

Abstract
BRCA1 is a checkpoint and DNA damage repair gene that secures genome integrity. We have previously shown that mice lacking full-length Brca1 (Brca1Δ11/Δ11) die during embryonic development. Haploid loss of p53 completely rescues embryonic lethality, and adult Brca1Δ11/Δ11p53+/− mice display cancer susceptibility and premature aging. Here, we show that reduced expression and/or the absence of Chk2 allow Brca1Δ11/Δ11 mice to escape from embryonic lethality. Compared to Brca1Δ11/Δ11p53+/− mice, lifespan of Brca1Δ11/Δ11Chk2−/− mice was remarkably extended. Analysis of Brca1Δ11/Δ11Chk2−/− mice revealed that p53-dependent apoptosis and growth defect caused by Brca1 deficiency are significantly attenuated in rapidly proliferating organs. However, in later life, Brca1Δ11/Δ11Chk2−/− female mice developed multiple tumors. Furthermore, haploid loss of ATM also rescued Brca1 deficiency-associated embryonic lethality and premature aging. Thus, in response to Brca1 deficiency, the activation of the ATM–Chk2–p53 signaling pathway contributes to the suppression of neoplastic transformation, while leading to compromised organismal homeostasis. Our data highlight how accurate maintenance of genomic integrity is critical for the suppression of both aging and malignancy, and provide a further link between aging and cancer.
Keywords: aging, apoptosis, breast cancer, cell senescence, G1/S cell cycle checkpoint
Introduction
Defects in DNA damage repair and cell cycle checkpoints cause genetic instability, resulting in the accumulation of mutations and eventual cancer development. The ATM–Chk2–p53 pathway is involved in the DNA damage repair network that is activated in response to DNA damage or errors in cell cycle events (Bartek and Lukas, 2003; Kastan and Bartek, 2004; Deng, 2006). Upon the occurrence of double-stranded DNA breaks (DSBs), ATM directly phosphorylates p53 on Ser-15 and Thr-68 on Chk2, which, in turn, phosphorylates p53 on Ser-20, thereby helping to regulate the cell cycle and apoptosis (Matsuoka et al, 1998; Hirao et al, 2000). Thus, Chk2 works as both a transducer acting in the ATM–Chk2–p53 cascade and a candidate tumor suppressor. Indeed, Chk2 mutations are found in some hereditary malignancies, such as Li-Fraumeni Syndrome (Bell et al, 1999).
The tumor suppressor BRCA1 acts as both a checkpoint and a DNA damage repair gene that secures genome integrity (Deng and Wang, 2003; Deng, 2006). Brca1-null embryos were embryonic lethal at days 7–8 (E7–E8), and could be partially rescued by p53 or p21 deficiency (Hakem et al, 1997; Ludwig et al, 1997; Shen et al, 1998). We showed that over 98% of mutant embryos carrying a hypomorphic mutation of Brca1 (Brca1Δ11/Δ11) died at E12–E18, and haploid loss of p53 could completely suppress embryonic lethality (Xu et al, 2001). The Brca1Δ11/Δ11p53+/− female mice developed tumors in multiple organs with loss of heterozygosity (LOH) of p53. As p53 mutations are present in about 50% of human cancers and in 90% of BRCA1-associated mammary tumors (Schuyer and Berns, 1999), it is important to identify other genetic alterations that allow BRCA1 mutant cells to survive and undergo malignant transformation. On the other hand, the Brca1Δ11/Δ11p53+/− male mice exhibited premature senescence and aging due to p53 hyperactivity (Cao et al, 2003). These results suggest that the absence of full-length Brca1 results in genomic instability/DNA damage, which leads to p53 activation. It is not clear, however, how p53 becomes activated in Brca1-deficient mice and leads to premature aging.
Several mechanisms are thought to contribute to the progression of aging, including apoptosis and senescence (Hasty et al, 2003; Campisi, 2005; Lombard et al, 2005). Current research suggests that compromised organismal homeostasis is an important pathogenic factor in accelerated aging (Wong et al, 2003). Tissue stem/progenitor cells govern organismal homeostasis in somatic tissues through self-renewal and differentiation of stem/progenitor cells. This is important in the face of detrimental genetic and environmental factors in order to avoid aging and tumorigenesis. Apoptosis induced by DNA damage and genetic instability may impair organ function. Cell cycle arrest may then affect the self-renewal of tissue stem/progenitor cells and impair their ability to replace these damaged cells.
Recently, inactivation of Chk2 was found to rescue thymic T-cell growth defects caused by Brca1 mutation, and enhance tumorigenesis in thymus and mammary glands, with specific targeting of Brca1 (McPherson et al, 2004). In this study, we use a candidate approach to identify the genes involved in p53 activation. Our data revealed that ATM–Chk2–p53 DNA damage response (DDR) is activated in the absence of Brca1 during embryonic development and that DDR acts as a natural barrier to eliminate cells carrying DNA damage. Consistent with this, we demonstrated that either the loss or haploid loss of Chk2 or ATM enables Brca1Δ11/Δ11mice to survive to adulthood. These mice displayed delayed onset of aging and tumor formation at lower frequency than the Brca1Δ11/Δ11p53+/− mice. These observations point to the requirement of the ATM–Chk2–p53 signaling in governing development and aging, as well as preventing malignancy in late adulthood associated with Brca1 mutation.
Results
A genetic candidate approach to identify p53 pathway regulation factors in response to Brca1 deficiency
During our initial study of Brca1Δ11/Δ11 embryos, we found that about 1–2% of them, primarily male mice, survived to adulthood (Xu et al, 2001). We followed 15 Brca1Δ11/Δ11 male mice for 2 years, and found that the average lifespan of these mice was 15 months. The Brca1Δ11/Δ11p53+/− male mice, however, had an average age of only 7.5 months. We hypothesize that some genetic modifiers may account for the prolonged survival. To investigate this, we used a candidate approach to explore expression changes of genes that are involved in the DNA damage repair network and/or p53-mediated genotoxic stress pathway. We found that the expression of Chk2 was downregulated in these mice (Figure 1A). Interestingly, our further analysis revealed that the expression of p53 was also downregulated to an extent roughly correlated with the level of Chk2 (Figure 1A); this finding suggests that the Brca1Δ11/Δ11 mice escaped from embryonic lethality, possibly owing to impaired Chk2 and/or p53 signaling. We therefore first tested whether inactivation of Chk2 could rescue the embryonic lethality of Brca1Δ11/Δ11 mice by introducing a Chk2-null mutation (Takai et al, 2002) into these mice. We found that the absence of Chk2 completely rescued embryonic lethality caused by Brca1 deficiency (Figure 1B and Supplementary Table I). In addition, about 32% of Brca1Δ11/Δ11Chk2+/− mice also survived to adulthood (Supplementary Table I).
Figure 1.
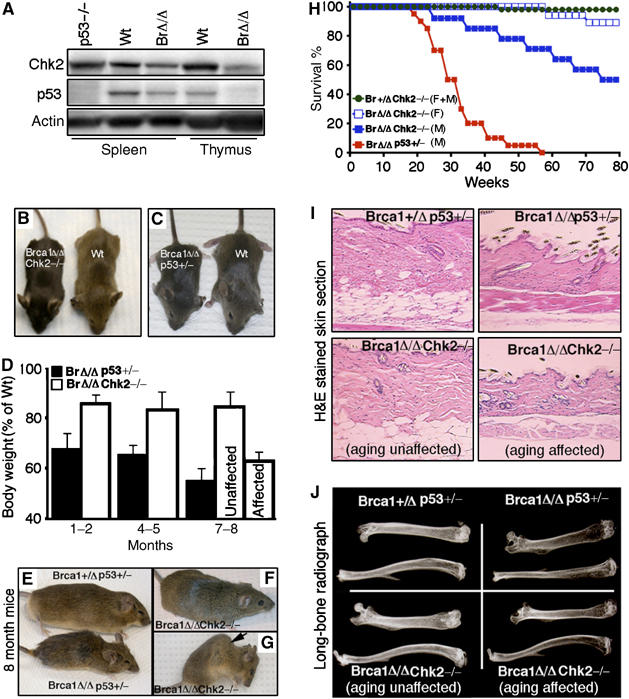
Inactivation of Chk2 rescues embryonic lethality and premature aging caused by Brca1 deficiency. (A) Western blot analysis of spleen and thymus from natural-survived Brca1Δ11/Δ11 mice for Chk2 and p53 expression (n=6). (B, C) Photograph of postnatal 1-month-old wild-type, Brca1Δ11/Δ11Chk2−/− and Brca1Δ11/Δ11p53+/− mice. (D) Body weights of Brca1Δ11/Δ11Chk2−/− and Brca1Δ11/Δ11p53+/− male mice (each time point, n=10; aging-affected Brca1Δ11/Δ11Chk2−/− mice, n=6). (E) Photograph of 8-month-old Brca1+/Δ11p53+/− and Brca1Δ11/Δ11p53+/− mice. (F, G) Photograph of 8-month-old aging-unaffected (F) and aging-affected (G) Brca1Δ11/Δ11Chk2−/− mice. An arrow in (G) points to severe kyophosis. (H) Lifespan of Brca1+/Δ11Chk2−/− male and female mice (n=15), Brca1Δ11/Δ11Chk2−/− female male (n=30), Brca1Δ11/Δ11Chk2−/− male mice (n=20) and Brca1Δ11/Δ11p53+/− male mice (n=14) (F: female; M: Male). (I) Skin histologic section of 8-month-old Brca1+/Δ11p53+/−, Brca1Δ11/Δ11p53+/− and Brca1Δ11/Δ11Chk2−/− male mice (n=4). (J) Long-bone radiograph of 8-month-old Brca1+/Δ11p53+/−, Brca1Δ11/Δ11p53+/− and Brca1Δ11/Δ11Chk2−/− male mice (n=4).
Next, we tested Chk1, which is involved in the ATR–Chk1–p53 signaling pathway at the DNA replication checkpoint. Chk1-null embryos died at E4.5 (Liu et al, 2000); therefore, we chose to test whether haploinsufficiency of Chk1 could rescue embryonic lethality caused by Brca1 deficiency. Our data indicated that heterozygosity of the Chk1 mutation could partially rescue Brca1Δ11/Δ11 embryos, as about 80% Brca1Δ11/Δ11Chk1+/− mice were found alive at birth and died within 24 h (Supplementary Table II). We have also tested the effect of loss of function mutations of p19ARF, Pten, Bax, p21, Gadd45a and Parp1, which are involved in various aspects of p53 functions, such as its stability, DNA damage repair pathway, apoptosis and/or cell cycle regulation, but this failed to yield any live Brca1Δ11/Δ11 mice above background levels (data not shown).
Chk2 inactivation delays premature aging caused by Brca1 deficiency
The Brca1Δ11/Δ11Chk2−/− mice survived to adulthood, enabling us to explore the possible connections among Brca1 deficiency, elevated p53 activity and the premature aging phenotype of Brca1-deficient mice. We followed cohorts of tumor-free Brca1+/Δ11Chk2−/−, Brca1Δ11/Δ11Chk2−/− and Brca1Δ11/Δ11p53+/− male mice. The young Brca1Δ11/Δ11Chk2−/− and Brca1Δ11/Δ11p53+/− mice were healthy, although they were infertile and smaller (Figure 1B and C). The body weights of Brca1Δ11/Δ11p53+/− and Brca1Δ11/Δ11Chk2−/− mice were 70 and 85% of control mice (Brca1+/Δ11Chk2−/−), respectively, at ages younger than 6 months. At 7–8 months of age, 80% of Brca1Δ11/Δ11p53+/− mice exhibited signs of premature aging and showed a substantially decreased body weight compared to control mice (Figure 1D and E). In contrast, only 10% (6/60) of the Brca1Δ11/Δ11Chk2−/− mice showed a further decrease in body weight in this time period (aging affected, Figure 1D). The majority of the Brca1Δ11/Δ11p53+/− mice died within 8 months, whereas the majority of the Brca1Δ11/Δ11Chk2−/− mice were healthy at this age (Figure 1F and H). By 18 months of age, about 47% (9/19) of Brca1Δ11/Δ11Chk2−/− male and 12.5% (2/16) of female mice showed morphological signs of premature aging, such as kyphosis (Figure 1G). Thus, although some Brca1Δ11/Δ11Chk2−/− mice exhibited premature aging, their lifespan was remarkably extended compared to the Brca1Δ11/Δ11p53+/− mice.
We next studied aging-related pathological changes, such as reductions in dermal thickness, subcutaneous adipose tissue of skin and osteoporosis in the long bones (Supplementary Table III). Most Brca1Δ11/Δ11p53+/− male mice displayed decreased skin thickness (Figure 1I) and reduced bone density, as revealed by decreased intensity of X-ray images (Figure 1J) at 8 months of age, whereas only the aging-affected Brca1Δ11/Δ11Chk2−/− male mice showed similar changes at this time (Figure 1J). This suggests that inactivation of Chk2 attenuates aging-related pathological changes and delays premature aging caused by Brca1 deficiency compared with the p53+/− mutation.
Chk2 inactivation represses p53-mediated apoptosis in Brca1-deficient mice
It has been suggested that apoptosis is a major factor that causes premature aging by the disruption of normal organismal homeostasis (Zhang and Herman, 2002). We therefore performed histological analysis of both young and elderly mouse organs, focusing on the gastrointestinal tract, which is dependent on a highly renewable epithelium. By H&E staining, 80% of 8-month-old Brca1Δ11/Δ11p53+/− mice showed villous atrophy in the small intestine (Figure 2A, right upper panel), but fewer than 10% of the Brca1Δ11/Δ11Chk2−/− mice showed this abnormality (Figure 2A, right lower panel). To see if apoptosis contributed to these aging-related changes, we examined the degree of apoptosis in the small intestine by TUNEL assay. We found that massive apoptosis occurred in the Brca1Δ11/Δ11p53+/− mouse intestinal villi (Figure 2B, right upper panel), but not in the crypts containing rapidly dividing cells. In contrast, there was less apoptosis in the Brca1Δ11/Δ11Chk2−/− mice (Figure 2B, right lower panel). In addition, we found that p53 was highly expressed in the intestinal villi from Brca1Δ11/Δ11p53+/− mice (Figure 2C, right upper panel), but not in Brca1Δ11/Δ11Chk2−/− mice (Figure 2C, lower panel). These results suggest that apoptosis induced by Brca1 deficiency is p53-dependent; however, it can be inhibited by Chk2 inactivation. These data also suggest that the absence of Chk2 might block p53 hyperactivity caused by Brca1 deficiency.
Figure 2.

The absence of Chk2 inhibits p53 activity and p53-mediated apoptosis caused by Brca1 deficiency. (A) Small intestine histologic section of 8-month-old Brca1+/Δ11p53+/−, Brca1Δ11/Δ11p53+/−, Brca1Δ11/Δ11Chk2−/− (aging unaffected) and Brca1Δ11/Δ11Chk2−/− (aging affected) male mice (n=5). (B) TUNEL-assay on small intestine from 8-month-old Brca1+/Δ11p53+/−, Brca1Δ11/Δ11p53+/−, Brca1Δ11/Δ11Chk2−/− (aging-unaffected) and Brca1Δ11/Δ11Chk2−/− (aging affected) male mice (n=4). (C) p53 expression detected by immunohistochemical staining using an antibody to p53 in small intestine from 8-month-old Brca1+/Δ11p53+/−, Brca1Δ11/Δ11p53+/−, Brca1Δ11/Δ11Chk2−/− (aging-unaffected) and Brca1Δ11/Δ11Chk2−/− (aging-affected) male mice (n=4).
To address this further, we studied ionizing radiation (γ-IR)-induced apoptosis in thymocytes, which is known to be mediated by p53 (Clarke et al, 1993). We found that Brca1 deficiency resulted in a p53-dependent decrease of thymocyte viability under normal culture condition (Supplementary Figure 1A) and p53-dependent increase of apoptosis upon γ-IR treatment (Supplementary Figure 1B). Significantly, the absence of Chk2 restored the viability and apoptotic levels of Brca1Δ11/Δ11 cells to wild-type levels (Supplementary Figure 1A and B). Our data also indicated that Chk2–p53 signaling is activated in response to Brca1 deficiency and that the absence of Chk2 attenuated p53 induction (Supplementary Figure 1C). These results suggested that the increased apoptosis in Brca1-deficient mice is mediated by Chk2–p53 signaling pathway, and that the absence of Chk2 blocks p53-dependent apoptosis. Thus, the reduction of Chk2–p53 signaling-mediated apoptosis may prevent loss of organismal homeostasis caused by Brca1 deficiency-associated DNA damage.
ATM–Chk2–p53 pathway is activated in response to Brca1 deficiency-associated DNA damage
As our data indicated that Brca1 deficiency triggers Chk2 activation and p53-dependent apoptosis, we hypothesized that the Chk2–p53 signaling pathway might be activated in response to DNA damage and/or genomic instability associated with Brca1 deficiency. To test this hypothesis, we examined whether Brca1 deficiency could induce DNA damage and activate p53 in vivo. We stained embryonic tissues of wild-type, Brca1Δ11/Δ11 and Brca1Δ11/Δ11Chk2−/− embryos using an anti-γ-H2AX antibody. Phosphorylation of histone H2AX (γ-H2AX) is among the earliest ATM-dependent responses to DNA double-strand breaks (Rogakou et al, 1998; Burma et al, 2001). Our analysis detected massive γ-H2AX foci in Brca1Δ11/Δ11 (Figure 3B and I), Brca1Δ11/Δ11Chk2−/− (Figure 3C and I) and Brca1Δ11/Δ11p53−/− (data not shown) embryonic brain tissue, but not in controls (Figure 3A and I). These results indicated an accumulation of un-repaired DNA double-strand breaks in Brca1 mutants, but not in control embryos. Next, we tested whether p53 was activated in the Brca1 mutant embryos by using an antibody specific to phosphorylated Ser23 of p53, which is dependent on Chk2 activation (Matsuoka et al, 1998; Hirao et al, 2000). We detected markedly increased levels of p53-Ser23 phosphorylation in Brca1Δ11/Δ11 embryonic brain tissues (Figure 3F and I) compared with the controls (Figure 3E and I). In contrast, the level of p53-Ser23 phosphorylation was significantly lower in Brca1Δ11/Δ11Chk2−/− embryonic brain tissue (Figure 3G and I).
Figure 3.

Activation of the ATM–Chk2–p53 signaling pathway in response to Brca1 deficiency in embryogenesis. (A–D) γ-H2AX staining on brain tissues of wild-type, Brca1Δ11/Δ11, Brca1Δ11/Δ11Chk2−/− and Brca1Δ11/Δ11Atm−/− E12.5 embryos (n=3). (E–H) p53-Ser23 staining on brain tissues of wild-type, Brca1Δ11/Δ11, Brca1Δ11/Δ11Chk2−/− and Brca1Δ11/Δ11Atm−/− E12.5 embryos (n=3). (I) Quantification of the γ-H2AX and p53-Ser23-positive cells. (J) Western blot analysis of Atm, Chk2 and p53 expression of passage 1 MEF cells of wild type, Brca1Δ11/Δ11p53−/− and Brca1Δ11/Δ11Atm−/− in treated or untreated with γ–irradiation.
In our crosses between Brca1 mutant mice and other mutant strains, interestingly, we found that haploid loss or complete loss of ATM could also overcome embryonic lethality caused by Brca1 deficiency (Supplementary Table IV). Using this strain of mice, we investigated whether Chk2–p53 activation in Brca1 mutant cells is ATM dependent. We showed that despite the formation of γ-H2AX foci in Brca1Δ11/Δ11Atm−/− embryonic brain tissue (Figure 3D and I), p53-Ser23 foci were diminished (Figure 3H and I). In addition, Western blot analysis revealed Chk2 phosphorylation in Brca1Δ11/Δ11p53−/− mouse embryonic fibroblast (MEF) cells, but not in Brca1Δ11/Δ11Atm−/− MEF cells (Figure 3J). These observations provide compelling evidence that the activation of Chk2 in Brca1-mutant cells is ATM dependent.
Recent studies indicated that overexpression of certain oncogenes could cause stalled or collapsed replication forks, which activate the ATR/ATM-regulated checkpoint, followed by DNA double-strand breaks leading to ATM-Chk2 activation (Bartek and Lukas, 2003; Kastan and Bartek, 2004; Bartkova et al, 2005; Gorgoulis et al, 2005). To investigate whether the activation of DDR in Brca1-deficient cells was also caused by replication errors, we performed further studies using a number of molecular markers. Our analyses on cell proliferation using BrdU labeling (Supplementary Figure 2A–D) and immunohistochemical staining using an antibody to the proliferation cell nuclear antigen (PCNA), which binds to replication forks, failed to reveal a significant difference between Brca1Δ11/Δ11 and wild-type embryos (Supplementary Figure 2E). We also detected no apparent alterations in expression/activation of ATR and Chk1 in Brca1 mutant embryonic tissues and MEF cells (Supplementary Figure 2F–H). These observations provide compelling evidence that the ATM–Chk2 pathway is specifically activated owing to DNA damage caused by Brca1 deficiency. The activation of ATM–Chk2 signaling, in turn, triggers phosphorylation of p53, leading to p53-dependent apoptosis.
Consistent with this, by TUNEL assay, we detected high levels of apoptosis in Brca1Δ11/Δ11 embryos, but not in Brca1Δ11/Δ11Chk2−/−, Brca1Δ11/Δ11Atm−/− and Brca1Δ11/Δ11p53−/− embryos (Supplementary Figure 2I–L and data not shown). Moreover, although the Brca1Δ11/Δ11Atm−/− mice died of lymphoma before 6 months of age similar to Atm−/− mice (Barlow et al, 1996), the lifespan of Brca1Δ11/Δ11Atm+/− mice was extended. In a total of 15 Brca1Δ11/Δ11Atm+/− mice followed up to 15 months of age, none developed the premature aging phenotypes yet. We believe that the apoptosis occurring in highly renewable adult organs and in the rapidly growing embryos may be caused by p53 activation. Hence, the absence of Chk2 or ATM attenuates p53-dependent apoptosis and delays the onset of premature aging caused by Brca1 deficiency.
Chk2 inactivation attenuates stem/progenitor cell growth arrest in aging-affected Brca1-deficient mice
Aging is characterized by a gradual decline in organ function and may be due, in part, to impaired self-renewal of stem/progenitor cells. To address the effect of Brca1 deficiency in tissue stem/progenitor cells, we performed a BrdU labeling assay in transit amplifying (TA) cells to reflect the stem/progenitor cell self-renewal capacity. The small intestine is the most rapidly renewing organ with the stem cells located in the intestinal crypts. The crypt stem cells divide by self-renewal to produce TA cells. The TA cells, in turn, undergo a number of rapid cell divisions, and differentiate into the mature epithelial cell types (Foulkes et al, 2003). Fewer BrdU positive cells were seen in the Brca1Δ11/Δ11p53+/− mice (Figure 4A, right upper panel than in control mice), although no difference was observed in younger mice (data not shown). In contrast, in age-matched Brca1Δ11/Δ11Chk2−/− mice, the majority showed no difference (Figure 4A, left lower panel), and only some aging-affected Brca1Δ11/Δ11Chk2−/− mice showed decrease of BrdU-positive TA cells (Figure 4A, right lower panel).
Figure 4.
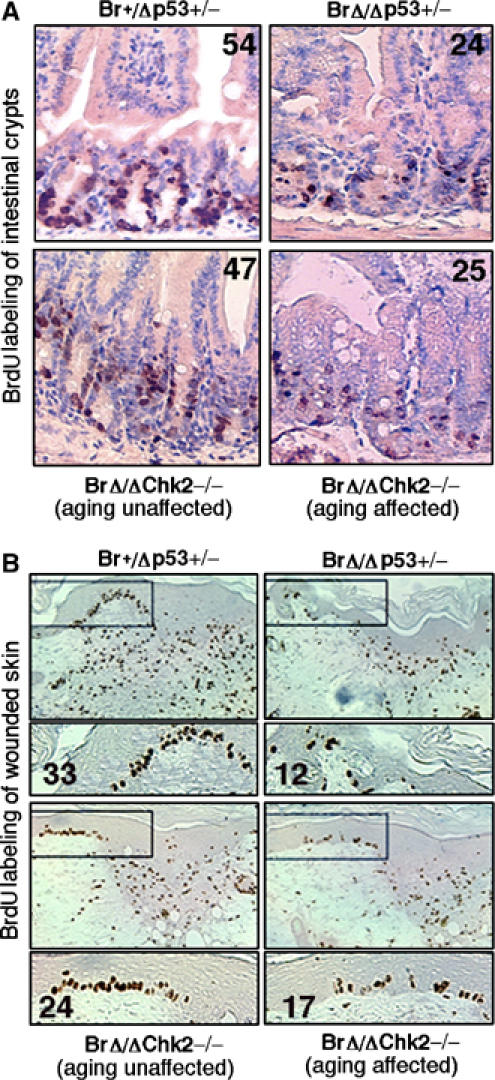
Activation of Chk2–p53 signaling in transit amplifying stem cell proliferation. (A) BrdU-labeling assay for transit amplifying cells of small intestine crypts from 8-month-old Brca1+/Δ11p53+/−, Brca1Δ11/Δ11p53+/−, Brca1Δ11/Δ11Chk2−/− (aging unaffected) and Brca1Δ11/Δ11Chk2−/− (aging affected) male mice (n=4). (B) BrdU-labeling assay for transit amplifying cells of skin keratinocyte from 8-month-old Brca1+/Δ11p53+/−, Brca1Δ11/Δ11p53+/−, Brca1Δ11/Δ11Chk2−/− (aging unaffected) and Brca1Δ11/Δ11Chk2−/− (aging affected) male mice after wounded 3 days (n=4). Number of BrdU-positive cells in these areas were counted and shown in each panel.
Skin is another rapidly renewable organ. Under normal physiological conditions, the process of cell renewal is not as quick as it is in the intestine. Upon wounding/injury, TA cells rapidly proliferate and differentiate to generate more natural cells to repair the wound (Lehrer et al, 1998). Skin wound-healing assays, followed by BrdU-labeling of the TA cells, showed that fewer BrdU-positive TA cells were seen in Brca1Δ11/Δ11p53+/− (Figure 4B, right upper panel) and in aging-affected Brca1Δ11/Δ11Chk2−/− mice than in wild-type mice (Figure 4B, right lower panel). On the other hand, aging-unaffected Brca1Δ11/Δ11Chk2−/− mice did not show a decrease in BrdU-positive TA cells (Figure 4B, left lower panel). These results suggest that Chk2 activation caused by Brca1 deficiency impairs tissue stem cell self-renewal capacity and may account for the decline in organ function seen in aging. Taken together, again our data suggested that aging might be caused by multiple factors such as apoptosis and tissue stem cell depletion, which can be induced by DNA damage and/or genomic instability.
Chk2–p53 signaling pathway-mediated apoptosis suppresses Brca1-associated tumor formation in a tissue-specific manner
We next studied tumor development in Brca1Δ11/Δ11Chk2−/− mice. Our data indicated that 72% (16/22) of the Brca1Δ11/Δ11Chk2−/− female mice developed mammary tumors by 16 months of age with a mean age of 12 months (Figure 5A and Supplementary Figure 3A). Previously, we have reported that only 10% of the mice carrying a Cre/LoxP-mediated mammary-specific deletion of Brca1 exon 11 (Brca1Co/Co;MMTV-Cre) developed mammary tumors by 18 months of age (Xu et al, 1999). The comparison between Brca1Co/Co;MMTV-Cre and Brca1Δ11/Δ11Chk2−/− mice indicates that the absence of Chk2 enhances Brca1-associated mammary tumor, underscoring an important role for Chk2–p53 signaling in suppression of Brca1-associated mammary tumor formation.
Figure 5.
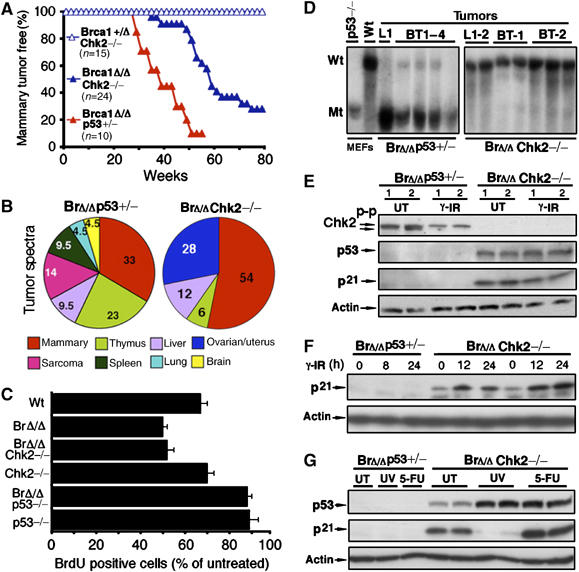
Incidence, latency, spectrum and p53 status in tumors from Brca1Δ11/Δ11p53+/− and Brca1Δ11/Δ11Chk2−/− female mice. (A) Onset of Brca1-associated mammary tumors of Brca1+/Δ11Chk2−/−, Brca1Δ11/Δ11Chk2−/− and Brca1Δ11/Δ11p53+/− female mice. (B) Tumor spectra of Brca1Δ11/Δ11p53+/− and Brca1Δ11/Δ11Chk2−/− female mice. (C) Analysis of the G1/S checkpoint of passage 1 wild-type, Brca1Δ11/Δ111, Brca1Δ11/Δ11Chk2−/−, Chk2−/−, Brca1Δ11/Δ11p53−/− and p53−/− MEF cells (n=3) upon 10 Gy γ-IR. The percentage of BrdU-positive cells after γ-IR relative to the unirradiated controls was shown. (The profiles of FACS were shown in Supplementary Figure 3B and C.) (D) Southern blot analysis of p53 on breast tumors (BT) and lymphomas (L) from Brca1Δ11/Δ11p53+/− and Brca1Δ11/Δ11Chk2−/− mice by EcoRI digestion. Wild-type (Wt) band (16 kb) and mutant (Mt) bands (8 kb) were indicated. (E) Western blot analysis for p53 and p21 of cell lines derived from Brca1Δ11/Δ11p53+/− and Brca1Δ11/Δ11Chk2−/− mice mammary tumors without (UT) and with 10 Gy γ-IR after 6 h. (F) Time point of p21 expression of tumor cell lines from Brca1Δ11/Δ11p53+/− and Brca1Δ11/Δ11Chk2−/− mice after 10 Gy of γ-IR. (G) Western blot analysis of p53 and p21 expression of tumor cell lines from Brca1Δ11/Δ11p53+/− and Brca1Δ11/Δ11Chk2−/− mice by without (UT), 100 J/m2 of UV-irradiation and 50 μg/ml of 5-fluorouracil (5-FU) treatment for 6 h.
We also compared the tumor spectrum between Brca1Δ11/Δ11Chk2−/− and Brca1Δ11/Δ11p53+/− female mice. The Brca1Δ11/Δ11p53+/− female mice developed multiple tumor types, which is similar to those seen in Li-Fraumeni Syndrome, including mammary tumor, sarcoma, lymphoma, lung cancer, liver cancer and brain tumors (Kleihues et al, 1997). In contrast, the Brca1Δ11/Δ11Chk2−/− mice developed mostly mammary and ovarian tumors (Figure 5B). As p53 is also required for controlling the G1/S checkpoint after DNA damage, we compared the p53-dependent G1 cell cycle arrest of wild-type, Brca1Δ11/Δ11, Brca1Δ11/Δ11Chk2−/−, Chk2−/−, Brca1Δ11/Δ11p53−/− and p53−/− MEF cells in response to γ-IR. Upon γ-IR, wild-type cells exhibited G1 arrest as indicated by 66% reduction in BrdU-positive cells compared with untreated cells. Brca1Δ11/Δ11 and Brca1Δ11/Δ11Chk2−/− cells exhibited a more significant decrease of BrdU-positive cells than wild-type cells, suggesting that these cells not only have an intact G1/S checkpoint but also display a stronger checkpoint than wild-type cells. In contrast, only a slight reduction in the BrdU-positive population was observed in p53−/− and Brca1Δ11/Δ11p53−/− MEF cells (Figure 5C and Supplementary Figure 3B and C). Taken together, these data again suggest that Chk2-p53 pathway-mediated apoptosis acts as important factor in the suppression of Brca1-associated mammary tumor formation. The intact p53-dependent G1/S checkpoint in Brca1Δ11/Δ11Chk2−/− mice may contribute to the more limited spectrum, lower frequency and late onset of tumors observed in these mice.
In human, p53 mutations are found in about 50% of sporadic breast cancer and about 90% of BRCA1-associated breast cancer (Schuyer and Berns, 1999). To further address whether the additional p53 mutations are required for tumorigenesis in Brca1Δ11/Δ11Chk2−/− tumors, we performed Southern blot analysis for p53 on tumors from both Brca1Δ11/Δ11Chk2−/− and Brca1Δ11/Δ11p53+/− mice. All tumors isolated from Brca1Δ11/Δ11p53+/− mice lost the remaining wild-type allele of p53. In contrast, all tumors from Brca1Δ11/Δ11Chk2−/− mice had intact p53 (Figure 5D). Moreover, Western blot analysis showed that Brca1Δ11/Δ11p53+/− tumor cell lines lost both p53 and p53-mediated induction of p21 (Figure 5E). In contrast, p53 and p21 were expressed in Brca1Δ11/Δ11Chk2−/− tumor cell lines, although they failed to be induced at 6 h after γ-IR (Figure 5E). p21, however, could be induced 12 h after γ-IR (Figure 5F). This observation suggests that Chk2 plays a role in transient p21 induction after γ-IR (up to 6 h); however, its induction is not dependent on Chk2 function in the later phase after γ-IR. To test whether p53 could be induced by other genotoxic stresses, we treated these cells with UV or 5-FU. We found that p53 was induced under both conditions (Figure 5G), whereas p21 expression was induced upon 5-FU treatment. The expression of p21 was decreased upon UV treatment, perhaps owing to UV-induced degradation (Bendjennat et al, 2003). These data indicated that the p53 function was completely lost in Brca1Δ11/Δ11p53+/− tumors owing to LOH, whereas in Brca1Δ11/Δ11Chk2−/− tumors, which showed no alterations of p53 at genomic level, Chk2-mediated p53 function was attenuated only partially. Our data highlight the importance of p53-dependent apoptosis and the G1/S cell cycle checkpoint in suppression of Brca1-associated tumorigenesis. In addition, our data distinguish the role of p53-dependent apoptosis and the G1/S cell cycle checkpoint in tumor onset and tumor spectrum.
We also performed cytogenetic characterization of mammary tumors derived from Brca1Δ11/Δ11p53+/− and Brca1Δ11/Δ11Chk2+/− mice. We detected more extensive genetic instability in tumors derived from Brca1Δ11/Δ11p53+/− mice than in tumors from Brca1Δ11/Δ11Chk2−/− mice (Supplementary Figure 4A and B). Tumors developed from Brca1Δ11/Δ11Chk2−/− mice were less diverse in their histopathology and showed a higher mitotic index that was accompanied by increased expression of several oncogenes, including cyclin D1 and c-myc (Supplementary Figure 4C–F). These data suggest that p53-mediated G1/S checkpoint serves as a barrier against most alterations/mutations caused by Brca1 deficiency. However, some alterations/mutations, that is those with significant survival and/or proliferation potential, may overcome this barrier and eventually result in tumorigenesis in Brca1Δ11/Δ11Chk2−/− mice.
Senescence in aging and mammary tumorigenesis
Cellular senescence, which acts as a potential anticancer mechanism, can be induced by multiple conditions, including DNA damage, telomere shortening and oncogene expression (Braig et al, 2005; Chen et al, 2005; Collado et al, 2005; Michaloglou et al, 2005). Our previous observation that Brca1 deficiency causes both premature aging in adult mice and senescence in cultured cells (Cao et al, 2003) suggests that cellular senescence might be linked to premature aging. To investigate this, we examined the replicative capacity of wild-type, Brca1Δ11/Δ11, Chk2−/− and Brca1Δ11/Δ11Chk2−/− MEFs by using a standard 3T3 immortalization protocol (Todaro and Green, 1963). We found that Brca1Δ11/Δ11 MEFs displayed a profound proliferation defect, which could be rescued partially by the absence of Chk2 (Brca1Δ11/Δ11Chk2−/−) at passage 1 (Supplementary Figure 3D) but not later passages (data not shown). Using senescence-associated acidic β-galactosidase (SA-β-gal) activity assay, we found that Brca1Δ11/Δ11 and Brca1Δ11/Δ11Chk2−/− MEFs displayed similar premature senescence phenotype starting at passage 2. By passage 4, more than 50% of these cells, but not wild-type and Brca1Δ11/Δ11p53−/− MEFs, showed SA-β-gal activity (Figure 6A). To understand the mechanisms underlying cellular senescence in Brca1Δ11/Δ11 and Brca1Δ11/Δ11Chk2−/− cells, we examined the status of Chk2 and p53 in these cells. We found that Chk2 phosphorylation was increased in passage 4 of Brca1Δ11/Δ11, and more dramatically in Brca1Δ11/Δ1p53−/− MEF cells, but not in wild-type MEF cells (Figure 6B). These data suggest that senescence may trigger Chk2 activation as about 50% Brca1Δ11/Δ11 MEF cells had already undergone senescence. Notably, p53 accumulation was also found in Brca1Δ11/Δ11Chk2−/−cells, although it was slightly lower than in Brca1Δ11/Δ11 MEF cells (Figure 6B). Thus, under cell culture conditions, p53 can still be activated in the absence of Chk2, perhaps by other upstream kinases, such as ATM. This activation may be responsible for the reduced replicative capacity and increased senescence in Brca1Δ11/Δ11Chk2−/− cells.
Figure 6.

Senescence in aging and tumorigenesis. (A) SA-β-gal staining of passage 4 MEF cells from wild-type, Brca1Δ11/Δ11, Brca1Δ11/Δ11p53−/− and Brca1Δ11/Δ11Chk2−/− E14.5 embryos. (B) Western blot analysis for Chk2 and p53 expression of passage 4 MEF cells from wild-type, Brca1Δ11/Δ11, Brca1Δ11/Δ11p53−/− and Brca1Δ11/Δ11Chk2−/− E14.5 embryos. (C) SA-β-gal staining of kidney from 8-month-old wild-type, Chk2−/−, Brca1Δ11/Δ11p53+/− and Brca1Δ11/Δ11Chk2−/− mice (aging affected). (D) SA-β-gal staining of brain from 8-month-old wild-type, Chk2−/−, Brca1Δ11/Δ11p53+/− and Brca1Δ11/Δ11Chk2−/− mice (aging affected). (E) SA-β-gal staining of normal and hyperplasia mammary tissue, and mammary tumor Brca1Δ11/Δ11Chk2−/− female mice.
Next, we studied the relationship between cell senescence and premature aging on tissues of aging-affected Brca1Δ11/Δ11p53+/− and Brca1Δ11/Δ11Chk2−/− mice. We detected SA-β-gal activity in kidney and brain in aging-affected mutant mice but not in wild-type and mutant mice before aging onset (Figure 6C and D). Next, we examined the effects of senescence in Brca1-associated mammary tumor formation. Our data detected increased SA-β-gal-positive staining in mammary tissues of aged Brca1Δ11/Δ11Chk2−/− mice at premalignant stages (Figure 6E). This observation suggests that Brca1 mutation also induces cellular senescence in this tissue in a manner similar to other tissues. Interestingly, more pronounced SA-β-gal-positive staining was detected in tissues associated with hyperplasia, whereas the staining became negative in the tumors (Figure 6E). These observations demonstrate, in principle, that activation of DDR due to Brca1 deficiency induces cell senescence, which serves as a barrier to prevent cancer formation. Further alterations associated with Brca1 deficiency may eventually inactivate DDR and overcome senescence, leading to tumor formation in late developmental stages.
Discussion
In this study, we showed that DNA damage and genetic instability caused by BRCA1 deficiency-activated ATM–Chk2–p53 mediated DDR, leading to apoptosis, senescence and cell cycle arrest through distinct pathways. At the same time, BRCA1 deficiency also impairs the DDR, resulting in tumorigenesis (Figure 7). The ATM-Chk2 signaling acts in the pathway involved in p53-dependent apoptosis in response to genotoxic stress associated with BRCA1 deficiency. In embryonic development, the ATM–Chk2–p53-dependent apoptosis acts as a natural selection to eliminate mutations in individuals. In adult tissues, Chk2–p53 signaling pathway acts as a ‘security gate' to prevent neoplastic transformation as manifested by the finding that the absence of either Chk2 or p53 results in tumorigenesis in Brca1 mutant mice. However as a side effect, the hyperactivity of the Chk2–p53 pathway causes apoptosis and tissue stem cell depletion. This may also compromise organism homeostasis, causing premature aging.
Figure 7.
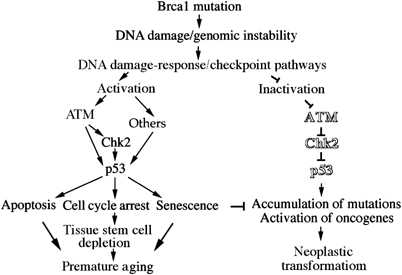
Model of the ATM–Chk2–p53 signaling pathway upon Brca1 mutation-associated premature aging and tumorigenesis. ATM–Chk2–p53 signaling pathway senses DNA damage/genomic instability and acts as a gatekeeper to eliminate mutations, but, as a side effect, it may also lead to premature aging.
BRCA1 plays critical roles in DNA damage repair and cell cycle regulation through interacting with many proteins, including the BRCA1-associated genome surveillance complex (BASC) and several Fanconi anemia (FA)-associated proteins (Wang et al, 2000; Rothfuss and Grompe, 2004; Deng, 2006). Our data indicate that the absence of Brca1 results in the accumulation of un-repaired DNA damage, as revealed by markedly elevated γH2AX foci formation, which may be responsible for activation of ATM–Chk2–p53 signaling. Recent studies demonstrated that oncogenic stress (overexpression of oncogenes, such as cyclin-E or ras) could activate the ATR/ATM-regulated checkpoint through deregulation of DNA replication and DNA damage (Bartkova et al, 2005; Gorgoulis et al, 2005). Specifically, it was proposed that oncogenic stress might cause abnormalities in prereplication complex maturation and/or stalled or collapsed replication forks, which activate the ATR-H2AX/Chk1 cascade (Bartek and Lukas, 2003; Kastan and Bartek, 2004). The collapsed replication forks, consequently, result in double-strand breaks, leading to the activation of the ATM-H2AX/Chk2-p53 signaling (Bartkova et al, 2005; Gorgoulis et al, 2005). Our analysis of Brca1 mutant cells failed to detect ATR and Chk1 activation, suggesting that the DNA damage observed in Brca1 mutant cells is caused by a different mechanism rather than by replication errors. The activation of the ATM–Chk2–p53 signaling pathway, in turn, acts as a natural barrier to eliminate mutations during embryonic development and to prevent neoplastic transformation in late life. However, the ‘hyperactivation' of the ATM–Chk2–p53 signaling in response to genotoxic stress induces apoptosis and stem cell depletion, which may compromise organism survival and result in premature aging. This study reveals the importance of ‘genomic integrity' and the balance of ATM–Chk2–p53 signaling pathway regulation in both repressing tumorigenesis and maintaining organism homeostasis.
Biochemical studies have shown that defects in Chk2–p53 checkpoint cascades might cause accumulation of mutations and chromosomal aberrations, which is one of the hallmarks of cancer cells (Bartek and Lukas, 2003; Motoyama and Naka, 2004). We have previously shown that p53-deficiency could rescue embryonic lethality of Brca1Δ11/Δ11 embryos and allow tumor formation in adult mice (Xu et al, 2001). These findings suggest that suppression of apoptosis caused by Brca1 deficiency is a part of the mechanism underlying Brca1-associated tumorigenesis. The abolishing of apoptosis in Brca1-deficient cells may give cells a chance to survive and accumulate mutations, causing activation of oncogenes and/or inactivation of tumor suppressor genes, eventually leading to tumorigenesis. In this study, by crossing mice carrying targeted mutations of genes that are involved in p53 activation, p53 stability and p53 downstream functions, we showed that the loss of functions of either ATM or Chk2 can substitute for p53 inactivation in Brca1-associated embryonic lethality by inhibition of p53-mediated apoptosis. Brca1 mutant mice in Chk2-deficient background escape from embryonic lethality and develop normally, but they are at risk for neoplastic transformation later in life. Chk2 deficiency cooperates with Brca1 mutation in mammary tumors even in the presence of wild-type p53. However, the latency of mammary tumors in Brca1Δ11/Δ11Chk2−/− mice is much longer and the frequency of thymic lymphoma formation is also much lower (Supplementary Figure 3E) than in Brca1Δ11/Δ11p53+/− and Brca1Δ11/Δ11p53−/− mice (Xu et al, 2001; Bachelier et al, 2003; Cao et al, 2003). Our study revealed that although the Brca1Δ11/Δ11Chk2−/− cells are defective in initiating apoptosis, they maintain an ability to induce cell cycle arrest. As this cell cycle arrest occurs only in Brca1Δ11/Δ11Chk2−/− cells, but not in Brca1Δ11/Δ11p53−/− cells, we may define the cell cycle arrest as p53-dependent and Chk2 independent. This observation highlights the importance of p53-dependent cell cycle arrest in suppression of thymic lymphoma in the absence of apoptosis.
On the other hand, we also found that, similar to Brca1Δ11/Δ11 MEF cells, Brca1Δ11/Δ11Chk2−/− MEF cells undergo p53-mediated senescence. These results indicate that the p53-mediated cell cycle checkpoint and senescence play an essential role in the suppression of tumorigenesis associated with BRCA1 mutations, thus highlighting distinct roles of p53-mediated apoptosis, cell cycle arrest and senescence in tumorigenesis. Therefore, our data support the theory that Chk2 acts as a tumor suppressor and distinguishes the role of p53-dependent apoptosis and G1/S cell cycle checkpoint in tumor onset and tumor development. These observations reveal a significant difference between Chk2 and p53 in Brca1- associated thymic lymphoma and mammary tumor development, and they suggest that the ATM–Chk2–p53 signaling pathway-mediated apoptosis suppresses Brca1-associated tumorigenesis in a tissue-specific manner.
We have previously shown that the Brca1Δ11/Δ11p53+/− male mice exhibit premature aging due to p53 hyperactivity triggered by Brca1 deficiency (Cao et al, 2003). However, the underlying mechanisms remain elusive. As Brca1Δ11/Δ11Chk2−/− mice could survive to adulthood, and inactivation of Chk2 only blocks p53-mediated apoptosis, but not the p53-dependent cell cycle checkpoint and senescence, this allowed us to further dissect the function of p53 signaling in premature aging. By comparing Brca1Δ11/Δ11p53+/− and Brca1Δ11/Δ11Chk2−/− male mice, we found that the premature aging phenotypes of Brca1Δ11/Δ11Chk2−/− male mice were significantly delayed. These mice also lost p53-dependent apoptosis and transiently amplified progenitor cell growth defects in adult renewable organs, but maintained the G1/S cell cycle checkpoint and senescence. These data demonstrate that DNA damage and/or tumor suppressor gene mutation causes tumor formation and/or premature aging (Vijg and Dolle, 2002; Campisi, 2003; Hasty et al, 2003). We believe that continuous DNA damage associated with BRCA1 deficiency activates the ATM–Chk2 pathway and triggers p53 hyperactivation, leading to apoptosis and cell growth defects and causing damage to individual cells and the depletion of stem cell renewal capacity of organs/tissues. This eventually results in the functional decline of organs and premature aging.
Materials and methods
Mice and MEF cells
ATM+/− (Barlow et al, 1996), Chk1+/− (Liu et al, 2000), Chk2+/− (Takai et al, 2002) or p53+/− (Donehower et al, 1992) mice were crossed with Brca1+/Δ11 (Xu et al, 2001) mice to generated double-mutant mice. MEF cells were derived from E14.5 embryos generated from intercrosses of Brca1/Δ11Chk2+/− mice. For proliferation analysis, 5 × 104 MEF cells were plated on six-well plates in DMEM supplemented with 10% FBS.
BrdU labeling
BrdU (100 mg/kg of body weight) was injected i.p. into mice for 2 h. The tissues were stained with BrdU staining bulk kit (ZYMED). The passage 1 MEF cells were labeled with BrdU for 30 min before stained by anti-BrdU (Becton-Dickinson) monoclonal antibody. The cells were incubated with Alexa Flour conjugated goat anti-mouse antibody (Molecular Probes), and propidium iodide (PI). Cellular fluorescence was measured using FACSCalibur.
Histologic and immunohistologic analysis
For histology, tissues were fixed in 10% formalin, blocked in paraffin, sectioned, stained with hematoxylin and eosin and examined by light microscopy. Antibodies for ATM (a gift from Eva Lee), p53-ser-20 (16G8, Cell Signaling), γ-H2AX (Upstate), PCNA (Ab-1, PC10, CALBIOCHM) Cyclin D1 (Santa Cruz) and C-myc (Upstate) were used in the immunohistologic analysis. Detection of primary antibodies was performed using the ZYMED Histomouse TM SP Kit according to the manufacturer's instruction.
Immunoblot analysis
Western blot analysis was accomplished according to standard procedures using ECL detection (Amersham). The following primary antibodies were used: p53 (Ab-7, Oncogene), p21WAF1 (Ab-6, Oncogene), Chk1 and Chk2 (Transduction Laboratories). Horseradish peroxidase-conjugated anti-rabbit, sheep, and anti-mouse antibodies (Amersham) were used as secondary antibodies.
Detection of apoptotic cells
For TUNEL assays of apoptotic cells on tissue sections, we used embryos at E12.5. For flow cytometric detection of apoptosis, thymocytes were isolated from 2–3-month-old animals fixed in 70% ethanol, stained with PI and analyzed for sub-G0/G1 events on a Becton-Dickinson FACSCalibur.
Senescence-associated acidic β-galactosidase activity assay
Acidic β-galactosidase activity was detected as described (Dimri et al, 1995). Cells were washed with PBS (pH 7.2), fixed with 0.5% glutaraldehyde in PBS (pH 7.2) for 5 min and then washed in PBS (pH 7.2). The frozen tissues were cut (5 μm) and fixed in 1% formalin in PBS for 1 min, then washed in PBS.
G1/S checkpoint analysis
G1/S checkpoint analysis in asynchronized population of cells was performed according to a method described (Takai et al, 2002). Briefly, 1 h after the MEF cells of passage 1 were treated with 10 Gy-IR, cells were labeled with BrdU for 30 min before they were stained with an antibody to BrdU and PI for DNA content, followed by flow cytometry using a Becton-Dickinson FACSCalibur.
Supplementary Material
Supplementary Figures
Supplementary Tables
Acknowledgments
We thank T Fishler for critically reading the manuscript and the editorial assistance of the NCI Fellows Editorial Board. This work was supported by the intramural research program of the National Institutes of Diabetes and Digestive Kidney Diseases, NIH, USA.
References
- Bachelier R, Xu X, Wang X, Li W, Naramura M, Gu H, Deng CX (2003) Normal lymphocyte development and thymic lymphoma formation in Brca1 exon-11-deficient mice. Oncogene 22: 528–537 [DOI] [PubMed] [Google Scholar]
- Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, Wynshaw-Boris A (1996) Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell 86: 159–171 [DOI] [PubMed] [Google Scholar]
- Bartek J, Lukas J (2003) Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 3: 421–429 [DOI] [PubMed] [Google Scholar]
- Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, Orntoft T, Lukas J, Bartek J (2005) DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434: 864–870 [DOI] [PubMed] [Google Scholar]
- Bell DW, Varley JM, Szydlo TE, Kang DH, Wahrer DC, Shannon KE, Lubratovich M, Verselis SJ, Isselbacher KJ, Fraumeni JF, Birch JM, Li FP, Garber JE, Haber DA (1999) Heterozygous germ line hCHK2 mutations in Li-Fraumeni syndrome. Science 286: 2528–2531 [DOI] [PubMed] [Google Scholar]
- Bendjennat M, Boulaire J, Jascur T, Brickner H, Barbier V, Sarasin A, Fotedar A, Fotedar R (2003) UV irradiation triggers ubiquitin-dependent degradation of p21(WAF1) to promote DNA repair. Cell 114: 599–610 [DOI] [PubMed] [Google Scholar]
- Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, Stein H, Dorken B, Jenuwein T, Schmitt CA (2005) Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 436: 660–665 [DOI] [PubMed] [Google Scholar]
- Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ (2001) ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem 276: 42462–42467 [DOI] [PubMed] [Google Scholar]
- Campisi J (2003) Cancer and ageing: rival demons? Nat Rev Cancer 3: 339–349 [DOI] [PubMed] [Google Scholar]
- Campisi J (2005) Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 120: 513–522 [DOI] [PubMed] [Google Scholar]
- Cao L, Li W, Kim S, Brodie SG, Deng CX (2003) Senescence, aging, and malignant transformation mediated by p53 in mice lacking the Brca1 full-length isoform. Genes Dev 17: 201–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, Cordon-Cardo C, Pandolfi PP (2005) Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 436: 725–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke AR, Purdie CA, Harrison DJ, Morris RG, Bird CC, Hooper ML, Wyllie AH (1993) Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature 362: 849–852 [DOI] [PubMed] [Google Scholar]
- Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, Benguria A, Zaballos A, Flores JM, Barbacid M, Beach D, Serrano M (2005) Tumour biology: senescence in premalignant tumours. Nature 436: 642. [DOI] [PubMed] [Google Scholar]
- Deng CX (2006) BRCA1: cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res 34: 1416–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng CX, Wang RH (2003) Roles of BRCA1 in DNA damage repair: a link between development and cancer. Hum Mol Genet 12 (Spec No 1): R113–R123 [DOI] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, Peacocke M, Campisi J (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA 92: 9363–9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr, Butel JS, Bradley A (1992) Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356: 215–221 [DOI] [PubMed] [Google Scholar]
- Foulkes WD, Stefansson IM, Chappuis PO, Begin LR, Goffin JR, Wong N, Trudel M, Akslen LA (2003) Germline BRCA1 mutations and a basal epithelial phenotype in breast cancer. J Natl Cancer Inst 95: 1482–1485 [DOI] [PubMed] [Google Scholar]
- Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA Jr, Kastrinakis NG, Levy B, Kletsas D, Yoneta A, Herlyn M, Kittas C, Halazonetis TD (2005) Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434: 907–913 [DOI] [PubMed] [Google Scholar]
- Hakem R, de la Pompa JL, Elia A, Potter J, Mak TW (1997) Partial rescue of Brca1 (5–6) early embryonic lethality by p53 or p21 null mutation. Nat Genet 16: 298–302 [DOI] [PubMed] [Google Scholar]
- Hasty P, Campisi J, Hoeijmakers J, van Steeg H, Vijg J (2003) Aging and genome maintenance: lessons from the mouse? Science 299: 1355–1359 [DOI] [PubMed] [Google Scholar]
- Hirao A, Kong YY, Matsuoka S, Wakeham A, Ruland J, Yoshida H, Liu D, Elledge SJ, Mak TW (2000) DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science 287: 1824–1827 [DOI] [PubMed] [Google Scholar]
- Kastan MB, Bartek J (2004) Cell-cycle checkpoints and cancer. Nature 432: 316–323 [DOI] [PubMed] [Google Scholar]
- Kleihues P, Schauble B, zur Hausen A, Esteve J, Ohgaki H (1997) Tumors associated with p53 germline mutations: a synopsis of 91 families. Am J Pathol 150: 1–13 [PMC free article] [PubMed] [Google Scholar]
- Lehrer MS, Sun TT, Lavker RM (1998) Strategies of epithelial repair: modulation of stem cell and transit amplifying cell proliferation. J Cell Sci 111 (Part 19): 2867–2875 [DOI] [PubMed] [Google Scholar]
- Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, Donehower LA, Elledge SJ (2000) Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev 14: 1448–1459 [PMC free article] [PubMed] [Google Scholar]
- Lombard DB, Chua KF, Mostoslavsky R, Franco S, Gostissa M, Alt FW (2005) DNA repair, genome stability, and aging. Cell 120: 497–512 [DOI] [PubMed] [Google Scholar]
- Ludwig T, Chapman DL, Papaioannou VE, Efstratiadis A (1997) Targeted mutations of breast cancer susceptibility gene homologs in mice: lethal phenotypes of Brca1, Brca2, Brca1/Brca2, Brca1/p53, and Brca2/p53 nullizygous embryos. Genes Dev 11: 1226–1241 [DOI] [PubMed] [Google Scholar]
- Matsuoka S, Huang M, Elledge SJ (1998) Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 282: 1893–1897 [DOI] [PubMed] [Google Scholar]
- McPherson JP, Lemmers B, Hirao A, Hakem A, Abraham J, Migon E, Matysiak-Zablocki E, Tamblyn L, Sanchez-Sweatman O, Khokha R, Squire J, Hande MP, Mak TW, Hakem R (2004) Collaboration of Brca1 and Chk2 in tumorigenesis. Genes Dev 18: 1144–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS (2005) BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 436: 720–724 [DOI] [PubMed] [Google Scholar]
- Motoyama N, Naka K (2004) DNA damage tumor suppressor genes and genomic instability. Curr Opin Genet Dev 14: 11–16 [DOI] [PubMed] [Google Scholar]
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM (1998) DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 273: 5858–5868 [DOI] [PubMed] [Google Scholar]
- Rothfuss A, Grompe M (2004) Repair kinetics of genomic interstrand DNA cross-links: evidence for DNA double-strand break-dependent activation of the Fanconi anemia/BRCA pathway. Mol Cell Biol 24: 123–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuyer M, Berns EM (1999) Is TP53 dysfunction required for BRCA1-associated carcinogenesis? Mol Cell Endocrinol 155: 143–152 [DOI] [PubMed] [Google Scholar]
- Shen SX, Weaver Z, Xu X, Li C, Weinstein M, Chen L, Guan XY, Ried T, Deng CX (1998) A targeted disruption of the murine Brca1 gene causes gamma-irradiation hypersensitivity and genetic instability. Oncogene 17: 3115–3124 [DOI] [PubMed] [Google Scholar]
- Takai H, Naka K, Okada Y, Watanabe M, Harada N, Saito S, Anderson CW, Appella E, Nakanishi M, Suzuki H, Nagashima K, Sawa H, Ikeda K, Motoyama N (2002) Chk2-deficient mice exhibit radioresistance and defective p53-mediated transcription. Embo J 21: 5195–5205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todaro GJ, Green H (1963) Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J Cell Biol 17: 299–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijg J, Dolle ME (2002) Large genome rearrangements as a primary cause of aging. Mech Ageing Dev 123: 907–915 [DOI] [PubMed] [Google Scholar]
- Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J (2000) BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev 14: 927–939 [PMC free article] [PubMed] [Google Scholar]
- Wong KK, Maser RS, Bachoo RM, Menon J, Carrasco DR, Gu Y, Alt FW, DePinho RA (2003) Telomere dysfunction and Atm deficiency compromises organ homeostasis and accelerates ageing. Nature 421: 643–648 [DOI] [PubMed] [Google Scholar]
- Xu X, Qiao W, Linke SP, Cao L, Li WM, Furth PA, Harris CC, Deng CX (2001) Genetic interactions between tumor suppressors Brca1 and p53 in apoptosis, cell cycle and tumorigenesis. Nat Genet 28: 266–271 [DOI] [PubMed] [Google Scholar]
- Xu X, Wagner KU, Larson D, Weaver Z, Li C, Ried T, Hennighausen L, Wynshaw-Boris A, Deng CX (1999) Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nat Genet 22: 37–43 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Herman B (2002) Ageing and apoptosis. Mech Ageing Dev 123: 245–260 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures
Supplementary Tables