficient function of PSII, Em(YZ) should lie between the Em values of P680 (+1110–1300 mV) (11,12) and water (+820 mV). Based on the equilibrium constants of the redox reactions involving YZ and YD and the pH-dependence of these redox reactions, the Em(YZ) and Em(YD) were estimated to be ∼+970 mV (13) and +720 to +760 mV (13,14), respectively. On the other hand, equilibrium constants measured for the redox reaction between P680 and YZ suggest that Em(YZ) operating in the ET process between P680 and YZ should be relatively high, yielding a value of +1200 mV, which is ∼100 mV below Em(P680) (3,15) (reviewed in (10)). However, the mechanism of how the YZ/D environment of PSII shifts Em(YZ/D) needs further investigations to be clarified (see discussion in (9)). To obtain a stable YZ radical, many investigations on its function were, for technical reasons, performed on Mn-depleted PSII, since under these circumstances spectroscopic signals originating from YZ/D could be detected under stationary conditions. However, it was suggested that Mn-depleted PSII samples could be subject to critical changes in conformation or H-bond network (see for instance (16,17)). Hence, an interpretation of these experiments involves uncertainties.
Recently, higher resolution crystal structures of PSII isolated from the thermophilic cyanobacterium Thermosynechococcus elongatus were obtained, revealing side-chain arrangements and their interactions with redox-active cofactors (1,18). Here, we report on Em(YZ/D) calculated by solving the linearized Poisson-Boltzmann (LPB) equation for the whole PSII complex based on the PSII crystal structure (1), taking into account the atomic coordinates of all amino-acid residues and bound cofactors. All computations presented here were performed under the same conditions and parameterization as in previous computations on PSII (for instance, (19,20)).
METHODS
Coordinates
In our computations, all atomic coordinates were taken from the crystal structure of PSII from thermophilic cyanobacterium T. elongatus at a 3.5 Å resolution (PDB: 1S5L) (1). Hydrogen atom positions were energetically optimized with CHARMM (21), while positions of all nonhydrogen atoms were fixed and all titratable groups were kept in their standard protonation states, i.e., acidic groups ionized and basic groups (including titratable histidines) protonated. Simultaneously, Chla, Pheoa, and QA/B were kept in the neutral charge redox states. Histidines that are ligands of Chla were treated as nontitratable with neutral total charge.
Atomic partial charges
Atomic partial charges of amino acids were adopted from the all-atom CHARMM22 (22) parameter set. To account implicitly for the presence of a proton, the charges of acidic oxygens were both increased symmetrically by +0.5 unit charges. Similarly, instead of removing a proton in the deprotonated state, all hydrogen charges of the basic groups of arginine and lysine were diminished symmetrically by a unit charge in total. The atomic charges for the redox-active tyrosine were adopted from Popovic et al. (23); deprotonated with negative charge (Y−); deprotonated radical with neutral charge (Y˙) in the redox pair Y˙/Y−; protonated with neutral charge (YH); and protonated radical with positive charge (YH˙+) in the redox pair YH·+/YH.
Molecular systems and dielectric media
Although a formidable and still unsolvable task, a complete description of a molecular system (as for instance, a protein immersed in solvent) can in principle be achieved by solving the Schrödinger equation for all atoms comprising electrons and nuclei for a statistical average of conformations. In such a complete description there is no space to introduce a continuum dielectric medium. The other extreme is to ignore the atomic structure and conformations of the molecular system completely. In this case the dielectric medium is necessary to describe polarization effects emerging from of the dipoles in the molecular system, which create a reaction field in response to explicit charges or dipoles. Polarization effects have contributions not only from distortions of electronic wave functions (electron polarization) and conformational changes of the nuclei (nuclear polarization), but also from nonhomogeneous distributions of charged groups and their variations (for instance, titratable groups in a protein).
In theoretical computations, often mixed models are used where part of the molecular structure is treated explicitly, while the nonexplicit parts are considered by an effective dielectric medium. Here as in a number of preceding studies (19,20,24–26), we consider the atomic partial charges and variations of charge pattern due to titratable groups explicitly, while we ignore electronic polarization and nuclear polarization. The latter is true, since we consider only a single conformation, which is directly related to the atomic coordinates of the protein crystal structure. Based on a number of studies (19,20,24–26) we obtained best results for redox-active and titratable groups with a dielectric constant of ɛP = 4 inside the protein and ɛW = 80 outside to model the polarization of bulk water. The same values were used in computational studies of other groups (27,28). If we ignore the influence of charged groups and their variations in a protein, the value of the dielectric constant would vary depending on the molecular system considered and exhibit spatial dependencies related to the distribution of titratable residues. It should be noted here that a dielectric constant used for such mixed models is a model parameter like atomic partial charges but not a quantity directly amenable to experimental measurement.
In other computational work, also larger values than ɛP = 4 were used for the protein dielectric constant, for instance ɛP = 20 (29). Since the dielectric constant accounts for the molecular components that are not explicitly modeled, a description with lower dielectric constant is superior as long as the agreement with experimental data is of the same level. In this respect it is interesting to note that considering conformational flexibility (i.e., including nuclear polarization) by energy minimization yields best results by lowering the dielectric constant from 4 to 2 to account for only electron polarization (30).
Experimental determinations of the static dielectric constant in a molecular system are generally based on the assumption to describe polarization effects solely by a dielectric continuum. In bacterial RC the dielectric constant was determined by measuring the Stark effect (31). In these measurements the influence of electrostatic interactions of a chromophore with the protein environment is probed in terms of a double difference in energies, namely ground and electronic excited state energies in presence and absence of a strong external electrostatic field. For such double differences the direct influence of charged groups cancels, if they do not change their charge state with the external electrostatic field. However, such changes are likely to occur for titratable residues. Interpreting such contributions in terms of an inhomogeneous dielectric medium would result in a spatially varying dielectric constant. The measured values of ɛP in the bacterial RC vary between 2.1 and 9.5 (31), presumably due to variations in the protonation pattern of titratable residues. However, the average of the measured dielectric constants is close to the value used in our electrostatic energy computations.
Protonation pattern and redox potential
Our computation is based on the electrostatic continuum model treated by solving the LPB equation with the program MEAD (32). To facilitate a direct comparison with previous computational results, we used uniformly the same computational conditions and parameters such as atomic partial charges and dielectric constants (for instance, (19,20)). To obtain absolute values of the Em(Y) in the protein, we calculated the electrostatic energy difference between the two redox states of tyrosine in a reference model system with known experimental Em. The shift of Em in the protein relative to the reference system was added to the known Em. As reference model system, the following solution Ems versus normal hydrogen electrode were used: Em(Y˙/Y−) = +680 mV for one-electron reduction; and Em(YH˙+/YH) = +1380 mV for one-electron oxidation in aqueous solution (10). Although the radical pair of YH˙+/YH could be also relevant for the radical state of YZ/D in PSII, in this study we did not focus on this radical pair (see Discussion). The redox states of all other cofactors (i.e., Car, Chla, Pheoa, quinone) were kept in their neutral charge state. Cytochrome b559 and cytochrome c550 were kept in the reduced state. Specifically, unless titrated, YZ and YD were kept in the neutral charge state. The ensemble of protonation patterns was sampled by a Monte Carlo method with our own program Karlsberg (B. Rabenstein, Karlsberg Online Manual, http://agknapp.chemie.fu-berlin.de/karlsberg/(1999)). All computations were performed at 300 K with pH 7.0 and an ionic strength of 100 mM. The LPB equation was solved using a three-step grid-focusing procedure with 2.5 Å, 1.0 Å, and 0.3 Å resolution. The Monte Carlo sampling yielded the probabilities [Aox] and [Ared] of the two redox states of molecule A. The Em was evaluated from the Nernst equation. A bias potential was applied to obtain an equal amount of both redox states ([Aox] = [Ared]), yielding the redox midpoint potential Em as the resulting bias potential. For convenience, the computed Em was given with mV accuracy, without implying that the last digit is significant. In general, a few 10 mV in Em is in a sufficiently reproducible range of our computational method (see, for instance, (33)). With the Henderson-Hasselbalch equation, the pKa can be calculated as the pH where the concentration of [−Adeprotonatel] and [Aprotonated] are equal. For further information about computational procedure and error estimate, see our previous work for Em (for instance, (19,20)) and pKa (for instance, (34,35)).
Modeling water between the Mn-cluster Ca2+ and YZ
The existence of a water molecule located in H-bond distance to YZ and D1-His190 has been proposed to play a significant role in the redox reaction of YZ for years. Although crystal structures of PSII could not reveal water molecules due to their limited resolutions, the crystal structure of PSII (1,6) indicates a possible water binding site X21 at Ca2+ of the Mn-cluster and YZ, which is in agreement with 18O isotope exchange measurements that pointed to a Ca-bound water (36). The exact mechanism of PSII water oxidation as mediated by YZ and the Mn-cluster remains a matter of debate, but the majority of the proposed mechanisms seem to place at least one water molecule in the neighborhood of YZ and the Mn-cluster (37,38).
We modeled this water molecule in the vicinity of YZ and optimized its geometry energetically with CHARMM (21) while all other atomic coordinates except YZ/D and D1-His190/D2-His189 were fixed. Both YZ/D were treated as 1), negatively charged deprotonated tyrosine (Y− model, Fig. 2, a and c) (17,39,40); or 2), neutral protonated tyrosine (YH model, Fig. 2, b and d)) (10,41,42). Alternatively, in a geometry optimization with positively charged tyrosine YH˙+, the side chains of YZ and D1-His190 moved apart, disrupting the common H-bond (data not shown), rendering this H-bond unstable. Hence YH˙+ might be relevant only as a transient state.
The exact PSII conformations for YZ/D radical states are so far unknown. One of the most relevant studies to refer to this issue is a structural refinement of Högbom et al. (43) for a tyrosine radical position in a single crystal of ribonucleotide reductase R2 protein by combined x-ray and high-field EPR crystallography (43). Although the protein environment of the tyrosine radical in the two proteins are different, in the R2 protein a displacement of the side chain was observed predominantly at this tyrosine upon generation of its radical state. Therefore, in our study we released the atomic coordinates only for the minimum set of relevant residues, i.e., YZ/D1-His190 (YD/D2-His189) pair. As a consequence of the geometry optimization, atom pair distances of tyrosines and histidines were changed by 0.1–0.8 Å with an RMS deviation of 0.63 Å for the Y− model and 0.51 Å for the YH model with respect to the side-chain atoms (excluding hydrogen and backbone atoms; see Table 1). These displacements of side chains are in the same range as those observed in the generation of tyrosine radicals in the R2 protein (PDB 1MXR; see also Fig. 5 in (43)). The energy-optimization procedure did not alter the OY–NɛHis distance for YZ-D1-His190. In contrast, the corresponding YD-His189 atom pair distance decreased by 0.4 Å to 2.6 Å, which is nearly identical to the distance of 2.7 Å for the YZ/D1-His190 pair. In the geometry-optimized PSII structure, the water molecule is 3.0 Å from the Ca2+ ion at the Mn-cluster, 2.6 Å from the hydroxyl oxygen atom of YZ, and 3.0 Å from the side-chain oxygen atom of D1-Gln165 (Fig. 2). This completely agrees with the suggestion of a Ca-bound water that is H-bonded to YZ (5,37,38). We obtained the H-bond formation between the modeled water and D1-Gln165, which agrees with a suggestion in McEvoy and Brudvig (37). The optimized atomic coordinates of YZ, D1-His190, and the X21 water as well as YD and D2-His189 used in this study are available online as Supplementary Material (files YZ_deprot.pdb and YD_deprot.pdb for Y− model, and YZ_prot.pdb and YD_prot.pdb for YH model, respectively).
TABLE 1.
Geometry of the H-bond network at YZ/D after energy optimization of YZ/D and D1-His190/D2-His189 side chains in the presence of negatively charged tyrosine
| Distance [Å]
|
||
|---|---|---|
| Atom pair | Original* | Optimized† |
| YZ | ||
| NɛHis‡-OηYZ§ | 2.7 | 2.7 |
| NδHis‡-OδAsn¶ | 3.3 | 4.1 |
| Obicarbonate-OηYZ§ | 3.4 | 2.9 |
| OH2O-OηYZ§ | (2.7)‖ | (2.6)‖ |
| OH2O-NɛHis‡ | (3.4)‖ | (3.0)‖ |
| YD | ||
| NɛHis‡-OηYD§ | 3.0 | 2.6 |
| NδHis‡-NɛArg** | 2.4 | 3.2 |
| NδHis‡-NηArg** | 3.2 | 3.5 |
Based on the atomic coordinates of the PSII crystal structure (1).
Based on energy optimized atomic coordinates of PSII with CHARMM (see text).
D1-His190 for YZ and D2-His189 for YD.
Hydroxyl oxygen atom of YZ/D.
Side-chain oxygen atom of D1-Asn298.
The H2O molecule was placed in the PSII crystal structure between the Mn-cluster Ca2+ and YZ. It was first energetically optimized alone and subsequently together with YZ and the D1-His190 side chains.
Side-chain nitrogen atom of D2-Arg294.
The dielectric constant for the explicit modeled water X21 was set to ɛP = 4, i.e., to the same value as in the protein volume, in contrast to ɛW = 80 used for bulk water. With higher dielectric constant, electrostatic interactions are shielded more efficiently, as it is for instance the case in bulk water. The screening ability of electrostatic interactions caused by a single, explicitly modeled water molecule buried in the protein can be understood by reorientation of its dipole to stabilize atomic charges in the protein. One focus in this study is to investigate explicit H-bond pattern of water X21 to protein cofactors. Atomic coordinates of this modeled water near YZ/D were energetically optimized to form explicit H-bonds with the protein environment, which can establish sufficient electrostatic screening of protein charges in the neighborhood. However, the electrostatic screening is effective only if the water volume possesses a low dielectric constant as, for instance, ɛ = 4.
Evaluation of Em(YZ/D) with/without energy minimization
When we used the original atomic coordinates from the crystal structure, we calculated 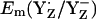 and
and 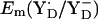 to be +944 mV and +586 mV at pH 6 for the S2 state, respectively. After energy minimization of the side chains of YZ/D and D1-His190/D2-His189, we obtained at pH 6 for the S2 state
to be +944 mV and +586 mV at pH 6 for the S2 state, respectively. After energy minimization of the side chains of YZ/D and D1-His190/D2-His189, we obtained at pH 6 for the S2 state 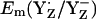 = +985 mV and
= +985 mV and 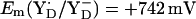 (see main text). The Em difference between YZ and YD was formerly estimated to be +240 meV from the equilibrium constant for YZ/YD (14) or +210 meV from the equilibrium constants for YZ/S2/1 and YD/S2/1 (13). From this Em difference, Vass and Styring (13) estimated
(see main text). The Em difference between YZ and YD was formerly estimated to be +240 meV from the equilibrium constant for YZ/YD (14) or +210 meV from the equilibrium constants for YZ/S2/1 and YD/S2/1 (13). From this Em difference, Vass and Styring (13) estimated 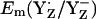 and
and 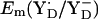 to be ∼+970 mV and +760 mV, respectively (13). The Em difference Em(YZ) − Em(YD) of +243 mV as well as the absolute values of Em(YZ/D) obtained in our computations using the energy-minimized coordinates are in good agreement with the estimated values of Vass and Styring (13). On the other hand, using the original atomic coordinates from the PSII crystal structure, the calculated
to be ∼+970 mV and +760 mV, respectively (13). The Em difference Em(YZ) − Em(YD) of +243 mV as well as the absolute values of Em(YZ/D) obtained in our computations using the energy-minimized coordinates are in good agreement with the estimated values of Vass and Styring (13). On the other hand, using the original atomic coordinates from the PSII crystal structure, the calculated 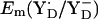 is lower by ∼150 mV than by using the energy-minimized coordinates. In contrast, the calculated
is lower by ∼150 mV than by using the energy-minimized coordinates. In contrast, the calculated 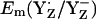 is nearly the same in the two atomic coordinate sets. The resulting Em difference of +358 mV between YZ and YD in the original atomic coordinates is by ∼100 mV larger than the corresponding values from the estimate of Vass and Styring (13) and our computations using the atomic coordinates from the energy-minimized model. Analyzing the two structures in atomic detail, it becomes evident that the low value of
is nearly the same in the two atomic coordinate sets. The resulting Em difference of +358 mV between YZ and YD in the original atomic coordinates is by ∼100 mV larger than the corresponding values from the estimate of Vass and Styring (13) and our computations using the atomic coordinates from the energy-minimized model. Analyzing the two structures in atomic detail, it becomes evident that the low value of 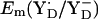 in the original PSII structure originates mainly from the relatively large NɛHis-OηYD distance of 3.0 Å in the PSII crystal relative to the smaller distance of 2.6 Å in the geometry-optimized SPII structure (Table 1). The same distance of 2.6 Å was also obtained by DFT computations of Faller et al. (44) considering the same H-bond pattern between YD and D2-His189. Indeed, the distance of 2.6 Å is nearly identical to the corresponding distance of 2.7 Å in the YZ side of the PSII crystal structure (Table 1). Although the NɛHis-OηTyr in the YD side was thus affected, it is quite remarkable that the same energy-minimization process did not alter the corresponding distance in the YZ side. Despite the alteration in Em(YD) by the energy minimization, the other features (e.g., protonation pattern in PSII) remained unchanged in our computation. Therefore, in the following part, we focused on the computation based on the atomic coordinates with these side chains being energetically optimized.
in the original PSII structure originates mainly from the relatively large NɛHis-OηYD distance of 3.0 Å in the PSII crystal relative to the smaller distance of 2.6 Å in the geometry-optimized SPII structure (Table 1). The same distance of 2.6 Å was also obtained by DFT computations of Faller et al. (44) considering the same H-bond pattern between YD and D2-His189. Indeed, the distance of 2.6 Å is nearly identical to the corresponding distance of 2.7 Å in the YZ side of the PSII crystal structure (Table 1). Although the NɛHis-OηTyr in the YD side was thus affected, it is quite remarkable that the same energy-minimization process did not alter the corresponding distance in the YZ side. Despite the alteration in Em(YD) by the energy minimization, the other features (e.g., protonation pattern in PSII) remained unchanged in our computation. Therefore, in the following part, we focused on the computation based on the atomic coordinates with these side chains being energetically optimized.
RESULTS AND DISCUSSION
Em(YZ˙/YZ−)
At pH 6 in the S2 state, the calculated 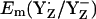 is +985 mV, which agrees with the measured Em(YZ) of ∼+970 mV, as reported by Vass and Styring (13). This value is also close to Em(Y˙/Y) = +970 mV measured for the neutral tyrosine radical in aqueous solution at pH 5.0 (10,45). At pH 7, the calculated
is +985 mV, which agrees with the measured Em(YZ) of ∼+970 mV, as reported by Vass and Styring (13). This value is also close to Em(Y˙/Y) = +970 mV measured for the neutral tyrosine radical in aqueous solution at pH 5.0 (10,45). At pH 7, the calculated 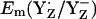 are +926 mV (+959 mV) in the S2 (S3) state (Table 2). EPR signals from a tyrosine radical in PSII were assigned to YZ interacting with the Mn-cluster in the S2 state and its pH-dependence observed in these experiments was attributed to a pH-dependent Em(YZ) (46). In our computations, we observed pH-dependence of Em(Y˙/Y−) for YZ and YD with a slope of ∼–60 mV/pH in the S2 state.
are +926 mV (+959 mV) in the S2 (S3) state (Table 2). EPR signals from a tyrosine radical in PSII were assigned to YZ interacting with the Mn-cluster in the S2 state and its pH-dependence observed in these experiments was attributed to a pH-dependent Em(YZ) (46). In our computations, we observed pH-dependence of Em(Y˙/Y−) for YZ and YD with a slope of ∼–60 mV/pH in the S2 state.
TABLE 2.
Computed Em(YZ/D) in [mV] at pH 7.0
| Redox couple | Mn-cluster S-state | Em(YZ) | Em(YD) |
|---|---|---|---|
| Y˙/Y−* | S0 | +932 | +686 |
| S1 | +931 | +689 | |
| S2 | +926 | +694 | |
| S3 | +959 | +702 | |
| Mn-depleted† | +780 | +685 | |
| Y˙+/Y* | S2 | +1669 | +1907 |
For the Em values used as reference model system, see Tommos and Babcock (10).
All the titratable groups in PSII were titrated, as was done in computations with intact Mn-cluster.
When we calculated 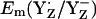 in the S2 state at pH 7.0 (i.e., at [Y˙] = [Y−] = 0.5), D1-His190 was to 50% protonated, implying a neutral charge state of the residue pair YZ/D1-His190. On the other hand, a number of experimental studies proposed that D1-His190 is protonated (i.e., positively charged) before relaxing to the S2 state (reviewed in (47)). In our computation, at pH 7 D1-His190 was fully protonated if YZ was fully reduced as tyrosinate (
in the S2 state at pH 7.0 (i.e., at [Y˙] = [Y−] = 0.5), D1-His190 was to 50% protonated, implying a neutral charge state of the residue pair YZ/D1-His190. On the other hand, a number of experimental studies proposed that D1-His190 is protonated (i.e., positively charged) before relaxing to the S2 state (reviewed in (47)). In our computation, at pH 7 D1-His190 was fully protonated if YZ was fully reduced as tyrosinate (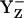 ) for all S-states investigated.
) for all S-states investigated.
Considering protonated D1-His190, which presumably prevails in a thermally activated state of PSII, we obtained 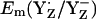 = +1216 mV. Quite interestingly, the Em(YZ) value obtained in presence of a fully protonated D1-His190 is nearly as high as Em(P680) of ∼+1200 to +1300 mV (11,12). The proximity between Em(YZ) and Em(PD1/D2) is consistent with a small Em difference between P680 and YZ, governing the ET between these cofactors in the nanosecond time domain before possible structural relaxation (3,15). This Em difference is time-dependent and increases in the microsecond time regime, yielding finally under equilibrium conditions in the S2 state Em(YZ) = +970 mV (13) (Em(YZ) = +985 mV computed in this study). Thus, in our computational model, a change in protonation at D1-His190 (using the same atomic coordinates from the PSII crystal structure (1)) has a predominant impact on Em(YZ), yielding an upshift of 260–290 mV. This mechanism may be necessary to reduce P680+ by YZ quickly in the nanosecond time regime (48).
= +1216 mV. Quite interestingly, the Em(YZ) value obtained in presence of a fully protonated D1-His190 is nearly as high as Em(P680) of ∼+1200 to +1300 mV (11,12). The proximity between Em(YZ) and Em(PD1/D2) is consistent with a small Em difference between P680 and YZ, governing the ET between these cofactors in the nanosecond time domain before possible structural relaxation (3,15). This Em difference is time-dependent and increases in the microsecond time regime, yielding finally under equilibrium conditions in the S2 state Em(YZ) = +970 mV (13) (Em(YZ) = +985 mV computed in this study). Thus, in our computational model, a change in protonation at D1-His190 (using the same atomic coordinates from the PSII crystal structure (1)) has a predominant impact on Em(YZ), yielding an upshift of 260–290 mV. This mechanism may be necessary to reduce P680+ by YZ quickly in the nanosecond time regime (48).
Em(YD˙/YD−)
At pH 6 in the S2 state, the calculated 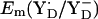 is +742 mV. At pH 7 we calculated
is +742 mV. At pH 7 we calculated 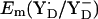 to be +694 mV in the S2 state and +702 mV in the S3 state (Table 2). The experimentally estimated
to be +694 mV in the S2 state and +702 mV in the S3 state (Table 2). The experimentally estimated 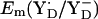 lies between +720 and +760 mV according to redox titration with strong oxidants (14) or from determination of the Em difference, Em(YZ) – Em(YD) (13). Interestingly, these estimated
lies between +720 and +760 mV according to redox titration with strong oxidants (14) or from determination of the Em difference, Em(YZ) – Em(YD) (13). Interestingly, these estimated 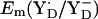 in PSII (13,14) are very similar to the Em(Y˙/Y−) value in aqueous solution where it was measured to be +680 mV for pH >9.9 independent of pH (10) although the very polar solvent environment differs considerably from the PSII environment. The different redox behavior of the two symmetry-equivalent tyrosines seems to be mainly due to the asymmetric location of the Mn-cluster being close to YZ and distant to YD with edge-to-edge distances of 5 Å and 28 Å, respectively. As observed in
in PSII (13,14) are very similar to the Em(Y˙/Y−) value in aqueous solution where it was measured to be +680 mV for pH >9.9 independent of pH (10) although the very polar solvent environment differs considerably from the PSII environment. The different redox behavior of the two symmetry-equivalent tyrosines seems to be mainly due to the asymmetric location of the Mn-cluster being close to YZ and distant to YD with edge-to-edge distances of 5 Å and 28 Å, respectively. As observed in 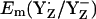 , we found also
, we found also 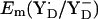 to be pH-dependent with ∼–60 mV/pH in our electrostatic energy computations.
to be pH-dependent with ∼–60 mV/pH in our electrostatic energy computations.
Model of ET from YZ to P680+ in intact PSII
The calculated Em(YH˙+/YH) were +1576 mV for YZ and +1804 mV for YD at pH 7 in the S2 state, which indicates less stability of YH˙+/YH relative to the deprotonated redox pair Y˙/Y−. In agreement with our computed results on Em(YH˙+/YH), Tommos and Babcock (10) also estimated Em(YH˙+/YH) for the PSII protein environment to be +1680 mV to +1830 mV (10). These large values for YZ/D are not only far from +970 mV (+985 mV) obtained by Vass and Styring (13) (computed in this study for 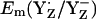 ), but also far from
), but also far from 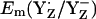 = +1216 mV, computed for protonated D1-His190.
= +1216 mV, computed for protonated D1-His190.
Although we obtained for Em(YZ) values as low as +985 and +1216 mV only by considering the redox pair 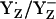 , it does not exclude the possibility that YZ is protonated (YZH) before its radical reaction, as found in Fourier-transform infrared (FTIR) studies (41,42). Hence, our computations suggest that
, it does not exclude the possibility that YZ is protonated (YZH) before its radical reaction, as found in Fourier-transform infrared (FTIR) studies (41,42). Hence, our computations suggest that 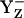 relative to
relative to 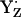 is energetically more stabilized than YH·+ relative to YZH, such that the redox pair
is energetically more stabilized than YH·+ relative to YZH, such that the redox pair 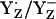 is likely more relevant than YZH˙+/YZH.
is likely more relevant than YZH˙+/YZH.
Tommos and Babcock (10) argued that 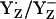 is not the functional redox pair in PSII unless positive charges near YZ lower the pKa of YZH to values <5, concluding that
is not the functional redox pair in PSII unless positive charges near YZ lower the pKa of YZH to values <5, concluding that 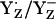 is unlikely to be the relevant redox pair (10). However, we remark that not only positive charges but also appropriate H-bonds with the titratable redox-active YZ are able to lower the pKa and stabilize its anionic form. A number of studies suggest that, in the YZ side, a Ca-bound water exists (1,6,37,38). Indeed, the fixation of a water at the Ca2+ of the Mn-cluster results in a strong H-bond between this water and the hydroxyl oxygen of YZ (Fig. 2 a and Table 3). Due to the H-bond with this Ca-bound water, we obtained a pKa of 7.4 for YZH (Fig. 3, panels 1 and 4), which is relatively low with respect to the pKa of 9.6 for tyrosine in aqueous solution. However, this pKa value for YZ is in agreement with the pKa values of 7.0–8.3 derived by Ahlbrinck et al. (3) and Diner et al. (40) from the ET rate involving YZ and P680+. A further support for this pKa is the FTIR result of Berthomieu et al. (42), which observed the disappearance of a band at 1238–1255 cm−1 with an apparent pKa of ∼8–9, corresponding to a δ(COH) tyrosine-bending mode (cited as personal communication in (49)). When YZ is equilibrated in the
is unlikely to be the relevant redox pair (10). However, we remark that not only positive charges but also appropriate H-bonds with the titratable redox-active YZ are able to lower the pKa and stabilize its anionic form. A number of studies suggest that, in the YZ side, a Ca-bound water exists (1,6,37,38). Indeed, the fixation of a water at the Ca2+ of the Mn-cluster results in a strong H-bond between this water and the hydroxyl oxygen of YZ (Fig. 2 a and Table 3). Due to the H-bond with this Ca-bound water, we obtained a pKa of 7.4 for YZH (Fig. 3, panels 1 and 4), which is relatively low with respect to the pKa of 9.6 for tyrosine in aqueous solution. However, this pKa value for YZ is in agreement with the pKa values of 7.0–8.3 derived by Ahlbrinck et al. (3) and Diner et al. (40) from the ET rate involving YZ and P680+. A further support for this pKa is the FTIR result of Berthomieu et al. (42), which observed the disappearance of a band at 1238–1255 cm−1 with an apparent pKa of ∼8–9, corresponding to a δ(COH) tyrosine-bending mode (cited as personal communication in (49)). When YZ is equilibrated in the 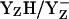 states then we calculated pKa of D1-His190 to be 6.6 (Fig. 3, panel 1). Hence, the Ca-bound water plays a role in stabilizing transient YZ states such as YZδ− (where YZ and D1-His190 share a proton, thus being in the neutral charge state) or
states then we calculated pKa of D1-His190 to be 6.6 (Fig. 3, panel 1). Hence, the Ca-bound water plays a role in stabilizing transient YZ states such as YZδ− (where YZ and D1-His190 share a proton, thus being in the neutral charge state) or 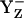 , which otherwise would be very unstable.
, which otherwise would be very unstable.
TABLE 3.
Influence of PD1PD2 and D2-Arg180 charge states on Em(YZ/D) at pH 7.0 in the S2 state
| Charge state | Em(YZ) [mV] | Em(YD) [mV] |
|---|---|---|
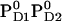 |
+926 | +694 |
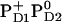 |
+918 | +732 |
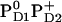 |
+924 | +802 |
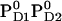 with D2-Arg180 deprot. with D2-Arg180 deprot. |
+926 | +564 |
FIGURE 3.

Scheme for ET from YZ to P680. Y and B stand for YZ and D1-His190, respectively, and the calculated pKa values of Y and B for these residues at each step are listed in the scheme.
We interpreted our results for 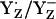 by the following redox reaction scheme of YZ/D visualized in Fig. 3:
by the following redox reaction scheme of YZ/D visualized in Fig. 3:
In the initial state, YZH dissociates the phenolic proton (Fig. 3, panel 1).
The presence of the Ca-bound water stabilizes the
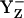 state transiently with fully protonated D1-His190 (BH+) (Em(YZ) = +1216 mV) (Fig. 3, panel 2). This step is particularly unstable and should therefore exist only transiently, since according to our calculated pKa YZ and B are an extremely strong acid and base, respectively. Since in the initial state proton release of YZH, producing
state transiently with fully protonated D1-His190 (BH+) (Em(YZ) = +1216 mV) (Fig. 3, panel 2). This step is particularly unstable and should therefore exist only transiently, since according to our calculated pKa YZ and B are an extremely strong acid and base, respectively. Since in the initial state proton release of YZH, producing 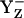 (Fig. 3, panel 2), is immediately quenched by an oxidation process where
(Fig. 3, panel 2), is immediately quenched by an oxidation process where 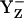 decays to the neutral radical state
decays to the neutral radical state 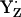 , the intermediate reaction state
, the intermediate reaction state 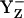 displayed in Fig. 3, panel 2, is often not considered explicitly. Thus, the resulting net redox reaction of these two steps involves
displayed in Fig. 3, panel 2, is often not considered explicitly. Thus, the resulting net redox reaction of these two steps involves 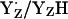 (Fig. 3, panels 1–3).
(Fig. 3, panels 1–3).For the neutral radical state
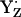 , the pKa of B is 2.2, still considerably low (Fig. 3, panel 3). As a consequence, the protonated BH+ becomes unstable, donating its proton to the neutral radical
, the pKa of B is 2.2, still considerably low (Fig. 3, panel 3). As a consequence, the protonated BH+ becomes unstable, donating its proton to the neutral radical 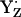 . In this proton transfer step, the residue pair YZ and B remains positively charged.
. In this proton transfer step, the residue pair YZ and B remains positively charged.This energetically unfavorable charge state relaxes by charge compensation receiving an electron from the Mn-cluster, which in turn changes from state Sn to Sn+1 (Fig. 3, panel 4).
It is a matter of debate whether the H-bond between the Ca-bound water and YZ remains unchanged even in this state (Fig. 3, panel 4), but if it does, then it leads to a protonated YZ and a deprotonated B, which are H-bonded [B···HYZ] or alternatively to a partially polarized (but also uncharged) species [B-Hδ+···YZδ−] where the proton is delocalized in the H-bond, yielding Em(YZ) = +926 for the S2 state (+959 mV for the S3 state) (Fig. 3, panel 4). Hereby, our computations yielded a partially protonated YZ (∼0.7 H+), which as a consequence carries a fractional negative charge. This fractional negative charge is practically completely neutralized by a complementary fractional protonation of D1-His190 (∼0.3 H+). This polarized H-bond resembles the tyrosinate intermediate model (17,39,40). Hence, in FTIR measurements the predominantly protonated YZ species may still be detected as protonated YZH (41,42).
Em(YZH˙+/YZH) in the Mn-depleted PSII model
Although it has been suggested that Mn-depleted PSII may possess considerably different H-bond pattern around YZ compared to those in intact PSII (16,17), we tentatively modeled an Mn-depleted PSII by simply removing the Mn-cluster, the attached bicarbonate, Ca2+, and water, without considering any conformational change of the protein environment nearby. To distinguish this model from the actual Mn-depleted PSII in vitro, we denote this model as Mn-depleted PSII model. This model possesses different H-bond pattern from those in intact PSII mainly due to the removal of the Ca-bound water, which was H-bonded to the YZ hydroxyl oxygen (OH2O–OTyr distance of 2.6 Å) in the intact PSII. In contrast to the enormously high potential of +1576 mV for Em(YZH˙+/YZH) in intact PSII, the YZH˙+/YZH calculated in the Mn-depleted PSII model was lower, yielding +1264 mV. This value is also lower than +1380 mV for YH˙+/YH measured in the aqueous solution (10), indicating that YH˙+ is dramatically stabilized in the Mn-depleted PSII model with respect to intact PSII or aqueous solution. Furthermore, the calculated Em(YZH˙+/YZH) = +1264 mV in the Mn-depleted PSII model is approximately as high as Em(P680) of ∼+1200 to +1300 mV (11,12), implying that YZH˙+/YZH may serve as functional electron donor to P680+ in the Mn-depleted PSII. The lack of electron donor for YZ (i.e., the Mn-cluster) in the Mn-depleted PSII might energetically, thus, be able to accumulate YZH˙+. Indeed, an interpretation that YZH˙+ may be the functional species in PSII was made based on EPR spectroscopy on Mn-depleted PSII (50,51). To investigate the stability of YZH˙+, we calculated the pKaox for YZH˙+. Indeed, in the Mn-depleted PSII model the calculated pKaox was 0.0, being increased by two units from the aqueous solution value of –2.0. Nevertheless, this value is still too low to maintain a stable YZH˙+ in the Mn-depleted PSII under physiological conditions, implying that this species is likely to occur only transiently also in the Mn-depleted PSII model. An alternative interpretation for these EPR signals would be that another titratable residue, whose position is ∼5 Å from YZ, is responsible for the positively charged intermediate (50). According to this interpretation of EPR studies, the involvement of this unidentified residue occurs specifically after the S2 state (50). Since the distance estimate of 5 Å is ambiguous, the involved titratable residue may be different from D1-His190, as discussed in the next section. In the following, we will focus our discussion on the redox couple Y˙/Y− for YZ/D in intact PSII. Accordingly, the notation Em(YZ/D) refers to “Em(Y˙/Y−) for YZ/D” unless otherwise stated.
Influence of CP43-Arg357 on Em(YZ˙/YZ−)
Surprisingly, our computations yielded very similar values for 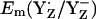 in the S0 state (+932 mV) and the S2 state (+926 mV), while for the S3 state we calculated
in the S0 state (+932 mV) and the S2 state (+926 mV), while for the S3 state we calculated 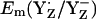 = 959 mV, which is ∼30 mV higher than for the S2 state (Table 2). In general, the presence of two additional positive unit charges in the S2 state of the Mn-cluster relative to the S0 state should upshift
= 959 mV, which is ∼30 mV higher than for the S2 state (Table 2). In general, the presence of two additional positive unit charges in the S2 state of the Mn-cluster relative to the S0 state should upshift 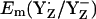 considerably. Considering the calculated difference of 30 mV for
considerably. Considering the calculated difference of 30 mV for 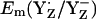 between the S2 and S3 states, we estimate
between the S2 and S3 states, we estimate 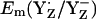 to be as low as +860 mV in S0 state but the actual calculated
to be as low as +860 mV in S0 state but the actual calculated 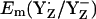 for the S0 state is +932 mV. The apparent insensitivity of
for the S0 state is +932 mV. The apparent insensitivity of 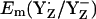 to changes of Mn-cluster charges is due to charge compensation by a protonation change of titratable residues nearby. In this connection, we found that CP43-Arg357 deprotonates as the S-state shifts from S0 to S2. This residue was observed to be fully protonated in the S0 state, partially deprotonated in the S1 state, and fully deprotonated in the S2 state. Indeed, when we forced CP43-Arg357 to be fully deprotonated also in the S0 state, the calculated
to changes of Mn-cluster charges is due to charge compensation by a protonation change of titratable residues nearby. In this connection, we found that CP43-Arg357 deprotonates as the S-state shifts from S0 to S2. This residue was observed to be fully protonated in the S0 state, partially deprotonated in the S1 state, and fully deprotonated in the S2 state. Indeed, when we forced CP43-Arg357 to be fully deprotonated also in the S0 state, the calculated 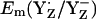 decreased by 108 mV to +824 mV. Similarly, when we forced CP43-Arg357 to be fully protonated in the S2 state, the calculated
decreased by 108 mV to +824 mV. Similarly, when we forced CP43-Arg357 to be fully protonated in the S2 state, the calculated 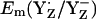 increased by 63 mV to +989 mV in the S2 state. Thus, mainly CP43-Arg357 is responsible for the S-state independence of
increased by 63 mV to +989 mV in the S2 state. Thus, mainly CP43-Arg357 is responsible for the S-state independence of 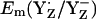 observed in our computations. Hence, the variable protonation of CP47-Arg357 seems to function as buffer for
observed in our computations. Hence, the variable protonation of CP47-Arg357 seems to function as buffer for 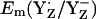 during S0–S2 state transitions.
during S0–S2 state transitions.
CP43-Arg357 is at an edge-to-edge distance of 4 Å from the Mn-cluster, close to one of the three possible substrate water binding sites, X22, which differs from the suggested Ca-bound water X21 proximal to YZ (1,5,6). In this location, an intermediate oxygen product before O=O bond formation of the final product O2 might be stabilized by H-bonds with CP43-Arg357 as proposed in Ferreira et al. (1). These suggestions are in line with the mutation study from CP43-Arg357 to Ser that resulted in severely inhibited O2-evolution (52). Based on these arguments, McEvoy and Brudvig (37) suggested that CP43-Arg357 is protonated in the S-states below S2 and becomes deprotonated in the higher oxidized S-states due to its proximity to the positively charged Mn-cluster (37). This is in perfect agreement with our results on CP43-Arg357 protonation. Thus, as they suggested, CP43-Arg357 may play a central role in organizing the S-state-dependent flow of protons from the Mn-cluster, which may transfer protons to D1-Asp61 belonging to the proton exit pathway (37). Instead of the proton pathway consisting mainly in YZ and D1-His190 as proposed by Hoganson and Babcock (2), an alternative pathway not directly via YZ and D1-His190 was proposed by Haumann and Junge (4). The deprotonation of CP43-Arg357 observed here favors these alternative proton exit pathways.
Factors determining the Em difference between YZ and YD
The Em difference, Em(YZ) – Em(YD), was estimated to be +240 meV from the equilibrium constant for YZ/YD (14) or +210 meV from the equilibrium constants for YZ/S2/1 and YD/S2/1 (13). The Em(YZ) – Em(YD) of +232 meV in the S2 state (+257 meV in the S3 state) obtained in our computations are in good agreement with these experimental estimates. The main factors contributing to the difference between Em(YZ) and Em(YD) are differences in 1), the distance between the Mn-cluster and YZ/D; and 2), the H-bond pattern involving the histidines D1-His190 and D2-His189, respectively (see Fig. 4).
FIGURE 4.

H-bond network around (a) YZ and( b) YD. Only residue side chains are shown. Dotted lines indicate H-bonds or salt-bridges. Only protons belonging to YZ/D are shown in the figure. H(+) denotes the proton whose presence depends on the redox state of YZ.
According to the PSII crystal structure (1) the edge-to-edge distances of the Mn-cluster to YZ and to YD are 5 Å and 28 Å, respectively, which is quite different. The removal of the Mn-cluster from PSII in vitro could result in drastic changes of protein environment, especially around YZ (16,17). However, solely for the purpose of distinguishing the direct influence of Mn-cluster on Em(YZ/D) from other influences, we performed electrostatic energy computations in the absence of the Mn-cluster (removing also Ca2+ and bicarbonate) without considering any conformational change of the remaining protein. To elucidate the actual electrostatic influence in the computation, this treatment is more appropriate. Hereby, Em(YZ) was downshifted by 150 mV, relative to the S2 state, to be now +780 mV (Table 2). The corresponding decrease in Em(YD) was only 10 mV due to the much larger distance between the Mn-cluster and YD. Thus, the main contribution to the Em difference is due to the asymmetric location of the Mn-cluster relative to the D1/D2 proteins. But, there still remains an Em difference of ∼100 mV between YZ and YD.
For the computation of Em(YZ/D), the total net charge of YZ-D1-His190/YD-D2-His189 was found to be +0.5/0.0. Thus, the radical pair Y˙/Y− is stabilized more for YZ than for YD. In the PSII crystal structure (1), both YZ and YD form H-bonds with Nɛ of D1-His190 and D2-His189, respectively. Nδ of D1-His190 is close to the side-chain oxygen of D1-Asn298 (Fig. 4). The NδHis–OAsn distance is 3.3:4.1 Å before/after constrained optimization of the atomic coordinates of the YZ-D1-His190 pair in the reduced state. This contrasts with D2-His189, whose Nδ forms a strong H-bond of salt-bridge character with the guanidinium nitrogens of the positively charged D2-Arg294 (NδHis–NɛArg distance of 2.4 Å/3.2 Å and NδHis–NηArg distance of 3.2 Å/ 3.5 Å before/after geometry optimization). The more distant Nη of this arginine is also involved in a strong salt-bridge with the acidic oxygen of CP47-Glu364 (NηArg–OGlu distance of 2.9 Å) from the neighboring antenna complex CP47. Although the total net charge of the salt-bridged pair, D2-Arg294 and CP47-Glu364 at YD vanishes, the two H-bonds from D2-Arg294 to Nδ at D2-His189 do not allow this Nδ to be protonated.
Only for deprotonated D2-Arg294, Nδ of D2-His189 could have a chance to protonate. But, due to the salt-bridge with CP47-Glu364, D2-Arg294 remains positively charged. Indeed, the calculated pKa are ∼15–17 for D2-Arg294 and ∼0–2 for CP47-Glu364, independent of the YD redox state. Thus, protonation of D2-His189 at Nδ is very unlikely. In fact, D2-Arg294 was fully protonated even when we removed the CP47 subunit. Contrary to D2-His189, we observed Nδ of D1-His190 to be permanently protonated, forming an H-bond with D1-Asn298. To estimate the influence of the positively charged D2-Arg294 on Em(YD), we also considered D2-Arg294 in its deprotonated neutral charged state and obtained Em(YD) = +823 mV in the S2 state. The resulting upshift of 129 mV in Em(YD) demonstrates the significant role of the positively charged D2-Arg294 in lowering Em(YD), which may explain the remaining Em difference of 100 mV between YZ and YD.
Although there was no report about an explicit role of D2-Arg294 in YD redox function, the importance of D2-Arg294 for stability and function of PSII was suggested from random mutagenesis studies of PSII from Synechocystis PCC 6803 (53). PSII mutated from D2-Arg294 to Trp was still capable of O2 evolution but with a four-times-smaller initial rate, very sensitive to light, and rapid inhibition (53). An enhanced light-sensitivity, photoinhibition, and a limited ability to evolve O2 were also observed in the CP47-Glu364 mutant for PsbV-depleted PSII, where D2-Arg294 is unable to form the salt-bridge with the residue Glu364 in CP47 (54). Among cyanobacteria and higher plants, the three residues D1-Asn298, D2-Arg294, and CP47-Glu364 are highly conserved. Thus, the asymmetry in protein sequence and structure of D1/D2 and CP43/CP47 proteins differentiates the function of YZ and YD by the H-bond and salt-bridge pattern.
Electrostatic interactions between YZ/D and PD1/D2
So far all computations were performed for the neutral charged state of PD1/D2 i.e., 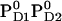 . Here we report the influence of the other charge states at PD1/D2 on Em(YZ/D). In general, an increase of the net charge in the proximity of a redox-active group upshifts its Em. Thus, formation of
. Here we report the influence of the other charge states at PD1/D2 on Em(YZ/D). In general, an increase of the net charge in the proximity of a redox-active group upshifts its Em. Thus, formation of 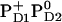 or
or 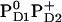 is expected to upshift Em(YZ/D). Nevertheless, the calculated Em(YZ) remains almost unchanged among these PD1/D2 redox states, yielding values of +918 mV and +924 mV in the states
is expected to upshift Em(YZ/D). Nevertheless, the calculated Em(YZ) remains almost unchanged among these PD1/D2 redox states, yielding values of +918 mV and +924 mV in the states 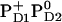 and
and 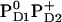 , respectively (Table 3). In contrast, Em(YD) showed a remarkable upshift of 38 mV for
, respectively (Table 3). In contrast, Em(YD) showed a remarkable upshift of 38 mV for 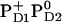 and 108 mV for
and 108 mV for 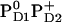 . Thus, YD is much more sensitive to the redox state of PD1/D2 than YZ with a strong electrostatic link to PD2.
. Thus, YD is much more sensitive to the redox state of PD1/D2 than YZ with a strong electrostatic link to PD2.
The stronger interaction between YD and PD2 is likely due to the hydrophobic environment at YD as opposed to YZ, the latter having the polar Mn-cluster in its proximity (1). Furthermore, a cluster of titratable residues extends from the Mn-cluster to the luminal surface (involving D1-Asp61, D1-Glu65, D2-Lys317, and D2-Glu312, and six residues of the PsbO protein, which are AspO158, AspO222, AspO223, AspO224, His-O228, and GluO229), forming a hydrophilic channel with D1-Asp61 at its mouth (5). On the other hand, the space near YD, which is equivalent to the position of the Mn-cluster at YZ, is filled with bulky hydrophobic residues (D2-Phe168, D2-Phe184, D2-Phe185, D2-Phe188, CP47-Phe362, and CP47-Phe363) (5). These nontitratable residues cannot compensate for charges induced at YD or PD2. Furthermore, the region between YD and PD2 seems to be relatively rigid. Thus, the low dielectric medium at YD/PD2 does not screen electrostatic interactions between YD and PD2.
Faller et al. (7) measured the ET rate from YD to P680+ with a half-time of 180 ns (7). Their observed ET was much faster than that from earlier estimates (reviewed in (55)). To explain the kinetics of this ET process, they suggested that the cationic state of P680 should be localized on PD2 (7), where it can be pushed onto PD1 electrostatically if YD is in the oxidized state, thereby accelerating ET from YZ to P680+ (7,9). The specifically strong electrostatic interaction between YD and 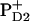 observed in our computations may corroborate these suggestions. Hence, presence of nontitratable residues in the neighborhood of YD/PD2 and their absence near to YZ/PD1 may be necessary to favor ET from YZ to P680+.
observed in our computations may corroborate these suggestions. Hence, presence of nontitratable residues in the neighborhood of YD/PD2 and their absence near to YZ/PD1 may be necessary to favor ET from YZ to P680+.
Influence of D2-Arg180 on Em(YD)
D2-Arg180 is suggested to have an impact not only on P680 (12,56) but also on YD, because mutation of this residue led to a loss of EPR signal originated from the oxidized state of YD (57). To investigate the influence of this residue, we calculated Em(YZ/D) in the S2 state in forcing D2-Arg180 to be deprotonated. The resulting Em(YD) is +564 mV, significantly downshifted by 130 mV (Table 3). Surprisingly, Em(YZ) did not shift upon deprotonation of D2-Arg180, confirming the EPR studies of D2-Arg180 mutants (57) and thereby suggesting that this arginine has a predominant impact on YD but not YZ. To explain this large effect, it was previously proposed that D2-Arg180 was capable of accepting a proton released from YD or stabilizing the proton on D2-His189 (57). However, our computation yielded a pKa of 11.3 for D2-Arg180, which is standard for arginine. Hence, this arginine residue is generally protonated regardless of the redox states for PD1/D2 and YZ/D.
Indeed, the persistence of the D2-Arg180 protonation is also one of the reasons that in our computation deprotonation/protonation by redox change at PD1/D2 and YD occurs predominantly at D2-His61. Interestingly, the symmetrical counterpart of D2-His61 is the acidic D1-Asp61, which is very close to the Mn-cluster, belonging to the hydrophilic channel leading to the luminal bulk surface (1,5,6). Both D1-Asp61 and D2-His61 are highly conserved among cyanobacteria and higher plants.
In the absence of light, the majority of PSII is adapted to the S1 state, not the lowest oxidized state S0 (58). This is probably due to the oxidative ability of YD (9,59). The corresponding Em(S1/S0) is estimated to be ∼+700 mV (10,13). Since with deprotonated D2-Arg180 the computed Em(YD) is only +564 mV, in a PSII mutant with depleted D2-Arg180, YD might not be able to oxidize the S0 state to S1 efficiently. Hence, we suggest that the loss of EPR signal from YD in mutant PSII with depleted D1-Arg180 can be associated with the inability of YD to oxidize the S0 to the S1 state. Therefore, YD in the wild-type PSII could play a redox-active role in photoassembly of the Mn-cluster as suggested in Rutherford et al. (9), and the ability to oxidize S0 to S1 by YD could depend on the value of Em(YD) with respect to Em(S1/S0).
CONCLUDING REMARKS
The calculated Em(Y˙/Y−) were +926 mV/+694 mV for YZ/YD in S2 state of the Mn-cluster in agreement with former estimates (13,14). The differences in the distances of YZ and YD from the Mn-cluster as well as those in the H-bond network of YZ (YZ/D1-His190/D1-Asn298) and YD (YD/D2-His189/D2-Arg294/CP47-Glu364) are responsible for the differences in Em. If D1-His190 is protonated, 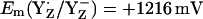 is nearly as high as Em(P680). Em(YZH˙+/YZH) calculated in the Mn-depleted PSII model was +1264 mV, lower than that of +1576 mV calculated in intact PSII, implying that YZH˙+/YZH may serve as functional electron donor to P680+ in the Mn-depleted PSII as suggested by EPR spectroscopy on Mn-depleted PSII (50,51). CP43-Arg357 deprotonates as the S-state shifts from S0 to S2, suggesting its participation in water oxidation and proton translocation. A strong electrostatic link was found specifically between YD and PD2. This is probably because of the presence of bulky hydrophobic residues (D2-Phe168, D2-Phe184, D2-Phe185, D2-Phe188, CP47-Phe362, and CP47-Phe363) in the YD side. These residues cannot compensate for charges induced at YD or PD2, in contrast to the hydrophilic residues likely serving as proton channel in the YZ side (D1-Asp61, D1-Glu65, D2-Lys317, D2-Glu312, and several residues of the PsbO protein). This difference in protein environment may explain the former suggestion that the cationic state of P680 should be localized on PD2 and can be pushed onto PD1 electrostatically, thereby accelerating ET from YZ to P680+ (7,9). The positively charged D2-Arg180 upshifts Em(YD) significantly. Upshifting of Em(YD) by D2-Arg180 might play a significant role in maintaining the redox ability of YD in intact PSII, as demonstrated by mutational studies (57).
is nearly as high as Em(P680). Em(YZH˙+/YZH) calculated in the Mn-depleted PSII model was +1264 mV, lower than that of +1576 mV calculated in intact PSII, implying that YZH˙+/YZH may serve as functional electron donor to P680+ in the Mn-depleted PSII as suggested by EPR spectroscopy on Mn-depleted PSII (50,51). CP43-Arg357 deprotonates as the S-state shifts from S0 to S2, suggesting its participation in water oxidation and proton translocation. A strong electrostatic link was found specifically between YD and PD2. This is probably because of the presence of bulky hydrophobic residues (D2-Phe168, D2-Phe184, D2-Phe185, D2-Phe188, CP47-Phe362, and CP47-Phe363) in the YD side. These residues cannot compensate for charges induced at YD or PD2, in contrast to the hydrophilic residues likely serving as proton channel in the YZ side (D1-Asp61, D1-Glu65, D2-Lys317, D2-Glu312, and several residues of the PsbO protein). This difference in protein environment may explain the former suggestion that the cationic state of P680 should be localized on PD2 and can be pushed onto PD1 electrostatically, thereby accelerating ET from YZ to P680+ (7,9). The positively charged D2-Arg180 upshifts Em(YD) significantly. Upshifting of Em(YD) by D2-Arg180 might play a significant role in maintaining the redox ability of YD in intact PSII, as demonstrated by mutational studies (57).
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
We thank Drs. Donald Bashford and Martin Karplus for providing the programs MEAD and CHARMM22, respectively. We thank Drs. Wolfram Saenger, Jacek Biesiadka, and Bernhard Loll for useful discussions.
This work was supported by the Deutsche Forschungsgemeinschaft grant No. SFB 498, Projects A5, Forschergruppe Project No. KN 329/5-1/5-2, grant No. GRK 80/2, grant No. GRK 268, and grant No. GRK 788/1. H.I. was supported by the Deutscher Akademischer Austauschdienst.
H. Ikishita's present address is Dept. of Chemistry, The Pennsylvania State University, 104 Chemistry Building, University Park, PA 16802.
References
- 1.Ferreira, K. N., T. M. Iverson, K. Maghlaoui, J. Barber, and S. Iwata. 2004. Architecture of the photosynthetic oxygen-evolving center. Science. 303:1831–1838. [DOI] [PubMed] [Google Scholar]
- 2.Hoganson, C. W., and G. T. Babcock. 1997. A metalloradical mechanism for the generation of oxygen from water in photosynthesis. Science. 277:1953–1956. [DOI] [PubMed] [Google Scholar]
- 3.Ahlbrink, R., M. Haumann, D. Cherepanov, O. Bogershausen, A. Mulkidjanian, and W. Junge. 1998. Function of tyrosine Z in water oxidation by photosystem II: electrostatical promoter instead of hydrogen abstractor. Biochemistry. 37:1131–1142. [DOI] [PubMed] [Google Scholar]
- 4.Haumann, M., and W. Junge. 1999. Photosynthetic water oxidation: a simplex-scheme of its partial reactions. Biochim. Biophys. Acta. 1411:86–91. [DOI] [PubMed] [Google Scholar]
- 5.Barber, J., K. Ferreira, K. Maghlaoui, and S. Iwata. 2004. Structural model of the oxygen-evolving centre of photosystem II with mechanistic implications. Phys. Chem. Chem. Phys. 6:4737–4742. [Google Scholar]
- 6.Iwata, S., and J. Barber. 2004. Structure of photosystem II and molecular architecture of the oxygen-evolving centre. Curr. Opin. Struct. Biol. 14:447–453. [DOI] [PubMed] [Google Scholar]
- 7.Faller, P., R. J. Debus, K. Brettel, M. Sugiura, A. W. Rutherford, and A. Boussac. 2001. Rapid formation of the stable tyrosyl radical in photosystem II. Proc. Natl. Acad. Sci. USA. 98:14368–14373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diner, B. A., and F. Rappaport. 2002. Structure dynamics, and energetics of the primary photochemistry of photosystem II of oxygenic photosynthesis. Annu. Rev. Plant Biol. 53:551–580. [DOI] [PubMed] [Google Scholar]
- 9.Rutherford, A. W., A. Boussac, and P. Faller. 2004. The stable tyrosyl radical in photosystem II: why D? Biochim. Biophys. Acta. 1655:222–230. [DOI] [PubMed] [Google Scholar]
- 10.Tommos, C., and G. T. Babcock. 2000. Proton and hydrogen currents in photosynthetic water oxidation. Biochim. Biophys. Acta. 1458:199–219. [DOI] [PubMed] [Google Scholar]
- 11.Rappaport, F., M. Guergova-Kuras, P. J. Nixon, B. A. Diner, and J. Lavergne. 2002. Kinetics and pathways of charge recombination in photosystem II. Biochemistry. 41:8518–8527. [DOI] [PubMed] [Google Scholar]
- 12.Ishikita, H., B. Loll, J. Biesiadka, W. Saenger, and E. W. Knapp. 2005. Redox potentials of chlorophylls in the photosystem II reaction center. Biochemistry. 44:4118–4124. [DOI] [PubMed] [Google Scholar]
- 13.Vass, I., and S. Styring. 1991. pH-dependent charge equilibria between tyrosine-D and the S states in photosystem II. Estimation of relative midpoint redox potentials. Biochemistry. 30:830–839. [DOI] [PubMed] [Google Scholar]
- 14.Boussac, A., and A. L. Etienne. 1982. Oxido-reduction kinetics of signal II slow in Tris-washed chloroplasts. Biochem. Biophys. Res. Commun. 109:1200–1205. [DOI] [PubMed] [Google Scholar]
- 15.Brettel, K., E. Schlodder, and H. T. Witt. 1984. Nanosecond reduction kinetics of photooxidized chlorophyll-aII (P-680) in single flashes as a probe for the electron pathway, H+-release and charge accumulation in the O2-evolving complex. Biochim. Biophys. Acta. 766:403–415. [Google Scholar]
- 16.Tommos, C., X.-S. Tang, K. Warncke, C. W. Hoganson, S. Styring, J. McCracken, B. A. Diner, and G. T. Babcock. 1995. Spin-density distribution, conformation, and hydrogen bonding of the redox-active tyrosine YZ in photosystem II from multiple-electron magnetic-resonance spectroscopies: implications for photosynthetic oxygen evolution. J. Am. Chem. Soc. 117:10325–10335. [Google Scholar]
- 17.Haumann, M., A. Mulkidjanian, and W. Junge. 1999. Tyrosine-Z in oxygen-evolving photosystem II: a hydrogen-bonded tyrosinate. Biochemistry. 38:1258–1267. [DOI] [PubMed] [Google Scholar]
- 18.Biesiadka, J., B. Loll, J. Kern, K.-D. Irrgang, and A. Zouni. 2004. Crystal structure of cyanobacterial photosystem II at 3.2 Å resolution: a closer look at the Mn-cluster. Phys. Chem. Chem. Phys. 6:4733–4736. [Google Scholar]
- 19.Ishikita, H., and E. W. Knapp. 2005. Control of quinone redox potentials in photosystem II: electron transfer and photoprotection. J. Am. Chem. Soc. 127:14714–14720. [DOI] [PubMed] [Google Scholar]
- 20.Ishikita, H., and E. W. Knapp. 2005. Oxidation of the non-heme iron complex in photosystem II. Biochemistry. 44:14772–14783. [DOI] [PubMed] [Google Scholar]
- 21.Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM: a program for macromolecular energy minimization and dynamics calculations. J. Comput. Chem. 4:187–217. [Google Scholar]
- 22.MacKerell, A. D., Jr., D. Bashford, R. L. Bellott, R. L. Dunbrack, Jr., J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher III, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin, and M. Karplus. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- 23.Popovic, D. M., A. Zmiric, S. D. Zaric, and E.-W. Knapp. 2002. Energetics of radical transfer in DNA photolyase. J. Am. Chem. Soc. 124:3775–3782. [DOI] [PubMed] [Google Scholar]
- 24.Rabenstein, B., G. M. Ullmann, and E.-W. Knapp. 1998. Energetics of electron-transfer and protonation reactions of the quinones in the photosynthetic reaction center of Rhodopseudomonas viridis. Biochemistry. 37:2488–2495. [DOI] [PubMed] [Google Scholar]
- 25.Popovic, D. M., S. D. Zaric, B. Rabenstein, and E.-W. Knapp. 2001. Artificial cytochrome b: computer modeling and evaluation of redox potentials. J. Am. Chem. Soc. 123:6040–6053. [DOI] [PubMed] [Google Scholar]
- 26.Voigt, P., and E. W. Knapp. 2003. Tuning heme redox potentials in the cytochrome c subunit of photosynthetic reaction centers. J. Biol. Chem. 278:51993–52001. [DOI] [PubMed] [Google Scholar]
-
27.Alexov, E. G., and M. R. Gunner. 1999. Calculated protein and proton motions coupled to electron transfer: electron transfer from
 to QB in bacterial photosynthetic reaction centers. Biochemistry. 38:8253–8270. [DOI] [PubMed] [Google Scholar]
to QB in bacterial photosynthetic reaction centers. Biochemistry. 38:8253–8270. [DOI] [PubMed] [Google Scholar] - 28.Zhu, Z., and M. R. Gunner. 2005. Energetics of quinone-dependent electron and proton transfers in Rhodobacter sphaeroides photosynthetic reaction centers. Biochemistry. 44:82–96. [DOI] [PubMed] [Google Scholar]
- 29.Sugita, Y., N. Miyashita, M. Ikeguchi, A. Kidera, and C. Toyoshima. 2005. Protonation of the acidic residues in the transmembrane cation-binding sites of the Ca2+ pump. J. Am. Chem. Soc. 127:6150–6151. [DOI] [PubMed] [Google Scholar]
- 30.Rabenstein, B., G. M. Ullmann, and E.-W. Knapp. 1998. Calculation of protonation patterns in proteins with structural relaxation and molecular ensembles—application to the photosynthetic reaction center. Eur. Biophys. J. 27:626–637. [Google Scholar]
- 31.Steffen, M. A., K. Lao, and S. G. Boxer. 1994. Dielectric asymmetry in the photosynthetic reaction center. Science. 264:810–816. [DOI] [PubMed] [Google Scholar]
- 32.Bashford, D., and M. Karplus. 1990. pKa's of ionizable groups in proteins: atomic detail from a continuum electrostatic model. Biochemistry. 29:10219–10225. [DOI] [PubMed] [Google Scholar]
- 33.Ishikita, H., B. Loll, J. Biesiadka, A. Galstyan, W. Saenger, and E.-W. Knapp. 2005. Tuning electron transfer by ester-group of chlorophylls in bacterial photosynthetic reaction center. FEBS Lett. 579:712–716. [DOI] [PubMed] [Google Scholar]
- 34.Ishikita, H., and E. W. Knapp. 2005. Induced conformational change upon Cd2+ binding at photosynthetic reaction centers. Proc. Natl. Acad. Sci. USA. 102:16215–16220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ishikita, H., D. Stehlik, J. H. Golbeck, and E.-W. Knapp. 2006. Electrostatic influence of PsaC protein binding to the PsaA/PsaB heterodimer in photosystem I. Biophys. J. 90:1081–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hendry, G., and T. Wydrzynski. 2003. 18O isotope exchange measurements reveal that calcium is involved in the binding of one substrate-water molecule to the oxygen-evolving complex in photosystem II. Biochemistry. 42:6209–6217. [DOI] [PubMed] [Google Scholar]
- 37.McEvoy, J. P., and G. W. Brudvig. 2004. Structure-based mechanism of photosynthetic water oxidation. Phys. Chem. Chem. Phys. 6:4754–4763. [Google Scholar]
- 38.Rutherford, A. W., and A. Boussac. 2004. Water photolysis in biology. Science. 303:1782–1784. [DOI] [PubMed] [Google Scholar]
- 39.Candeias, L. P., S. Turconi, and J. H. A. Nugent. 1998. Tyrosine YZ and YD of photosystem II: comparison of optical spectra to those of tyrosine oxidised by pulsed radiolysis. Biochim. Biophys. Acta. 1363:1–5. [DOI] [PubMed] [Google Scholar]
- 40.Diner, B. A., D. A. Force, D. W. Randall, and R. D. Britt. 1998. Hydrogen bonding, solvent exchange, and coupled proton and electron transfer in the oxidation and reduction of redox-active tyrosine YZ in Mn-depleted core complexes of photosystem II. Biochemistry. 37:17931–17943. [DOI] [PubMed] [Google Scholar]
- 41.Noguchi, T., Y. Inoue, and X.-S. Tang. 1997. Structural coupling between the oxygen-evolving Mn cluster and a tyrosine residue in photosystem II as revealed by Fourier transform infrared spectroscopy. Biochemistry. 36:14705–14711. [DOI] [PubMed] [Google Scholar]
- 42.Berthomieu, C., R. Hienerwadel, A. Boussac, J. Breton, and B. A. Diner. 1998. Hydrogen bonding of redox-active tyrosine Z of photosystem II probed by FTIR difference spectroscopy. Biochemistry. 37:10547–10554. [DOI] [PubMed] [Google Scholar]
- 43.Högbom, M., M. Galander, M. Andersson, M. Kolberg, W. Hofbauer, G. Lassmann, P. Nordlund, and F. Lendzian. 2003. Displacement of the tyrosyl radical cofactor in ribonucleotide reductase obtained by single-crystal high-field EPR and 1.4-Å x-ray data. Proc. Natl. Acad. Sci. USA. 100:3209–3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Faller, P., C. Goussias, A. W. Rutherford, and S. Un. 2003. Resolving intermediates in biological proton-coupled electron transfer: a tyrosyl radical pair to proton movement. Proc. Natl. Acad. Sci. USA. 100:8732–8735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tommos, C., J. J. Skalicky, D. L. Pilloud, A. J. Wand, and P. L. Dutton. 1999. De novo proteins as models of radical enzymes. Biochemistry. 38:9495–9507. [DOI] [PubMed] [Google Scholar]
-
46.Geijer, P., F. Morvaridi, and S. Styring. 2001. The S3 state of the oxygen-evolving complex in photosystem II is converted to the
 state at alkaline pH. Biochemistry. 40:10881–10891. [DOI] [PubMed] [Google Scholar]
state at alkaline pH. Biochemistry. 40:10881–10891. [DOI] [PubMed] [Google Scholar] - 47.Petrouleas, V., D. Koulougliotis, and N. Ioannidis. 2005. Trapping of metalloradical intermediates of the S-states at liquid helium temperatures. Overview of the phenomenology and mechanistic implications. Biochemistry. 44:6723–6728. [DOI] [PubMed] [Google Scholar]
- 48.Hays, A.-M. A., I. R. Vassiliev, J. H. Golbeck, and R. J. Debus. 1998. Role of D1-His190 in proton-coupled electron transfer reactions in photosystem II: a chemical complementation study. Biochemistry. 37:11352–11365. [DOI] [PubMed] [Google Scholar]
- 49.Diner, B. A. 2001. Amino acid residues involved in the coordination and assembly of the manganese cluster of photosystem II. Proton-coupled electron transport of the redox-active tyrosines and its relationship to water oxidation. Biochim. Biophys. Acta. 1503:147–163. [DOI] [PubMed] [Google Scholar]
- 50.Astashkin, A. V., H. Mino, A. Kawamori, and T.-A. Ono. 1997. Pulsed EPR study of the S′3 signal in the Ca2+-depleted photosystem II. Chem. Phys. Lett. 272:506–516. [Google Scholar]
- 51.Mino, H., and A. Kawamori. 2001. EPR studies of the water oxidizing complex in the S1 and the higher S states: the manganese cluster and YZ radical. Biochim. Biophys. Acta. 1503:112–122. [DOI] [PubMed] [Google Scholar]
- 52.Knoepfle, N., T. M. Bricker, and C. Putnam-Evans. 1999. Site-directed mutagenesis of basic arginine residues 305 and 342 in the CP43 protein of photosystem II affects oxygen-evolving activity in Synechocystis 6803. Biochemistry. 38:1582–1588. [DOI] [PubMed] [Google Scholar]
- 53.Ermakova-Gerdes, S., Z. Yu, and W. Vermaas. 2001. Targeted random mutagenesis to identify functionally important residues in the D2 protein of photosystem II in Synechocystis sp. strain PCC 6803. J. Bacteriol. 183:145–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morgan, T. R., J. A. Shand, S. M. Clarke, and J. J. Eaton-Rye. 1998. Specific requirements for cytochrome c-550 and the manganese-stabilizing protein in photoautotrophic strains of Synechocystis sp. PCC 6803 with mutations in the domain Gly-351 to Thr-436 of the chlorophyll-binding protein CP47. Biochemistry. 37:14437–14449. [DOI] [PubMed] [Google Scholar]
- 55.Diner, B. A., and G. T. Babcock. 1996. Oxygenic Photosynthesis: The Light Reactions. D.R. Ort and C.F. Yocum, editors. Kluwer Academic Publishers, Dordrecht, Netherlands. 213–247.
- 56.Mulkidjanian, A. Y. 1999. Photosystem II of green plants: on the possible role of retarded protonic relaxation in water oxidation. Biochim. Biophys. Acta. 1410:1–6. [DOI] [PubMed] [Google Scholar]
- 57.Manna, P., R. LoBrutto, C. Eijckelhoff, J. P. Dekker, and W. Vermaas. 1998. Role of Arg180 of the D2 protein in photosystem II structure and function. Eur. J. Biochem. 251:142–154. [DOI] [PubMed] [Google Scholar]
- 58.Joliot, P., A. Joliot, and B. Kok. 1968. Analysis of the interactions between the two photosystems in isolated chloroplasts. Biochim. Biophys. Acta. 153:635–652. [DOI] [PubMed] [Google Scholar]
- 59.Styring, S., and A. W. Rutherford. 1987. In the oxygen-evolving complex of photosystem II the S0 state is oxidized to the S1 state by D+ (signal IIslow). Biochemistry. 26:2401–2405. [Google Scholar]