

Abstract
The discovery of RNA interference (RNAi) in C. elegans and in plants has revolutionized current approaches to biology and medicine. RNAi silences genes in a sequence-specific manner through the actions of small pieces of double-stranded RNAs (siRNAs and miRNAs). RNAi has been found as a widespread natural phenomenon in eukaryotic cells and is also being used as a powerful experimental tool to explore gene function. Most importantly, it has many potential therapeutic applications. Viral gene-specific siRNAs are theoretically very promising antiviral inhibitors and have been examined in a broad range of medically important viruses. However, many RNA viruses escape RNAi-mediated suppression by counteracting the RNAi machinery through mutation of the targeted region, by encoding viral suppressors, or both. DNA viruses also counteract the RNAi machinery, preferentially using viral suppressors. Cellular factors may also contribute to RNAi resistance; ADAR1 was the first cellular factor found to be responsible for editing-mediated RNAi resistance. Because siRNAs can be used as potent small-molecule inhibitors of any cellular gene, the best way for a cell to maintain expression of essential genes for its long-term survival is to develop a program to resist the detrimental effects of RNAi.
Keywords: RNA interference, siRNA, miRNA, gene expression, viral inhibitors, gene therapies
INTRODUCTION
RNA interference (RNAi) has recently been recognized as an important mechanism for controlling gene expression at a post-transcriptional level19,49 and regulating heterochromatin formation for genome stability 54,69. RNAi-mediated post-transcriptional gene silencing can restrict expression of certain genes during tissue development or cell differentiation. Small RNA duplexes, usually 21 to 23 nucleotides (nts) in size, are generated by cleavage of larger double-stranded (ds) RNAs by the ribonuclease Dicer 53,75. These small RNA duplexes trigger RNAi by sequence-specific cleavage and degradation of the homologous single-stranded mRNA or by suppression of mRNA translation 19,28,46.
Based on their origin and function, the small RNAs produced by Dicer cleavage are classified into two categories: small interfering RNAs (siRNAs) and microRNAs (miRNAs). Both siRNAs and miRNAs are ∼21- to 23-bp duplexes bearing 2-nt overhangs at the 3′ ends. They differ in that siRNAs originate in the cytoplasm from cleavage of long dsRNAs with perfect base-pairing26,73, whereas miRNAs are derived from cleavage of long, single-stranded primary transcripts containing imperfectly matched hairpin-loop structures. In general, primary miRNA transcripts are cleaved first by the RNase III-like protein Drosha into ∼65 nt stem-loop pre-miRNAs in the nucleoplasm34,35 before being transported by Exportin-5 into the cytoplasm 44, where they are further processed by Dicer into ∼21- to 23-bp miRNA duplexes with imperfect base-pairing. For both siRNAs and miRNAs, one strand (antisense) is then selectively incorporated into a dsRNA-induced silencing complex (RISC) that contains Argonaute (eIF2C), SMN3, and several other proteins 27,46,47 and guides the selection of a complementary mRNA for cleavage or for translational inhibition. Recently, Ago 2 was shown to be a key component in cleavage of the substrate 27,55. The accumulated evidence so far indicates that siRNAs and miRNAs program RISCs equivalently and that they produce similar effects on mRNA expression despite their somewhat different origins 16,74.
RNAi can be induced by the introduction of synthetic ∼21- to 23-nt siRNA duplexes into cells, bypassing the requirement for processing of a long dsRNA mediated by Dicer 20. RNAi mediated by siRNAs or miRNAs artificially introduced into mammalian cells has been successfully used to knock down gene expression and to suppress viral replication, although the outcome of individual RNAi events varies depending on the designed siRNAs and their targeted regions. Efficient silencing of a target using the RNAi approach also requires active cell metabolism, with the cells undergoing exponential growth. In most transient transfection assays of synthetic siRNA duplexes, one run of RNAi at an effective dose generally confers detectable suppression of the target for no more than 48-72 hours (Fig. 1). Several runs of RNAi may be needed to achieve more extensive or longer-lasting suppression. Although an individual siRNA’s half-life and its targeted region influence the proficiency of RNAi, active RISC function in cells is also necessary to make the suppression effective. Cells under exponentially active growth both confer efficient up-take of siRNA duplexes and have active RISC function. As shown in Fig. 1, cells at 96 hours in culture show no response to RNAi (compare lane 7 to lane 6) and thereby are not affected by siRNA-mediated p53 suppression. Although “RNAi resistance” usually refers to more active processes, as discussed below, care should be taken to study RNAi and RNAi resistance in cells at the appropriate phase of growth.
Fig. 1.
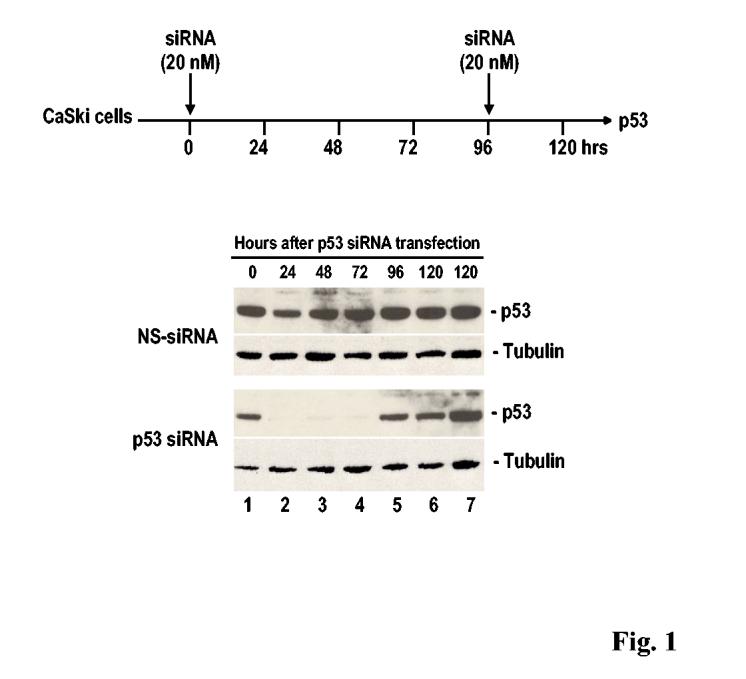
RNAi mediated by synthetic siRNA duplexes depends on active cell growth. Efficiencies of synthetic p53 siRNA and non-specific (NS) siRNA duplexes on cellular p53 expression were assayed in CaSki cells at 2 × 105 at 20-24 hours (0 time) after plating by transient transfection by using oligofactamine (Invitrogen). Cell samples were prepared at 24, 48, 72, 96, and 120 hours after transfection of each siRNA (20 nM) as diagramed above and examined by Western blot for p53 by using anti-p53 antibody (Oncogene). Cells with (lane 7) or without (lane 6) re-transfection with either NS or p53 siRNAs at 96 hours of the initial siRNA transfection, as indicated in diagram above, were further compared and showed no difference in responding to p53 siRNAs at 120 h post the initial transfection.
Although it has been known for a while in plant virology that many plant viruses have to counteract RNAi in plant cells in order to invade their hosts, the many reports of the development of RNAi resistance in mammalian cells came as a surprise. Approximately 22 silencing suppressors have been described in plant and insect viruses; these have been summarized in several excellent reviews 15,38,68,72. In this report, we emphasize the emergence of siRNA resistance as a mechanism that counteracts RNAi-mediated gene silencing in mammalian cells (Table 1).
Table 1.
Mechanisms in the development of resistance to RNAi in mammalian cells
Development of resistance to siRNA-mediated viral gene silencing in RNA virus infections
RNA viruses have an RNA genome that can be either single-stranded or double-stranded. Viral RNA replication in infected cells is mediated by a viral RNA-dependent RNA polymerase (RDRP), which produces a dsRNA intermediate phase even in single-stranded RNA viruses. Because it lacks proofreading activity, this viral RDRP replicates the RNA genome with low fidelity, resulting in a mutation rate on the order of ∼10-4 per incorporated nucleotide. That is, viral RDRP has an inherent error frequency on the order of 1 in 104, depending on the virus 17,36. Thus, it is conceivable that RNA viruses under selection pressure may escape siRNA-mediated suppression through mutation of the target regions, thus becoming siRNA-resistant viruses. This presumption has been supported by accumulating evidence from several elegant studies, as described below.
Using RNAi as an antiviral therapy has been considered for many viruses, including both DNA viruses and RNA viruses. Poliovirus was one of the first to be tested for siRNA-mediated sequence-specific inhibition. HeLa cells treated with an siRNA specific for a capsid protein or viral polymerase before infection with a virulent poliovirus produced only 1%-3% of the virus titer observed in control-treated cells and showed no virus plaque formation. However, the poliovirus readily escapes these highly effective siRNAs through unique point mutations within the targeted regions 23. The emergence of siRNA-resistant viruses begins 30-40 hours postinfection. Further analysis of these resistant mutants showed that RNAi recognition is sensitive to subtle point mutations within the central region and the 3′ end of the target RNA. Even a single point mutation resulting in a G:U mismatch is sufficient for the virus to escape siRNA inhibition 24. Using a pool of siRNAs to simultaneously target multiple sites in the virus genome might be a good choice to prevent the emergence of resistant viruses.
RNAi has been demonstrated to be a powerful tool for suppressing HIV infection in various transient, short-term assays 11,29 33,48. Recently, resistance to RNAi directed against HIV-1 has been noticed when siRNA duplexes targeting the viral tat 1 or nef 12 genes are stably expressed in human T cells for long-term inhibition. RNAi-resistant HIV-1 variants have been identified with deletions or substitutions in the siRNA target regions. Through these mutations, HIV-1 removes its RNA as a target and escapes from RNAi-mediated inhibition.
Resistance to siRNA-mediated suppression of La Crosse Virus (LACV) replication has also been noticed after only 72 hours of virus infection in 293T cells pre-treated with LACV siRNAs 61. Most of the resistant viruses recovered from culture supernatant show single or double nucleotide changes in the targeted regions, with a significantly high frequency in the portion of the S segment targeted by the siRNA LACS103. More interestingly, a viral nonstructural protein of LACV, NSs, appears to function as an RNAi suppressor protein. Cells expressing the NSs protein strongly suppress RNAi directed against both an LACV M segment and a host gene, GAPDH (glyeraldehyde-3-phosphate dehydrogenase) 61.
The Tas protein from the retrovirus primate foamy virus type 1 (PFV-1) has recently been identified as a suppressor of miR-32, an miRNA of cellular origin. The PFV-1 open reading frame 2 contains a target for miR-32. As a counterdefense to miR-32-mediated suppression, PFV-1 encodes Tas as a silencing suppressor that enables host cell invasion and virus replication 32.
Development of resistance to siRNA-mediated viral gene silencing in DNA virus infections
Compared to RNA viruses, which are designed to have a high rate of mutation during their RNA genome replication, the genomes of DNA viruses are relatively stable, with a low mutation rate of 10-8 to 10-11 per incorporated nucleotide. Thus, resistance to RNAi-mediated viral gene silencing though mutation of the target sequence has not been observed in DNA virus infection. However, siRNA may exert selection pressure on pre-existing resistant mutants, as exemplified by a recent report on hepatitis B virus (HBV) 70. HBV is a DNA virus, but its DNA replicates through a genomic RNA intermediate and utilizes a virally encoded reverse transcriptase. Consequently, a significant amount of diversity, similar to that seen in RNA viruses, occurs in the sequences of HBV isolates; these can be classified into at least 8 genotypes (A-H). A mutant C genotype that contains a mutation in the 3′ portion of the targeted region was isolated and was more resistant to HBVS1 short hairpin (sh) RNA-mediated RNAi than the wild-type. This mutant can be preferentially selected out in the presence of HBVS1 shRNA in Huh-7 cells co-transfected with wild-type and mutant HBV-C at a ratio of 9:1 at day 5 post-transfection 70.
Adenovirus infection may also result in inhibition of RNAi, perhaps through the actions of a virally encoded noncoding RNA, VA1. Adenovirus VA1 RNA is an ∼160-nt structured RNA that is highly expressed in adenovirus-infected cells (∼108 molecules/cell) and plays important roles in increasing viral mRNA translation 65 and stabilizing ribosome-bound mRNAs 63,64. VA1 RNA enhances viral mRNA translation by blocking activation of protein kinase R (PKR), which is usually induced by viral dsRNA produced during the adenoviral replication cycle. Cullen and colleagues recently used a reporter assay to show that VA1 RNA inhibits RNAi induced by shRNAs, but does not affect RNAi induced by synthetic siRNA duplexes. VA1 RNA-mediated inhibition of RNAi appears to occur through inhibition of nuclear export of shRNA, competition for Exportin-5, and inhibition of Dicer function by direct binding of Dicer 43.
Our recent study on shRNA-mediated oncogene silencing of human papillomavirus 16 (HPV16) in cervical cancer cells provides evidence on how a DNA virus might escape RNAi (Tang, S., et al., unpublished data). HPV16 contains two viral oncogenes that encode E6, which targets p53 for degradation, and E7, which targets pRB. The two oncogenes are transcribed as a single E6E7 bicistronic RNA and are essential genes for the survival of cervical cancer cells. Transfection of two HPV16-positive cervical cancer cell lines, CaSki and SiHa, with a synthetic siRNA duplex based on a sequence motif of 21 nts from the HPV16 E6E7 bicistronic RNA effectively suppressed both the E6 and E7 oncogenes, leading to stabilization of p53 and increased p21 expression. When stably expressed as a short hairpin RNA in these cells under selection pressure, however, the siRNA silencing of E6 and E7 expression was efficient only at early cell passages, and became inefficient with additional cell passages despite the continued expression of siRNA at the same level. This loss of the siRNA function could be duplicated in cells with stable p53 siRNA, but not in cells with stable lamin A/C siRNA, suggesting that it is target-selective. The siRNA-resistant cells retain normal siRNA processing, duplex unwinding and degradation of the unwound sense strand, and RISC formation, suggesting that the loss of siRNA function occurred at a later step. Surprisingly, the siRNA-resistant cells were found to contain a cytoplasmic protein of ∼50 kDa that specifically and characteristically interacted with the E6E7 siRNA. These data indicate that a potent siRNA targeting an essential or regulatory gene might induce a cell to develop siRNA resistance through the production of a specific protein.
Viral proteins from mammalian viruses function as suppressors of RNAi in non-mammalian cells and plants
Several viral proteins expressed from mammalian viruses, the interferon antagonist proteins of influenza virus NS1 (non-structural protein 1), reovirus σ3, and vaccinia virus E3L, have been examined for their suppressive effects on RNAi in non-mammalian cells.
Influenza viruses are a group of single-stranded RNA viruses and are divided into types A, B, and C. The influenza virus NS1 protein is a multifunctional, non-structural protein that contains three functional domains. Two of the domains are in the N-terminal half of NS1: one is responsible for translational enhancement of the influenza virus mRNAs through interactions with eIF4G, a eukaryotic translation initiation factor, and the other is essential for dsRNA-binding and antagonizing interferon induction by sequestering dsRNA from PKR binding and activation. The third domain, which inhibits mRNA processing and nuclear-cytoplasmic transport of host mRNAs, is in the C-terminal half of the protein 31. Using a plant silencing suppression assay, two groups simultaneously demonstrated that the influenza NS1 protein is an RNA silencing suppressor in plants and promotes plant virus pathogenicity of potato virus X in three different plant hosts 3,14. NS1 suppression of RNAi requires the dsRNA-binding domain of the protein to be functional and to interact with the siRNAs 3. These observations were further supported by independent evidence from the Ding and Palese groups showing that all three NS1 proteins from the influenza A, B, and C viruses are RNAi suppressors in a Drosophila cell-based, nodaviral silencing screen assay. This study showed that NS1 from influenza A viruses also suppresses RNAi in Drosophila cells through its N-terminal dsRNA-binding domain and its binding of siRNAs 39.
Reoviruses are a group of dsRNA viruses. Reovirus outer shell polypeptide σ3 is one of the best-characterized dsRNA binding proteins. Like influenza virus NS1, reovirus σ3 carries conserved dsRNA-binding motifs and binds dsRNAs in vitro and in vivo. Accordingly, reovirus σ3 protein sequesters dsRNA from PKR binding and thereby prevents activation by dsRNA. When tested in plant cells, σ3 showed strong RNAi suppression, although it failed to sequester miRNA precursors 40. Nevertheless, the data suggest that the reovirus σ3 protein is capable of counteracting RNAi-mediated gene silencing in addition to inhibiting PKR-mediated responses.
Vaccinia virus is a member of the poxvirus family and has a DNA genome that replicates in the cytoplasm during viral infection. The vaccinia E3L protein is a dsRNA-binding protein 13 that inhibits PKR by sequestering dsRNA from PKR, thus preventing binding 56,59. The C-terminus of the vaccinia virus E3L is responsible for binding to dsRNA and preventing it from activating the interferon pathway. A recent study demonstrated that the E3L protein is a functional suppressor of RNAi in Drosophila cells that inactivates the RNAi silencing-based antiviral response of the cells to flock house virus infection 39.
RNA editing plays a role in the development of siRNA resistance in mammalian cells
Double-stranded RNA induces the homology-dependent degradation of cognate mRNA in the cytoplasm via RNAi, but it is also a target for adenosine-to-inosine (A-to-I) RNA editing by adenosine deaminases acting on RNA (ADARs). RNA editing that affects siRNA-mediated RNAi in vitro was first reported by Chris Smith’s group 58, who showed that production of siRNAs could be progressively inhibited with increasing deamination of a long dsRNA. This initial observation was immediately supported by a study in C. elegans that showed that A-to-I editing of dsRNAs derived from both transgenes and endogenous genes indeed appeared to prevent their silencing by RNAi 30,67. Recent studies further demonstrated a direct interaction between three isoforms of ADARs and siRNA, two of which, ADAR1 and ADAR2, strongly bind siRNA without RNA editing. ADAR1p110, a short form of ADAR1 via an alternative translation initiation codon, and ADAR2 also bound a 19-bp siRNA, but their binding affinities were 15 and 50 times lower than that of ADAR1p150 (a full length ADAR1), respectively. ADAR3 bound longer dsRNAs, but failed to bind the 19-bp siRNA. All ADARs that were capable of binding the 19-bp siRNA (ADAR1p150 and p110 and ADAR2) also bound siRNAs containing either 15- or 23-bp dsRNA regions. Thus, the length of the siRNA determines whether the bound siRNA is edited or held in a stable complex without a change of sequence; the critical size threshold appears to be 30 bp 71.
The cytoplasmic full-length isoform of ADAR1 has the highest affinity for siRNA among known ADARs, with a subnanomolar dissociation constant. Gene silencing by siRNA is significantly more effective in mouse fibroblasts homozygous for an ADAR1 null mutation than in wild-type cells. This was further supported by the suppression of RNAi in fibroblast cells overexpressing functional ADAR1, but not in cells overexpressing mutant ADAR1 lacking double-stranded RNA-binding domains. The results provide convincing evidence that ADAR1 is a cellular factor that limits the efficacy of siRNA in mammalian cells 71.
Other factors that might lead to RNAi resistance in mammalian cells
As described above, resistance to RNAi during viral infection in mammalian cells has thus far been ascribed to two major mechanisms: mutations in the targeted regions and expression of suppressors (Table 1). One might wonder whether viruses have also evolved mechanism(s) to counteract the initiation of the RNAi pathway, rather than to block the pathway’s intermediate components.
This hypothesis has received some preliminary support from a hepatitis delta virus (HDV) study. Data from Taylor’s group indicate that HDV RNAs are resistant to Dicer activity 7. Dicer cleaves RNAs that are 100% double-stranded, as well as certain RNAs with extensive but <100% pairing, to release RNAs of ∼21 nt. The circular 1,679-nt genome of HDV and its exact complement, the antigenomic RNA, can fold into a rod-like structure with 74% pairing, but show resistance to Dicer cleavage. In an in vitro cleavage assay with purified recombinant Dicer, <0.2% of unit-length HDV RNAs were cleaved, even though Dicer was able to digest fragments of the genome. A 66-nt hairpin RNA with 79% pairing was digested with >80% cleavage activity, and a 66-nt hairpin derived from one end of the HDV genome was also digested. The data from these in vitro Dicer digestion experiments are consistent with the results that small RNAs of ∼21 nts cannot be detected during HDV replication in hepatocytes 7. That unit-length, highly structured HDV RNAs resist Dicer cleavage suggests the some viral RNAs have evolved to prevent initiation of RNAi by resisting Dicer. Although HDV itself can be edited by ADAR at adenosine 1012 position in the antigenome 6,52, its resistance to Dicer and RNAi appears unrelated to ADAR-mediated editing.
The same group also show that the 1,679-nt genomic and antigenomic RNAs of HDV are resistant to siRNA-mediated degradation, although its viral mRNAs are sensitive to synthetic siRNA duplexes. It was thought that the base-paring of HDV genomic and antigenomic RNAs within the nucleus might make them inaccessible to siRNA. However, further studies show that HDV circular RNAs transcribed from a DNA template by host RNA polymerase II in Huh-7 cells, which have a similar mRNA structure to mRNAs with a 5′ cap and a 3′ poly A tail, were also resistant to siRNA attack 8. The mechanism by which HDV becomes resistant to siRNA remains unknown.
Does siRNA activate the protein kinase PKR?
PKR, a mammalian dsRNA-dependent serine-threonine protein kinase, has an N-terminal dsRNA-binding domain that can interact non-sequence-specifically with long stretches of dsRNA, leading to activation. The activated PKR phosphorylates and inactivates the translation factor eIF2“, resulting in a generalized suppression of protein synthesis, followed by cell death via both apoptotic and nonapoptotic pathways. Activation of this nonspecific pathway could mask any sequence-specific effects that might result from the RNAi pathway. Nevertheless, successful application of RNAi has been reported in many mammalian tissue culture cell lines by using chemically synthesized siRNAs that bypass activation of PKR. It has been documented in the literature that a small blunt-ended dsRNA of less than 33 bp does not activate PKR 45. In addition, early studies demonstrated that siRNA-mediated inhibition of gene expression in mammalian cells is independent of non-specific interference pathways activated by larger dsRNAs 5. However, two recent studies indicate that, although the RNAi mechanism itself is independent of the interferon system, transfection of 21-bp siRNAs or expression of shRNAs from shRNA vectors does trigger an interferon response through PKR 2,60. Thus, these studies argue that siRNAs may have broad and complicating effects beyond the selective silencing of target genes when introduced into cells.
In an effort to evaluate these contradictory results and search for possible activators of PKR, truncated and full-length versions of PKR were used to select dsRNAs from a partially structured dsRNA library containing 1011 sequences 76. This study identified a minimal RNA motif for activation of PKR. This motif has a hairpin with a nonconserved, imperfect 16-bp dsRNA stem flanked by 11- to 15-nt single-stranded tails. Boundary experiments revealed that the single-stranded tails flanking the dsRNA core provide the critical determinant for activation. A minimum of 11 nts in the tails is needed for the activation, although the tails could be on the 5′-end, the 3′-end, or both 76. This group also compared dsRNAs with lengths of 24, 33, and 76 bp for PKR binding and activation and found that a 24-bp dsRNA was unable to activate PKR; instead, it inhibited the kinase. Resolution of the discrepancies among the findings of these different study groups will require additional biochemical experiments.
Closing remarks and perspectives
We are entering an RNAi era. The discovery of RNAi in C. elegans 21 and in plants 25 has revolutionized today’s approaches to biology and medicine. Through the actions of small pieces of dsRNAs (siRNAs and miRNAs), RNAi-mediated gene silencing has been found to be not only a widespread natural phenomenon in eukaryotes, but also a powerful experimental tool to explore gene function. Most importantly, it has many potential therapeutic applications. Although a great deal has been learned in the past few years about RNAi pathways and the functions of their components, much remains to be understood before we have a complete picture of how RNAi is regulated in mammalian cells. There is absolutely no doubt that RNAi pathways, just like many other pathways discovered in mammalian systems, will be subject to sophisticated regulation (Fig. 2).
Fig. 2.

RNAi pathways and RNAi resistance. Despite their different origins, both siRNA and microRNA have similar effects on mRNA expression through RISC-mediated activities. Development of RNAi resistance in mammalian cells could be because of any possible blocks (-) in RNAi pathways.
One RNAi function in mammalian cells is thought to be a nucleic acid-based antiviral immunity, as evidenced by the recent discovery that cellular miR-32 mediates antiviral activities in human cells 32. Identification of viral RNAi suppressors in medically important RNA and DNA viruses has heralded the beginning of exploration into existing anti-silencing mechanisms in virus infection. Many of the RNAi suppressors identified so far in various RNAi reporter assays turn out to be viral dsRNA binding proteins (DRBPs). Although fewer cellular DRBPs have been identified than viral DRBPs, one may assume that they will also regulate endogenous miRNA or siRNA functions for the cell whenever it is needed. About 300 miRNAs have been identified in the genomes of humans, viruses, and non-human eukaryotic species, some of which are very abundant (∼ 40,000 copies/cell) 4,41,42,50,51. The question is how to examine a DRBP’s suppressive function in a mammalian system. The suppressive functions of viral RNA suppressors in RNAi reporter assays have not been recapitulated in any in vivo viral infections in mammals. For example, NS1 of Influenza A virus behaves as an effective RNAi suppressor in Drosophila and plant RNAi reporters, but it seems to play no role in siRNA-mediated inhibition of animal influenza infection 22,66.
There are at least 388 known eukaryotic proteins with dsRNA-binding domains (DRBDs), 72 of which are human 37,62. More than 20 DRBPs have been identified and are reported to function in a diverse range of critically important roles in the cell, including some (such as Dicer and Ago-2) with essential roles in RNAi pathways 18,57. Since proteins harboring DRBDs have been reported to interact with as little as 11 bp of dsRNA, an event that is independent of nucleotide sequence arrangement 57, it is conceivable that many cellular DRBPs might actually be involved in switching on and off the function of a partially double-stranded miRNA that, on average, targets hundreds or more mRNAs through pairing of only 6-8 bases. ADAR1 is the only factor of this type that interacts with siRNAs in vitro and limits the efficacy of siRNAs in mammalian cells.
Perhaps the most intriguing question is why a natural siRNA has not been found in eukaryotic cells with or without virus infection. Some RNA viruses have a dsRNA genome and others carry a single-stranded RNA genome, but produce dsRNA intermediates during their replication. Theoretically, these viral dsRNAs could be substrates for Dicer digestion to produce siRNAs in the infected cells. However, viral dsRNAs appear to resist Dicer digestion, as exemplified by HDV. This may be because production of viral siRNAs to suppress viral gene expression would be harmful to replication of the virus. Similarly, endogenous siRNA production from the human genome would be expected, considering that over 20% of human genes produce antisense transcripts that might form sense-antisense pairs (dsRNA) 9, but production of endogenous siRNAs from these human transcripts would not be beneficial for human gene expression. Discovering how these dsRNAs escape Dicer digestion will be another challenge.
Understanding viral RNAi resistance has greatly enriched our knowledge of the regulation of RNAi pathways. It has also been greatly informative to know that some viruses escape RNAi-mediated inhibition through one or more counteracting mechanisms: mutations in the targeted region under siRNA selection pressure and/or production of RNAi suppressors. However, HPV16 seems to use another method to develop siRNA resistance, because the proteins that appear to be involved in siRNA resistance in cells expressing HPV16 E6 and E7 are neither viral proteins nor ADAR1. Identification and characterization of such proteins will be extremely challenging, but will definitely guide us to further understanding of how a cell counteracts siRNA-mediated events that would adversely affect its own survival. Since siRNAs can be used as potent small-molecule inhibitors to any cellular gene, the best way for a cell to keep expression of those genes that are essential for its survival is to develop a program to resist the detrimental effects of RNAi.
Reference List
- 1.Boden D, Pusch O, Lee F, Tucker L, Ramratnam B. Human immunodeficiency virus type 1 escape from RNA interference. J. Virol. 2003;77:11531–11535. doi: 10.1128/JVI.77.21.11531-11535.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bridge AJ, Pebernard S, Ducraux A, Nicoulaz AL, Iggo R. Induction of an interferon response by RNAi vectors in mammalian cells. Nat. Genet. 2003;34:263–264. doi: 10.1038/ng1173. [DOI] [PubMed] [Google Scholar]
- 3.Bucher E, Hemmes H, de Haan P, Goldbach R, Prins M. The influenza A virus NS1 protein binds small interfering RNAs and suppresses RNA silencing in plants. J. Gen. Virol. 2004;85:983–991. doi: 10.1099/vir.0.19734-0. [DOI] [PubMed] [Google Scholar]
- 4.Cai X, Lu S, Zhang Z, Gonzalez CM, Damania B, Cullen BR. Kaposi’s sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc. Natl. Acad. Sci. U. S. A. 2005;102:5570–5575. doi: 10.1073/pnas.0408192102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caplen NJ, Parrish S, Imani F, Fire A, Morgan RA. Specific inhibition of gene expression by small double-stranded RNAs in invertebrate and vertebrate systems. Proc. Natl. Acad. Sci. U. S. A. 2001;98:9742–9747. doi: 10.1073/pnas.171251798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Casey JL, Gerin JL. Hepatitis D virus RNA editing: specific modification of adenosine in the antigenomic RNA. J. Virol. 1995;69:7593–7600. doi: 10.1128/jvi.69.12.7593-7600.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang J, Provost P, Taylor JM. Resistance of human hepatitis delta virus RNAs to dicer activity. J. Virol. 2003;77:11910–11917. doi: 10.1128/JVI.77.22.11910-11917.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang J, Taylor JM. Susceptibility of human hepatitis delta virus RNAs to small interfering RNA action. J. Virol. 2003;77:9728–9731. doi: 10.1128/JVI.77.17.9728-9731.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen J, Sun M, Kent WJ, Huang X, Xie H, Wang W, Zhou G, Shi RZ, Rowley JD. Over 20% of human transcripts might form sense-antisense pairs. Nucleic Acids Res. 2004;32:4812–4820. doi: 10.1093/nar/gkh818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Child SJ, Hakki M, De Niro KL, Geballe AP. Evasion of cellular antiviral responses by human cytomegalovirus TRS1 and IRS1. J. Virol. 2004;78:197–205. doi: 10.1128/JVI.78.1.197-205.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coburn GA, Cullen BR. Potent and specific inhibition of human immunodeficiency virus type 1 replication by RNA interference. J. Virol. 2002;76:9225–9231. doi: 10.1128/JVI.76.18.9225-9231.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Das AT, Brummelkamp TR, Westerhout EM, Vink M, Madiredjo M, Bernards R, Berkhout B. Human immunodeficiency virus type 1 escapes from RNA interference-mediated inhibition. J. Virol. 2004;78:2601–2605. doi: 10.1128/JVI.78.5.2601-2605.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davies MV, Chang HW, Jacobs BL, Kaufman RJ. The E3L and K3L vaccinia virus gene products stimulate translation through inhibition of the double-stranded RNA-dependent protein kinase by different mechanisms. J. Virol. 1993;67:1688–1692. doi: 10.1128/jvi.67.3.1688-1692.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Delgadillo MO, Saenz P, Salvador B, Garcia JA, Simon-Mateo C. Human influenza virus NS1 protein enhances viral pathogenicity and acts as an RNA silencing suppressor in plants. J. Gen. Virol. 2004;85:993–999. doi: 10.1099/vir.0.19735-0. [DOI] [PubMed] [Google Scholar]
- 15.Ding SW, Li H, Lu R, Li F, Li WX. RNA silencing: a conserved antiviral immunity of plants and animals. Virus Res. 2004;102:109–115. doi: 10.1016/j.virusres.2004.01.021. [DOI] [PubMed] [Google Scholar]
- 16.Doench JG, Petersen CP, Sharp PA. siRNAs can function as miRNAs. Genes Dev. 2003;17:438–442. doi: 10.1101/gad.1064703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dougherty JP, Temin HM. Determination of the rate of base-pair substitution and insertion mutations in retrovirus replication. J. Virol. 1988;62:2817–2822. doi: 10.1128/jvi.62.8.2817-2822.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doyle M, Jantsch MF. New and old roles of the double-stranded RNA-binding domain. J. Struct. Biol. 2002;140:147–153. doi: 10.1016/s1047-8477(02)00544-0. [DOI] [PubMed] [Google Scholar]
- 19.Dykxhoorn DM, Novina CD, Sharp PA. Killing the messenger: short RNAs that silence gene expression. Nat. Rev. Mol. Cell Biol. 2003;4:457–467. doi: 10.1038/nrm1129. [DOI] [PubMed] [Google Scholar]
- 20.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 21.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 22.Ge Q, Filip L, Bai A, Nguyen T, Eisen HN, Chen J. Inhibition of influenza virus production in virus-infected mice by RNA interference. Proc. Natl. Acad. Sci. U. S. A. 2004;101:8676–8681. doi: 10.1073/pnas.0402486101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gitlin L, Karelsky S, Andino R. Short interfering RNA confers intracellular antiviral immunity in human cells. Nature. 2002;418:430–434. doi: 10.1038/nature00873. [DOI] [PubMed] [Google Scholar]
- 24.Gitlin L, Stone JK, Andino R. Poliovirus escape from RNA interference: short interfering RNA-target recognition and implications for therapeutic approaches. J. Virol. 2005;79:1027–1035. doi: 10.1128/JVI.79.2.1027-1035.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hamilton AJ, Baulcombe DC. A species of small antisense RNA in posttranscriptional gene silencing in plants. Science. 1999;286:950–952. doi: 10.1126/science.286.5441.950. [DOI] [PubMed] [Google Scholar]
- 26.Hammond SM, Bernstein E, Beach D, Hannon GJ. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature. 2000;404:293–296. doi: 10.1038/35005107. [DOI] [PubMed] [Google Scholar]
- 27.Hammond SM, Boettcher S, Caudy AA, Kobayashi R, Hannon GJ. Argonaute2, a link between genetic and biochemical analyses of RNAi. Science. 2001;293:1146–1150. doi: 10.1126/science.1064023. [DOI] [PubMed] [Google Scholar]
- 28.Hannon GJ. RNA interference. Nature. 2002;418:244–251. doi: 10.1038/418244a. [DOI] [PubMed] [Google Scholar]
- 29.Jacque JM, Triques K, Stevenson M. Modulation of HIV-1 replication by RNA interference. Nature. 2002;418:435–438. doi: 10.1038/nature00896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knight SW, Bass BL. The role of RNA editing by ADARs in RNAi. Mol. Cell. 2002;10:809–817. doi: 10.1016/s1097-2765(02)00649-4. [DOI] [PubMed] [Google Scholar]
- 31.Krug RM, Yuan W, Noah DL, Latham AG. Intracellular warfare between human influenza viruses and human cells: the roles of the viral NS1 protein. Virology. 2003;309:181–189. doi: 10.1016/s0042-6822(03)00119-3. [DOI] [PubMed] [Google Scholar]
- 32.Lecellier CH, Dunoyer P, Arar K, Lehmann-Che J, Eyquem S, Himber C, Saib A, Voinnet O. A cellular microRNA mediates antiviral defense in human cells. Science. 2005;308:557–560. doi: 10.1126/science.1108784. [DOI] [PubMed] [Google Scholar]
- 33.Lee NS, Dohjima T, Bauer G, Li H, Li MJ, Ehsani A, Salvaterra P, Rossi J. Expression of small interfering RNAs targeted against HIV-1 rev transcripts in human cells. Nat. Biotechnol. 2002;20:500–505. doi: 10.1038/nbt0502-500. [DOI] [PubMed] [Google Scholar]
- 34.Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, Kim S, Kim VN. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- 35.Lee Y, Jeon K, Lee JT, Kim S, Kim VN. MicroRNA maturation: stepwise processing and subcellular localization. EMBO J. 2002;21:4663–4670. doi: 10.1093/emboj/cdf476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leider JM, Palese P, Smith FI. Determination of the mutation rate of a retrovirus. J. Virol. 1988;62:3084–3091. doi: 10.1128/jvi.62.9.3084-3091.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Letunic I, Copley RR, Schmidt S, Ciccarelli FD, Doerks T, Schultz J, Ponting CP, Bork P. SMART 4.0: towards genomic data integration. Nucleic Acids Res. 2004;32:D142–D144. doi: 10.1093/nar/gkh088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li WX, Ding SW. Viral suppressors of RNA silencing. Curr. Opin. Biotechnol. 2001;12:150–154. doi: 10.1016/s0958-1669(00)00190-7. [DOI] [PubMed] [Google Scholar]
- 39.Li WX, Li H, Lu R, Li F, Dus M, Atkinson P, Brydon EW, Johnson KL, Garcia-Sastre A, Ball LA, Palese P, Ding SW. Interferon antagonist proteins of influenza and vaccinia viruses are suppressors of RNA silencing. Proc. Natl. Acad. Sci. U. S. A. 2004;101:1350–1355. doi: 10.1073/pnas.0308308100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lichner Z, Silhavy D, Burgyan J. Double-stranded RNA-binding proteins could suppress RNA interference-mediated antiviral defences. J. Gen. Virol. 2003;84:975–980. doi: 10.1099/vir.0.18987-0. [DOI] [PubMed] [Google Scholar]
- 41.Lim LP, Glasner ME, Yekta S, Burge CB, Bartel DP. Vertebrate microRNA genes. Science. 2003;299:1540. doi: 10.1126/science.1080372. [DOI] [PubMed] [Google Scholar]
- 42.Lim LP, Lau NC, Weinstein EG, Abdelhakim A, Yekta S, Rhoades MW, Burge CB, Bartel DP. The microRNAs of Caenorhabditis elegans. Genes Dev. 2003;17:991–1008. doi: 10.1101/gad.1074403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu S, Cullen BR. Adenovirus VA1 noncoding RNA can inhibit small interfering RNA and MicroRNA biogenesis. J. Virol. 2004;78:12868–12876. doi: 10.1128/JVI.78.23.12868-12876.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lund E, Guttinger S, Calado A, Dahlberg JE, Kutay U. Nuclear export of microRNA precursors. Science. 2004;303:95–98. doi: 10.1126/science.1090599. [DOI] [PubMed] [Google Scholar]
- 45.Manche L, Green SR, Schmedt C, Mathews MB. Interactions between double-stranded RNA regulators and the protein kinase DAI. Mol. Cell Biol. 1992;12:5238–5248. doi: 10.1128/mcb.12.11.5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martinez J, Patkaniowska A, Urlaub H, Luhrmann R, Tuschl T. Single-stranded antisense siRNAs guide target RNA cleavage in RNAi. Cell. 2002;110:563–574. doi: 10.1016/s0092-8674(02)00908-x. [DOI] [PubMed] [Google Scholar]
- 47.Mourelatos Z, Dostie J, Paushkin S, Sharma A, Charroux B, Abel L, Rappsilber J, Mann M, Dreyfuss G. miRNPs: a novel class of ribonucleoproteins containing numerous microRNAs. Genes Dev. 2002;16:720–728. doi: 10.1101/gad.974702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Novina CD, Murray MF, Dykxhoorn DM, Beresford PJ, Riess J, Lee SK, Collman RG, Lieberman J, Shankar P, Sharp PA. siRNA-directed inhibition of HIV-1 infection. Nat. Med. 2002;8:681–686. doi: 10.1038/nm725. [DOI] [PubMed] [Google Scholar]
- 49.Novina CD, Sharp PA. The RNAi revolution. Nature. 2004;430:161–164. doi: 10.1038/430161a. [DOI] [PubMed] [Google Scholar]
- 50.Pfeffer S, Sewer A, Lagos-Quintana M, Sheridan R, Sander C, Grasser FA, van Dyk LF, Ho CK, Shuman S, Chien M, Russo JJ, Ju J, Randall G, Lindenbach BD, Rice CM, Simon V, Ho DD, Zavolan M, Tuschl T. Identification of microRNAs of the herpesvirus family. Nat. Methods. 2005;2:269–276. doi: 10.1038/nmeth746. [DOI] [PubMed] [Google Scholar]
- 51.Pfeffer S, Zavolan M, Grasser FA, Chien M, Russo JJ, Ju J, John B, Enright AJ, Marks D, Sander C, Tuschl T. Identification of virus-encoded microRNAs. Science. 2004;304:734–736. doi: 10.1126/science.1096781. [DOI] [PubMed] [Google Scholar]
- 52.Polson AG, Bass BL, Casey JL. RNA editing of hepatitis delta virus antigenome by dsRNA-adenosine deaminase. Nature. 1996;380:454–456. doi: 10.1038/380454a0. [DOI] [PubMed] [Google Scholar]
- 53.Provost P, Dishart D, Doucet J, Frendewey D, Samuelsson B, Radmark O. Ribonuclease activity and RNA binding of recombinant human Dicer. EMBO J. 2002;21:5864–5874. doi: 10.1093/emboj/cdf578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Provost P, Silverstein RA, Dishart D, Walfridsson J, Djupedal I, Kniola B, Wright A, Samuelsson B, Radmark O, Ekwall K. Dicer is required for chromosome segregation and gene silencing in fission yeast cells. Proc. Natl. Acad. Sci. U. S. A. 2002;99:16648–16653. doi: 10.1073/pnas.212633199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rand TA, Ginalski K, Grishin NV, Wang X. Biochemical identification of Argonaute 2 as the sole protein required for RNA-induced silencing complex activity. Proc. Natl. Acad. Sci. U. S. A. 2004;101:14385–14389. doi: 10.1073/pnas.0405913101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Romano PR, Zhang F, Tan SL, Garcia-Barrio MT, Katze MG, Dever TE, Hinnebusch AG. Inhibition of double-stranded RNA-dependent protein kinase PKR by vaccinia virus E3: role of complex formation and the E3 N-terminal domain. Mol. Cell Biol. 1998;18:7304–7316. doi: 10.1128/mcb.18.12.7304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Saunders LR, Barber GN. The dsRNA binding protein family: critical roles, diverse cellular functions. FASEB J. 2003;17:961–983. doi: 10.1096/fj.02-0958rev. [DOI] [PubMed] [Google Scholar]
- 58.Scadden AD, Smith CW. RNAi is antagonized by A-->I hyper-editing. EMBO Rep. 2001;2:1107–1111. doi: 10.1093/embo-reports/kve244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sharp TV, Moonan F, Romashko A, Joshi B, Barber GN, Jagus R. The vaccinia virus E3L gene product interacts with both the regulatory and the substrate binding regions of PKR: implications for PKR autoregulation. Virology. 1998;250:302–315. doi: 10.1006/viro.1998.9365. [DOI] [PubMed] [Google Scholar]
- 60.Sledz CA, Holko M, de Veer MJ, Silverman RH, Williams BR. Activation of the interferon system by short-interfering RNAs. Nat. Cell Biol. 2003;5:834–839. doi: 10.1038/ncb1038. [DOI] [PubMed] [Google Scholar]
- 61.Soldan SS, Plassmeyer ML, Matukonis MK, Gonzalez-Scarano F. La Crosse virus nonstructural protein NSs counteracts the effects of short interfering RNA. J. Virol. 2005;79:234–244. doi: 10.1128/JVI.79.1.234-244.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stefl R, Skrisovska L, Allain FH. RNA sequence- and shape-dependent recognition by proteins in the ribonucleoprotein particle. EMBO Rep. 2005;6:33–38. doi: 10.1038/sj.embor.7400325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Strijker R, Fritz DT, Levinson AD. Adenovirus VAI-RNA regulates gene expression by controlling stability of ribosome-bound RNAs. EMBO J. 1989;8:2669–2675. doi: 10.1002/j.1460-2075.1989.tb08407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Svensson C, Akusjarvi G. A novel effect of adenovirus VA RNA1 on cytoplasmic mRNA abundance. Virology. 1990;174:613–617. doi: 10.1016/0042-6822(90)90116-9. [DOI] [PubMed] [Google Scholar]
- 65.Thimmappaya B, Weinberger C, Schneider RJ, Shenk T. Adenovirus VAI RNA is required for efficient translation of viral mRNAs at late times after infection. Cell. 1982;31:543–551. doi: 10.1016/0092-8674(82)90310-5. [DOI] [PubMed] [Google Scholar]
- 66.Tompkins SM, Lo CY, Tumpey TM, Epstein SL. Protection against lethal influenza virus challenge by RNA interference in vivo. Proc. Natl. Acad. Sci. U. S. A. 2004;101:8682–8686. doi: 10.1073/pnas.0402630101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tonkin LA, Bass BL. Mutations in RNAi rescue aberrant chemotaxis of ADAR mutants. Science. 2003;302:1725. doi: 10.1126/science.1091340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Voinnet O. Induction and suppression of RNA silencing: insights from viral infections. Nat. Rev. Genet. 2005;6:206–220. doi: 10.1038/nrg1555. [DOI] [PubMed] [Google Scholar]
- 69.Volpe TA, Kidner C, Hall IM, Teng G, Grewal SI, Martienssen RA. Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science. 2002;297:1833–1837. doi: 10.1126/science.1074973. [DOI] [PubMed] [Google Scholar]
- 70.Wu HL, Huang LR, Huang CC, Lai HL, Liu CJ, Huang YT, Hsu YW, Lu CY, Chen DS, Chen PJ. RNA interference-mediated control of hepatitis B virus and emergence of resistant mutant. Gastroenterology. 2005;128:708–716. doi: 10.1053/j.gastro.2004.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang W, Wang Q, Howell KL, Lee JT, Cho DS, Murray JM, Nishikura K. ADAR1 RNA deaminase limits short interfering RNA efficacy in mammalian cells. J. Biol. Chem. 2005;280:3946–3953. doi: 10.1074/jbc.M407876200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zamore PD. Plant RNAi: How a viral silencing suppressor inactivates siRNA. Curr. Biol. 2004;14:R198–R200. doi: 10.1016/j.cub.2004.02.021. [DOI] [PubMed] [Google Scholar]
- 73.Zamore PD, Tuschl T, Sharp PA, Bartel DP. RNAi: double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell. 2000;101:25–33. doi: 10.1016/S0092-8674(00)80620-0. [DOI] [PubMed] [Google Scholar]
- 74.Zeng Y, Yi R, Cullen BR. MicroRNAs and small interfering RNAs can inhibit mRNA expression by similar mechanisms. Proc. Natl. Acad. Sci. U. S. A. 2003;100:9779–9784. doi: 10.1073/pnas.1630797100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang H, Kolb FA, Brondani V, Billy E, Filipowicz W. Human Dicer preferentially cleaves dsRNAs at their termini without a requirement for ATP. EMBO J. 2002;21:5875–5885. doi: 10.1093/emboj/cdf582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zheng X, Bevilacqua PC. Activation of the protein kinase PKR by short double-stranded RNAs with single-stranded tails. RNA. 2004;10:1934–1945. doi: 10.1261/rna.7150804. [DOI] [PMC free article] [PubMed] [Google Scholar]