

Abstract
Background
SSeCKS is a major protein kinase C substrate with kinase scaffolding and metastasis-suppressor activity whose expression is severely downregulated in Src- and Ras-transformed fibroblast and epithelial cells and in human prostate, breast, and gastric cancers. We previously used NIH3T3 cells with tetracycline-regulated SSeCKS expression plus a temperature-sensitive v-Src allele to show that SSeCKS re-expression inhibited parameters of v-Src-induced oncogenic growth without attenuating in vivo Src kinase activity.
Methods
We use cDNA microarrays and semi-quantitative RT-PCR analysis to identify changes in gene expression correlating with i) SSeCKS expression in the absence of v-Src activity, ii) activation of v-Src activity alone, and iii) SSeCKS re-expression in the presence of active v-Src.
Results
SSeCKS re-expression resulted in the attenuation of critical Src-induced proliferative and pro-angiogenic gene expression including Afp, Hif-1α, Cdc20a and Pdgfr-β, and conversely, SSeCKS induced several cell cycle regulatory genes such as Ptpn11, Gadd45a, Ptplad1, Cdkn2d (p19), and Rbbp7.
Conclusion
Our data provide further evidence that SSeCKS can suppress Src-induced oncogenesis by modulating gene expression downstream of Src kinase activity.
Background
SSeCKS, originally identified as a transcriptionally-suppressed gene in v-src and ras-transformed rodent fibroblast cells [1], is the orthologue of human GRAVIN/AKAP12 gene that encodes a kinase-scaffolding protein [2] that is targeted as an autoantigen in some cases of myasthenia gravis [3]. SSeCKS/Gravin/AKAP12 expression is severely downregulated in human prostate, breast and gastric cancer, partially relating to the mapping of the human gene to 6q24-25.1 [4], a cancer deletion hotspot [5]. Re-expression of SSeCKS to physiologic levels in Src- or Ras-transformed fibroblasts or epithelial prostate cancer cells suppresses morphological transformation, anchorage- and growth factor-independent proliferation, and metastatic potential, while restoring normal actin-based cytoskeletal architecture and cell-cycle controls on cyclin D1 expression [4,6,7]. SSeCKS also seems to control the blood-brain barrier by suppressing astrocyte-expressed vascular endothelial growth factor (VEGF) during the switch to normoxic conditions after birth [8]. A recent study indicates that the ability of SSeCKS to suppress lung metastasis formation by MatLyLu prostate cancer cells correlates with its suppression of VEGF 165 and 121 isoforms [9]. Interestingly, SSeCKS does not grossly alter the Src-mediated tyrosine phosphorylation of cellular substrates in vivo [6], strongly suggesting that SSeCKS suppresses tumorigenicity by re-establishing controls on downstream cytoskeletal and signaling pathways. However, it remains unclear which pathways are regulated by SSeCKS during tumor or metastasis suppression.
In this report, we analyzed how SSeCKS re-expression affects v-Src-induced oncogenic gene expression patterns using oligonucleotide microarrays and semi-quantitative RT-PCR techniques. Our data show that SSeCKS suppresses several critical proliferation- and angiogenesis-associated genes while it induces differentiation and cell cycle control functions, strongly suggesting that SSeCKS is capable of reprogramming normal gene expression controls downstream of activated Src.
Methods
Cells
S2-6 cells are NIH3T3 cells that encode a tetracycline (tet)-regulated tTA transactivator (Tet-OFF), S24 cells are S2-6 cells encoding a tet-regulated rat SSeCKS cDNA, and S24/ts72v-Src cells express temperature-sensitive v-Src whose kinase activity is only active at the permissive temperature (PT = 35°C), as described previously [6]. Cell cultures were maintained in complete DMEM supplemented with 10% calf serum, penicillin/streptomycin/amphotericin B, 2 μg/ml puromycin (S24 and S24/ts72v-Src cells), 65 μg/ml G418 (S24/ts72v-Src cells) and 0.7 mg/ml tet (Sigma).
Oligonucleotide array analysis
1 μg of total RNA, isolated from comparable cell groups using TRIzol reagent (Invitrogen.), was reverse-transcribed into Cy-3- and Cy-5-labeled probes used to hybridize to Affymetrix A430 chips (Santa Clara, CA) according to the manufacturer's protocol. Fluorescence intensity for each chip was measured with an Affymetrix 428 Scanner. Data were derived from three independent microarray analyses performed for each cell type, and comparative analysis of resulting data was performed using software suites including GeneSpring v5.0 (Silicon Genetics), Data Mining Tool v3.0 (Affymetrix), GeneTraffic Uno (Iobion Informatics), dChip v1.1 (Harvard University) and SAM v1.15 (Stanford University) [10]. The mean hybridization signal for each sample was set as 1000 arbitrary units to normalize the signal values of all of the genes on the chip (global normalization) between different samples. The signal ratio of 2 or 0.5 was chosen as the criterion for induction or repression, respectively. In repeat experiments, most of the inter-experimental variation in gene expression (of the genes listed in Tables 3, 4, 5) was less than 2-fold, and only a few genes varied widely (e.g.- typically, 3.5- to 6-fold). However, these variations did not alter the trends in gene regulation (i.e.- up- or downregulation) by SSeCKS and/or v-Src.
Table 3.
Genes Regulated by SSeCKS in NIH3T3 fibroblastsa A value ≥ 2 represents induction; a value ≤ 0.5 represents repression
| Gene | Symbol | Modulation |
| Signal transduction | ||
| G protein-coupled receptor 56 | Gpr56 | 126.24* |
| BTB (POZ) domain containing 2 | Btbd2 | 10.85 |
| Guanine nucleotide binding protein (G protein), gamma 2 subunit | Gng2 | 9.58* |
| Ras association (RalGDS/AF-6) domain family 5 | Rassf5 | 8.75 |
| Calcium binding protein 2 | Cabp2 | 8.11 |
| Butyrate-induced transcript 1 | Ptplad1 | 4.08* |
| Signal-transducing adaptor protein-2 | Stam2 | 3.20 |
| Protein kinase C, epsilon | Prkce | 2.36 |
| Interferon-stimulated protein | G1p2 | 2.30 |
| Tnf receptor-associated factor 6 | Traf6 | 2.23 |
| RAB33B, member of RAS oncogene family | Rab33b | 2.17 |
| Insulin-like growth factor 2, binding protein 3 | Igf2bp3 | 0.49 |
| Myristoylated alanine rich protein kinase C substrate | Marcks | 0.48* |
| Insulin-like growth factor binding protein 4 | Igfbp4 | 0.47 |
| N-myc downstream regulated 1 | Ndrg1 | 0.41 |
| Paternally expressed 3 | Peg3 | 0.27 |
| Dual specificity phosphatase 9 | Dusp9 | 0.15 |
| Cell cycle and transcriptional regulator | ||
| Retinoblastoma 1 | Rb1 | 3.48 |
| c-myc binding protein | Mycbp | 3.41 |
| Histone 2, H2aa1 | Hist2h2aa1 | 3.07 |
| Histone 3, H2a | Hist3h2a | 2.99 |
| Zinc fingers and homeoboxes protein 1 | Zhx1 | 2.79 |
| Cyclin-dependent kinase inhibitor 2D (p19, inhibits CDK4) | Cdkn2d | 2.73* |
| CCAAT/enhancer binding protein (C/EBP), beta | Cebpb | 0.50 |
| Cyclin F | Ccnf | 0.48 |
| Cyclin A2 | Ccna2 | 0.47 |
| High mobility group box 1 | Hmgb1 | 0.46 |
| cell division cycle 20 homolog (S. cerevisiae) | Cdc20a | 0.46* |
| Cyclin B1 | Ccnb1 | 0.44 |
| LIM domain only 4 | Lmo4 | 0.44 |
| Fos-like antigen 1 | Fosl1 | 0.41 |
| Cyclin D1 | Ccnd1 | 0.37 |
| High mobility group box 3 | Hmgb3 | 0.24* |
| Nuclear autoantigenic sperm protein (histone-binding) | Nasp | 0.06 |
| Cytoskeleton | ||
| Synaptonemal complex protein 3 | Sycp3 | 8.34 |
| Microtubule-associated protein 7 | Mtap7 | 0.18 |
| Growth factor and receptor | ||
| Transforming growth factor, beta 1 | Tgfb1 | 2.33 |
| Nerve growth factor, beta | Ngfb | 2.28* |
| Transforming growth factor alpha | Tgfa | 2.27 |
| Platelet derived growth factor receptor, beta polypeptide | Pdgfrb | 0.30* |
| Interleukin 1 receptor-like 1 | Il1rl1 | 0.29* |
| Glial cell line derived neurotrophic factor family receptor alpha 1 | Gfra1 | 0.15* |
| Others | ||
| Alpha fetoprotein | Afp | 0.32* |
aS24 MEF, 37°C, Tet (-) vs (+)
* Similar effects after SSeCKS re-expression in v-Src transformed cells (Table 5).
Table 4.
Genes regulated by ts72v-Src activation A value ≥ 2 represents induction; a value ≤ 0.5 represents repression
| Modulation | |||
| Gene | Symbol | A | B |
| Signal Transduction | |||
| regulator of G-protein signaling 16 | Rgs16 | 10.13 | 12.70 |
| suppressor of cytokine signaling 3 | Socs3 | 3.89 | 3.00 |
| paternally expressed 3 | Peg3 | 3.22* | 3.50* |
| protein kinase, cGMP-dependent, type II | Prkg2 | 2.53 | 2.30 |
| insulin-like growth factor binding protein 4 | Igfbp4 | 0.15** | 0.42** |
| SH3-domain binding protein 5 (BTK-associated) | Sh3bp5 | NC | 0.29 |
| ARF-GAP, RHO-GAP, ankyrin repeat and plekstrin homology domains-containing protein 3 | Centd3 | 0.08 | 0.05 |
| Cell cycle and transcriptional regulator | |||
| v-maf musculoaponeurotic fibrosarcoma oncogene family, protein F | Maff | 17.03 | 3.00 |
| high mobility group AT-hook 2 | Hmga2 | 6.96 | 5.00 |
| runt related transcription factor 1, RUN X1/AML-1 | Runx1 | 6.77 | 5.50 |
| ets variant gene 4 (E1A enhancer binding protein, E1AF) | Etv4 | 6.68 | 8.30 |
| fos-like antigen 1 | Fosl1 | 4.47* | 2.80* |
| high mobility group box 3 | Hmgb3 | 3.73* | 3.30* |
| transcription factor EC | Tcfec | NC | 63.00 |
| inhibitor of DNA binding 4 | Id4 | 0.22 | NC |
| inhibitor of DNA binding 2 | Id2 | NC | 0.43 |
| basic helix-loop-helix transcription factor 6 | Atoh8 | 0.15 | 0.29 |
| Growth factor and receptor | |||
| glial cell line derived neurotrophic factor family receptor alpha 1 | Gfra1 | 4.17* | 7.00* |
| interleukin 1 receptor-like 1 | Il1rl1 | 4.20* | 3.50* |
| Others | |||
| pleckstrin homology, Sec7 and coiled-coil domains, binding protein | Pscdbp | 4.08 | 3.30 |
| alpha fetoprotein | Afp | 3.89* | 3.80* |
| spondin 2, extracellular matrix protein | Spon2 | 0.13 | 0.23 |
| carbonyl reductase 2 | Cbr2 | 0.11 | 0.07 |
A, S24/ts72v-Src cells, 35°C vs 39.5°C, + tet.
B, S24/ts72v-Src cells, 35°C vs S24 MEF 35°C, + tet.
*, opposite effect when compared to SSeCKS overexpression alone (Table 3).
**, similar effects when compared to SSeCKS overexpression alone (Table 3).
NC, no change.
Table 5.
Genes regulated by SSeCKS in v-Src transformed cellsa A value ≥ 2 represents induction; a value ≤ 0.5 represents repression
| Gene | Symbol | Modulation |
| Signal transduction | ||
| Cytokine inducible kinase | Plk3 | 52.35 |
| G protein-coupled receptor 56 | Gpr56 | 49.00* |
| Guanine nucleotide binding protein (G protein), gamma 2 subunit | Gng2 | 7.16* |
| Protein tyrosine phosphatase, non-receptor type 11 | Ptpn11 | 5.86 |
| Serine/threonine kinase 4 | Stk4 | 3.97 |
| RAB20, member RAS oncogene family | Rab20 | 3.92 |
| Phosphatidylinositol-4-phosphate 5-kinase, type 1 beta | Pip5k1b | 3.50 |
| Growth arrest and DNA-damage-inducible 45 alpha | Gadd45a | 3.43 |
| Butyrate-induced transcript 1 | Ptplad1 | 3.29* |
| Suppressor of cytokine signaling 3 | Socs3 | 2.07*** |
| Serine/arginine-rich protein specific kinase 2 | Srpk2 | 0.45 |
| Mitogen activated protein kinase kinase 6 | Map2k6 | 0.44 |
| Regulator of G-protein signaling 2 | Rgs2 | 0.38 |
| Ras and Rab interactor 2 | Rin2 | 0.33 |
| Dual specificity phosphatase 10 | Dusp10 | 0.17 |
| Myristoylated alanine rich protein kinase C substrate | Marcks | 0.12* |
| Cell cycle and transcriptional regulator | ||
| Histone 1, H2bp | Hist1h2bp | 3.61 |
| Cyclin-dependent kinase inhibitor 2D (p19, inhibits CDK4) | Cdkn2d | 2.46* |
| Retinoblastoma binding protein 7 | Rbbp7 | 2.39 |
| Histone 2, H3c2 | Hist2h3c2 | 2.28 |
| v-maf musculoaponeurotic fibrosarcoma oncogene family, protein F | Maff | 2.28*** |
| Inhibitor of DNA binding 2 | Id2 | 0.44*** |
| Transcription factor AP-4 (activating enhancer-binding protein 4) | Tcfap4 | 0.26 |
| Cell division cycle 20 homolog (S. cerevisiae) | Cdc2a | 0.25* |
| Basic helix-loop-helix transcription factor 6 | Atoh6 | 0.24*** |
| High mobility group box 3 | Hmgb3 | 0.24*/** |
| Hypoxia inducible factor 1, alpha subunit | Hif1a | 0.22 |
| Kruppel-like factor 7 (ubiquitous) | Klf7 | 0.10 |
| Growth factor and receptor | ||
| Chemokine (C-C motif) ligand 2 | Ccl2 | 4.26 |
| Nerve growth factor, beta | Ngfb | 2.38* |
| Glial cell line derived neurotrophic factor family receptor alpha 1 | Gfra1 | 0.33*/** |
| Interleukin 1 receptor-like 1 | Il1rl1 | 0.23*/** |
| Platelet derived growth factor receptor, beta polypeptide | Pdgfrb | 0.18* |
| Transforming growth factor, beta 2 | Tgfb2 | 0.14 |
| Others | ||
| S100 calcium binding protein A8 (calgranulin A) | S100a8 | 16.22 |
| Fatty acid desaturase 3 | Fads3 | 8.00 |
| Heat shock protein 4 | Hspa4 | 4.29 |
| Deubiquitinating enzyme 1 | Dub1 | 3.23 |
| alpha fetoprotein | Afp | 0.50*/** |
| Tissue inhibitor of metalloproteinase 3 | Timp3 | 0.33 |
| HIF-1 responsive RTP801 | Ddit4 | 0.26 |
| 5-azacytidine induced gene 1 | Azi1 | 0.24 |
| Growth differentiation factor 5 | Gdf5 | 0.18 |
| Growth arrest specific 1 | Gas1 | 0.18 |
| Caspase 7 | Casp7 | 0.18 |
| retinitis pigmentosa 1 homolog (human) | Rp1h | 0.12 |
| protocadherin 18 | Pcdh18 | 0.07 |
a, S24/ts72v-Src MEF, 35 °C, + vs – tet.
*, similar effects when compared to SSeCKS overexpression alone (Table 3).
**, opposite effects when compared to v-Src activation (Table 4).
***, similar effects when compared to v-Src activation (Table 4).
RT-PCR
1 μg of total RNA was primed with oligo-dT16, reverse transcribed into first strand cDNA and then amplified in the linear range using the SuperScript® III RTS One-Step RT-PCR Kit (Invitrogen), as described previously [1], using the primer sets described in Table 1. The primer sets for Gpr56, Maff, Socs3, Afp, Ngfb, Gadd45a, Marcks, Hif1a, AFP, PDGFRB and HIF1A were purchased as QuantiTect Primer Assays (Qiagen). Optimization of RT-PCRs for semi-quantitative analysis was carried out after normalization using the β-actin mRNA as an internal control. PCR products were electrophoresed in 1.6% agarose gels, stained with ethidium bromide, and digitally imaged using a Chemi-Genius2 BioImager (Syngene). Relative intensities of PCR bands were quantified using GeneTools image software analysis (Syngene).
Table 1.
Summary of primer sequences and positions
| Target gene | Sequence of forward and reverse primers | position |
| Mouse | ||
| Pdgfr-β | 5'-AGCTACATGGCCCCTTATGA-3' | 2749–2768 |
| 5'-GGATCCCAAAAGACCAGACA-3' | 3069–3115 | |
| Tgfβ-1 | 5'-CGGGGCGACCTGGGCA CCATCCATGAC-3' | 1597–1623 |
| 5'-CTGCTCCACCTTGGGCTTGCGACCCAC-3' | 1975–2001 | |
| Cdc20 | 5'-GCTGGTTCTGGTGACATCCT-3' | 757–776 |
| 5'-TGTTCCAACTGAGGGAGCTT-3' | 939–958 | |
| Ptpn11 | 5'-AGTCCAAAGTGACCCACGTC-3' | 623–643 |
| 5'-AGCGTCTCAAACTCTTCCCA-3' | 874–893 | |
| Id2 | 5'-GGACATCAGCATCCTGTCCT-3' | 383–402 |
| 5'-AACGGTATCACAGTCCAGGC-3' | 659–678 | |
| Il1rl1 | 5'-CGCTCGACTTATCCTGTGGA-3' | 299–318 |
| 5'-AGCTTGGCGGCTTTTTATGT-3' | 490–509 | |
| Cdkn2d | 5'AGCTTGGCGGCTTTTTATGT-3' | 212–229 |
| 5'-CGGTCCCATTACTTGTCAC-3' | 460–442 | |
| β- actin | 5'-TTCTTTGCAGCTCCTTCGTTGCCG-3' | 33–56 |
| 5'-TGGATGGCTACGTACATGGCTGGG-3' | 467–490 | |
| Gpr56 | Qiagen QuantiTect Assay, cat. #QT00178689 | |
| Maff | Qiagen QuantiTect Assay, cat. #QT00133224 | |
| Ngfb | Qiagen QuantiTect Assay, cat. #QT00093464 | |
| Socs3 | Qiagen QuantiTect Assay, cat. #QT00100331 | |
| Gadd45a | Qiagen QuantiTect Assay, cat. #QT00249655 | |
| Marcks | Qiagen QuantiTect Assay, cat. #QT00252973 | |
| Hif1a | Qiagen QuantiTect Assay, cat. #QT00182532 | |
| Afp | Qiagen QuantiTect Assay, cat. #QT00174020 | |
| Human | ||
| CDC20A | 5'-GGGTTCCTCTGCAGACATTC-3' | 1034–1053 |
| 5'-TGTAATGGGGAGACCAGAGG-3' | 1215–1234 | |
| PSCDBP | 5'-TCAATGCAGCAATTGGAGTC-3' | 741–761 |
| 5'-ATGTCAATGCACGTCAGCAT-3' | 920–940 | |
| FOSL1 | 5'-CCAAGCATCAACACCATGAG-3' | 302–321 |
| 5'-GGGCTGATCTGTTCACAAGG-3' | 473–492 | |
| HMGA2 | 5'-CGAAAGGTGCTGGGCAGCTCCGG-3' | 786–808 |
| 5'-CCATTTCCTAGGTCTGCCTCTTG-3' | 1086–1110 | |
| HMGB3 | 5'-ACAACCGAGACAAACCCTTG-3' | 1107–1126 |
| 5'-CCCCTTTGTCCACAGCTAAG-3' | 1290–1310 | |
| β- ACTIN | 5'-GCTCGTCGTCGACAACGGCTC-3' | 93–113 |
| 5'-CAAACATGATCTGGGTCATCTTCTC-3' | 445–421 | |
| AFP | Qiagen QuantiTect Assay, cat. #QT00085183 | |
| PDGFRB | Qiagen QuantiTect Assay, cat. #QT00082327 | |
| HIF1A | Qiagen QuantiTect Assay, cat. #QT00083644 |
Western blot
Blotting analysis was performed as described previously [11] using the following primary antibodies at 1:1000 dilutions: PAb anti-SSeCKS [12], MAb anti- β-actin (Sigma), anti-Src [poY418] (BioSource International), MAb anti-v-Src (Ab-1; Oncogene Sciences). Secondary PAbs were either horseradish peroxidase-conjugated anti-rabbit or -mouse Ig (1:2,500; Chemicon) followed by Lumi-Light chemiluminescence reagent (Roche).
Results and discussion
Conditional expression of SSeCKS and v-Src activation in mouse fibroblast cell lines
Our previous data indicated that SSeCKS overexpression in untransformed fibroblasts induces cell flattening as well as G1 phase growth arrest, the latter due to the suppression of growth factor-induced cyclin D1 expression [7]. The forced re-expression of SSeCKS could suppress v-Src-induced oncogenic growth in vitro without grossly affecting the ability of v-Src to phosphorylate cellular substrates [6]. Therefore, in order to identify genes differentially regulated by SSeCKS during G1 arrest or during suppression of Src-induced oncogenesis, we used NIH3T3-derived lines with tetracycline (Tet-OFF)-inducible SSeCKS expression ("S24 cells") as well as S24 cells expressing a temperature-sensitive v-Src allele ("S24/ts72v-Src"). The ectopic expression of SSeCKS was strongly induced (>25-fold) in the absence of tetracycline (tet), yet in the presence of tet, ts72v-Src activation (35°C) resulted in a >10-fold decrease in endogenous SSeCKS levels (Fig. 1A), confirming our initial identification of SSeCKS as a Src-suppressed gene [1]. Moreover, SSeCKS overexpression did not affect Src autophosphorylation activity, as monitored by phospho-Y416 levels (Fig. 1A). The relationship between increased Src activity levels and decreased SSeCKS levels is also maintained when comparing the metastatic human colon cancer cell line, HT29 (high Src activity, low Gravin), with the low metastatic line, HCT116 (low Src activity, high Gravin) (Figure 1B).
Figure 1.
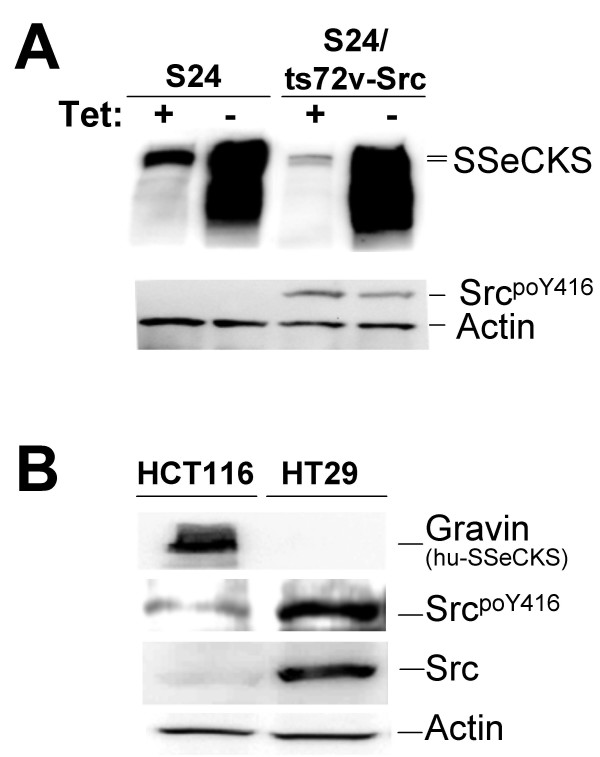
SSeCKS/Gravin/AKAP12 downregulation by activated Src. (A) Lysates from S24 and S24/ts72v-Src cells (grown at the PT) in the presence or absence of tet were immunoblotted for SSeCKS, activated Src (poY416) or actin. Note that SSeCKS overexpression (- tet) did not affect total ts72v-Src protein levels [6]. (B) A similar analysis as in panel A with lysates from HCT116 and HT29 human colon cancer cells with the addition of an immunoblot for total Src protein.
We previously showed that growth of S24/ts72v-Src cells in 0.04 μg/ml tet resulted in re-expression of ectopic SSeCKS to the physiologic levels found in parental NIH3T3 cells [6]. Therefore, as described in Table 2, we compared differential gene expression for the following conditions: i) SSeCKS overexpression in untransformed cells (S24 grown in + vs. – tet at 37°C), ii) v-Src activation without SSeCKS re-expression ([S24/ts72v-Src grown in the presence of tet at 35°C vs. 39.5°C] minus [S24 grown in the presence of tet at 35°C]), or iii) SSeCKS re-expression in the presence of activated v-Src (S24/ts72v-Src at 35°C grown in + vs. – tet).
Table 2.
The cell groups/conditions used to compare gene expression
| Cells/Growth condition | Comparison |
| NIH3T3/S24 cells, + vs. – tet, 37°C | SSeCKS overexpression |
| ([S24/ts72v-Src cells, 35°C vs. 39.5°C] minus S24 cells, 35°C), + tet (1 μg/ml) |
v-Src activation |
| S24/ts72v-Src cells, 35°C, 1.0 vs.0.04 μg/ml tet | SSeCKS re-expression plus v-Src activation |
Profile of SSeCKS-regulated gene expression
We identified 45 genes whose expression was altered 2-fold or more by SSeCKS overexpression using at least three independent Affymetrix microarray analyses, as described in Materials and Methods, of RNA from S24 cells grown at 37°C in the presence or absence of tet. We then subtracted gene expression effects induced by the tTA transactivator alone in the absence of SSeCKS expression, that is, RNA derived from S2-6 parental cells [13] grown in the absence of tet. Given our prior results that SSeCKS overexpression inhibits G1->S transition in untransformed fibroblasts [7], it was not unexpected that ectopic SSeCKS could induce tumor suppressor/cell cycle negative control genes such as those encoding Rb, p19INK4 or Rassf5, while suppressing cell cycle promoting genes, such as cyclins D, A, B and F, Fos-like antigen 1, Afp [14] and the CDC20 homolog, or neoplasia-promoting genes such as Hmgb3 [15] or Cebpb [16] (Table 3). Similarly, SSeCKS induction of Tgf-α and -β, and of Ngf-β, and the downregulation of Mtap7, is consistent with the pseudo-differentiated, non-polar phenotype displayed during SSeCKS-induced cell flattening [13]. The loss of PDGFR-β expression, which we have described elsewhere [17], likely relates to the ability of SSeCKS to suppress angiogenesis in the brain [8] and in tumor neovasculature. Of the signal transduction genes, the upregulation of Ptplad1, Traf6 [18] and Lmo4 [19] were consistent with the ability of SSeCKS to control cell cycle progression. Stam2 upregulation or Peg3 downregulation may facilitate SSeCKS-mediated anti-apoptosis based on reported roles for Stam2 role T cell survival signaling [20] and for Peg3 in neuronal death [21]. In contrast, SSeCKS unexpectedly upregulated several genes such as Gpr56, Prkce and Btbd2 which have been reported to be upregulated in human cancers [22,23] or involved in inducing cell cycle progression [24], whereas it downregulated several genes such as Nasp, known to antagonize cell cycle progression [25]. The role of other genes, such as Gng2, in SSeCKS regulation remain unclear although they are typically involved in specialized neuronal signaling responses [26]; upregulation might suggest that SSeCKS induces signaling pathways more associated with differentiated, especially neuronal, cells. Interestingly, we previously published that SSeCKS overexpression in S24 cells induces long axonal-like extensions which, when stained for SSeCKS, exhibit periodic, pearl-like enrichments of SSeCKS that are strikingly similar to SSeCKS-enriched structures in developing hippocampal axons [27]. The severe downregulation of Dusp9- which normally antagonizes insulin-mediated adipogenesis [28]- may reflect an ability by SSeCKS to induce glucose uptake and/or metabolism, though this has not been studied. Mycbp is known to inhibit PKA activation at various cell sites by preventing recruitment of the PKA catalytic subunit to the scaffolding proteins, S-AKAP84 and AKAP95 [29]. Thus, Mycbp upregulation by SSeCKS- also a known AKAP [2]-may reflect a higher level of temporal and/or spatial control of PKA activity by SSeCKS.
Profile of v-Src-regulated gene expression
24 genes were identified whose expression varied 2-fold or more by v-Src activation. Specifically, we compared microarray data from RNAs derived from S24/ts72v-Src cells grown in the presence of tet (i.e.- no ectopic SSeCKS) at either 35°C (the PT for Src kinase activity) or 39.5°C (the NPT) (Table 4, column A). To identify possible gene changes induced by incubation at 39.5°C (and not by the loss of Src activity), we compared microarray data derived from S24/ts72v-Src cells versus S24 cells grown in the presence of tet at 35°C (Table 4, column B). However, gene expression changes that occur under the latter conditions only (e.g.- Sh3bp5) might also reflect bona fide v-Src-induced changes that are negated at the NPT, and thus, we included both column A and B data in our overall analysis.
Six of the 24 genes (25%) were identified in Table 3 as being inversely regulated by SSeCKS, i.e.- if they were induced by SSeCKS, they were suppressed by v-Src, or vice-a-versa. These include Peg3, Fosl1, Hmgb3, Gfra1, Il1rl1 and Afp. In contrast, Igfbp4 is dowregulated both by SSeCKS overexpression and v-Src activation, lessening confidence that this gene plays a significant role in oncogenesis.
Rgs16, Id2 and Socs3 have paradoxical roles in that they play both positive and negative functions in cancer. For example, Rgs16- whose expression is induced by v-Src in our system- is a GTPase activating protein for Gαi [30], and Gαi is known to cooperate with Src in oncogenic transformation [31]. However, Rgs16 activity is required for retinoid-induced growth inhibition [32], suggesting that it could antagonize cancer progression. Similarly, Socs3- whose expression is also induced by v-Src, is known to inhibit insulin-mediated proliferation signaling [33] and to be downregulated in head and neck squamous cell carcinomas [34], yet it is upregulated in breast cancer [35]. Id2 plays positive roles in cancer, possibly by inhibiting parameters of differentiation [36], yet decreased Id2 levels correlate with increased invasive potential in human breast cancer cases [37].
The increased levels of the transcription factors Maff, Runx1 and Etv4, and the decreased levels of Centd3, Id4, Spon2, and Cbr2 by v-Src seem to directly relate to cancer progression. Specifically, Maff is overexpressed in some forms of cancer [38] and Maf proteins play direct roles in controlling v-Src-regulated gene expression [39]. Runx1 induces cell cycle progression [40] and increased Runx1 levels are found in chronic myelogenous leukemias [41]. Etv4 levels are increase in gastric cancers [42]. Centd2 is a GAP for RhoA whose activity is blocked by phosphorylation by Src-family kinases [43]. Id4 is downregulated in colon [44] and gastric cancers [45]. Spon2 and Cbr2 levels are decreased significantly in lung cancers [46,47]. In contrast, v-Src induction of Prkg2 does not make apparent sense given that higher Prkg2 levels correlate with increased patient survival rates for lung cancer [48]. Finally, it is unclear how the differential regulation of Sh3bp, Atoh6, Tcfec and Pscdbp would affect v-Src-induced oncogenesis.
Profile of gene expression in cells expressing activated Src plus physiologic SSeCKS levels
46 genes were identified whose expression was altered under conditions of activated v-Src and SSeCKS to physiologic levels (Table 5). 16 of these genes (36%) were identified in screens described in Tables 3 or 4. Of these, 12 genes (Gpr56, Gng2, Ptplad1, Marcks, Cdkn2d, Cdc2a, Hmgb3, Ngfb, Gfra1, Il1rl1, Pdgfrb and Afp) exhibited the same expression control (i.e.- up-or downregulation) as detected after SSeCKS overexpression alone (Table 3). This strongly suggests that these genes are markers for SSeCKS-mediated growth arrest and/or tumor suppression. The remaining 4 pre-identified genes (Socs3, Maff, Id2 and Atoh6) are regulated in the same manner as in v-Src-transformed cells (Table 4) and thus, it is likely that these genes remain controlled by v-Src yet are not sufficient for the oncogenic phenotype.
SSeCKS reverses the expression of several genes that might either antagonize v-Src-induced oncogenesis or function as markers for non-transformed cells. Specifically, SSeCKS induces the expression of Gadd45a- a gene known to inhibit progression at either S or G2/M phases [49] or to induce density-dependent G1 phase arrest [50], Cdkn2d, Rbbp7- which inhibits mitogen- and oncogene-induced c-Fos activation [51], and Dub1- whose overexpression induces G1 arrest [52]. In the same context, SSeCKS suppresses the expression of Map2kb- whose activity is increased in Ras-mediated invasiveness and metastatic potential [53], Rgs2- whose increased expression in mantle cell lymphomas correlates with increased metastatic potential [54], Tcfap4- a regulator of caspase-9 mediated apoptosis [55], Cdc2a, Hif1a- a mediator of tumor angiogenesis induced by v-Src [56,57] and activated c-Src [58,59], Ddit4- an Hif1a-inducible gene [60], and Pdgfrb. Interestingly, SSeCKS re-expression may facilitate increased immune surveillance of tumor cells by the induction of Hsp4a, a tumor antigen carrier that increases the immunogenicity of colon cancer cells in a murine model [61]. Although the decrease in Caspase-7 (Casp7) expression correlates with the decreased apoptotic index of v-Src cells re-expressing SSeCKS [6], it is unclear how the concomitant increase in Stk4- a pro-apoptotic serine/threonine kinase [62], affects cell survival after SSeCKS re-expression.
A set of genes seems to remain regulated by v-Src during SSeCKS re-expression, suggesting that their expression levels are not sufficient to induce or maintain oncogenic transformation in the presence of physiologic levels of SSeCKS. These include Ptpn11- which can activate Src-family kinases [63] and which is required for v-Src-induced morphological transformation [64], Pip5k1b- whose product is activated by Rho-family GTPases in cancer cells [65], Srpk2- a gene upregulated after retinoid-induced differentiation of HL60 leukemia cells [66], Dusp10- a negative regulator of MAP kinases and antagonist of prostate cancer cell proliferation [67], Marcks, Timp3- a gene downregulated in squamous cell carcinomas and prostate adenocarcinomas [68,69], Gdf5- a growth arrest inducer in mouse B cells [70], and Gas1- a gene downregulated in EGF-induced hepatocellular carcinoma [71] and in Ras-transformed cells [72]. The involvement of S100a8 is unclear because although its expression is induced in prostate cancer [73], its expression is downregulated in the metastatic breast cancer cell line, MDA-MB-231, compared to the non-metastatic MCF-7 [74].
In order to verify the gene expression changes identified in the microarray studies, several genes were assayed by semi-quantitative RT-PCR using primer sets shown in Table 1. Thus, RNAs derived from S24 or S24/ts72v-Src cells grown at the PT in the presence or absence of tet were amplified by RT-PCR as described in Materials and Methods and then the electrophoresed products were quantified by digital densitometry. Fig. 2A and Table 6 show a strong concordance between the regulation of Cdc2a, Il1rl1, Cdkn2d, Pdgfrb, Ptpn11, Tgfb1, Id2, Gpr56, Maff, Socs, Afp, Ngfb, Gadd45a, Marcks and Hif1a by either SSeCKS alone or SSeCKS re-expression in the presence of v-Src as gauged by both the microarray and RT-PCR assays. This strengthens the notion that the genes identified by our microarray comparisons reflect bona fide examples of differential expression due to SSeCKS overexpression or SSeCKS re-expression in the presence of active v-Src. Finally, we performed semi-quantitative RT-PCR on several genes shown to be induced by active v-Src (Table 3) using RNAs from HT29 and HCT116, representing cells with high and low levels of c-Src activity, respectively (Fig. 1B). Fig. 2B shows significantly higher levels of HMGA2, FOSL1, CDC20A, PSCDBP, AFP, PDGFRB and HIF1A expression in HT29 cells, which correlates with the v-Src-induced levels found for the mouse orthologues in Table 4 (for both cell comparisons A and B). In contrast, HMGB3 transcript levels were similar in HT29 and HCT116 cells, correlating with the finding that its mouse orthologue was likely not induced by v-Src (Table 4, column B). Taken together, these data strongly suggest that activated Src and SSeCKS/Gravin control similar sets of genes in both human and mouse fibroblasts and epithelial cells.
Figure 2.

Verification of differential gene expression using semi-quantitative RT-PCR. Equal amounts of total RNA isolated from S24 or S24/ts72v-Src cells (grown at the PT) in the presence or absence of tet (Panel A) or from HCT116 or HT29 (Panel B) were subjected to RT-PCR analysis as described in Materials and Methods using primer sets described in Table 1. These results are typical of at least two independent experiments.
Table 6.
Comparison of microarray and RT-PCR data
| Fold change (array) | Fold change (RT-PCR)a | |||
| Gene | SSeCKS Inductionb | SSeCKS induction plus v-Src activationc | SSeCKS induction | SSeCKS induction plus v-Src activation |
| Cdc2a | 0.46 | 0.25 | 0.27 | 0.25 |
| Il1rl1 | 0.29 | 0.23 | 0.31 | 0.36 |
| Cdkn2d | 2.73 | 2.46 | 3.10 | 3.50 |
| Pdgfrb | 0.30 | 0.18 | 0.34 | 0.30 |
| Ptpn11 | NC* | 5.86 | NC | 3.04 |
| Tgfb1 | 2.33 | NC | 2.50 | NC |
| Id2 | NC | 0.44 | NC | 0.33 |
| Gpr56 | 126.24 | 49.00 | 45.0 | 28.0 |
| Maff | NC | 2.28 | NC | 3.5 |
| Socs3 | NC | 2.07 | NC | 2.4 |
| Afp | 0.32 | 0.50 | 0.4 | 0.2 |
| Ngfb | 2.28 | 2.38 | 8.0 | 2.5 |
| Gadd45a | NC | 3.43 | 4.0 | 3.0 |
| Marcks | 0.48 | 0.12 | NC | 0.2 |
| Hif1a | NC | 0.22 | NC | 0.3 |
a, based on two independent experiments. SE < 0.005.
b, from Table 3.
c, from Table 5.
NC, no change.
Conclusion
We have identified sets of genes whose expression is controlled either by SSeCKS or v-Src alone, or only after re-expression of SSeCKS in the presence of activated v-Src. Our data suggest that some or all of the SSeCKS-regulated genes may either directly antagonize v-Src-induced oncogenesis or may serve as markers for non-oncogenic cells. Moreover, our analysis has identified a set of genes previously thought to contribute to the oncogenic phenotype yet which are insufficient to induce oncogenesis in the presence of physiologic levels of SSeCKS. In sum, our data indicate that SSeCKS may antagonize Src-induced oncogenesis through the normalization of functions controlling mitogenic signaling pathways, cell cycle progression, transcriptional regulation and apoptosis.
Abbreviations
SSeCKS, Src Suppressed C Kinase Substrate; PKC, protein kinase C; RT-PCR, reverse transcription-polymerase chain reaction; VEGF, vascular endothelial growth factor; PT, permissive temperature; NPT, non-permissive temperature; PAb, polyclonal antibody; MAb, monoclonal antibody; Ig, immunoglobulin; tet, tetracycline.
Competing interests
The author(s) declare that they have no competing interests.
Authors' contributions
YL performed the work as a postdoctoral fellow in the lab of IHG and thus, both contributed to the conceptual framework of this study. Both YL and IHG produced and approved the final written manuscript. LG performed many of the semi-quantitative RT-PCR assays.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Acknowledgments
Acknowledgements
We thank the RPCI Gene Expression CORE Facility for their assistance, and especially Leighton Stein for help in analyzing gene expression significance. This work was funded by grants from the National Cancer Institute (CA94108) and Department of Defense (PC040256) to I.H.G., and in part by NIH/NCI Cancer Center Support Grant 2P30 CA016056.
Contributor Information
Yongzhong Liu, Email: yoliu@mail.nih.gov.
Lingqiu Gao, Email: lingqiu.gao@roswellpark.org.
Irwin H Gelman, Email: irwin.gelman@roswellpark.org.
References
- Lin X, Nelson PJ, Frankfort B, Tombler E, Johnson R, Gelman IH. Isolation and characterization of a novel mitogenic regulatory gene, 322, which is transcriptionally suppressed in cells transformed by src and ras. Mol Cell Biol. 1995;15:2754–2762. doi: 10.1128/mcb.15.5.2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauert J, Klauck T, Langeberg LK, Scott JD. Gravin, an autoantigen recognized by serum from myasthenia gravis patients, is a kinase scaffolding protein. Current Biology. 1997;7:52–62. doi: 10.1016/s0960-9822(06)00027-3. [DOI] [PubMed] [Google Scholar]
- Gordon T, Grove B, Loftus JC, O'Toole T, McMillan R, Lindstrom J, Ginsberg MH. Molecular cloning and prelimnary characteriztion of a novel cytoplasmic antigen recognized by myasthenia gravis sera. J Clin Invest. 1992;90:992–999. doi: 10.1172/JCI115976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia W, Unger P, Miller L, Nelson J, Gelman IH. The src-suppressed C kinase substrate, SSeCKS, is a potential metastasis inhibitor in prostate cancer. Cancer Res. 2001;15:5644–5651. [PubMed] [Google Scholar]
- Gelman IH. The role of the SSeCKS/Gravin/AKAP12 scaffolding proteins in the spaciotemporal control of signaling pathways in oncogenesis and development. Front Biosci. 2002;7:1797. doi: 10.2741/A879. [DOI] [PubMed] [Google Scholar]
- Lin X, Gelman IH. Re-expression of the major protein kinase C substrate, SSeCKS, suppresses v-src- induced morphological transformation and tumorigenesis. Cancer Res. 1997;57:2304–2312. [PubMed] [Google Scholar]
- Lin X, Nelson P, Gelman IH. Regulation of G->S Progression by the SSeCKS Tumor Suppressor: Control of Cyclin D Expression and Cellular Compartmentalization. Mol Cell Biol. 2000;20:7259–7272. doi: 10.1128/mcb.20.19.7259-7272.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SW, Kim WJ, Choi YK, Song HS, Son MJ, Gelman IH, Kim YJ, Kim KW. SSeCKS regulates angiogenesis and tight junction formation in blood-brain barrier. Nat Med. 2003;9:900–906. doi: 10.1038/nm889. [DOI] [PubMed] [Google Scholar]
- Su B, Zheng Q, Vaughan MM, Bu Y, Gelman IH. SSeCKS metastasis-suppressing activity correlates with angiogenesis inhibition. Cancer Res. 2006. [DOI] [PubMed]
- Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moissoglu K, Gelman IH. v-Src Rescues Actin-Based Cytoskeletal Architecture and Cell Motility, and Induces Enhanced Anchorage-Independence During Oncogenic Transformation of FAK-Null Fibroblasts. J Biol Chem. 2003;278:47946–47959. doi: 10.1074/jbc.M302720200. [DOI] [PubMed] [Google Scholar]
- Lin X, Tombler E, Nelson PJ, Ross M, Gelman IH. A novel src- and ras-suppressed protein kinase C substrate associated with cytoskeletal architecture. J Biol Chem. 1996;271:430–28. doi: 10.1074/jbc.271.45.28430. [DOI] [PubMed] [Google Scholar]
- Gelman IH, Lee K, Tombler E, Gordon R, Lin X. Control of cytoskeletal architecture by the src-suppressed C kinase substrate, SSeCKS. Cell Motil Cytoskeleton. 1998;41:1–17. doi: 10.1002/(SICI)1097-0169(1998)41:1<1::AID-CM1>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Li MS, Li PF, Yang FY, He SP, Du GG, Li G. The intracellular mechanism of alpha-fetoprotein promoting the proliferation of NIH 3T3 cells. Cell Res. 2002;12:151–156. doi: 10.1038/sj.cr.7290121. [DOI] [PubMed] [Google Scholar]
- Fedele M, Battista S, Kenyon L, Baldassarre G, Fidanza V, Klein-Szanto AJ, Parlow AF, Visone R, Pierantoni GM, Outwater E, Santoro M, Croce CM, Fusco A. Overexpression of the HMGA2 gene in transgenic mice leads to the onset of pituitary adenomas. Oncogene. 2002;21:3190–3198. doi: 10.1038/sj.onc.1205428. [DOI] [PubMed] [Google Scholar]
- Zhu S, Yoon K, Sterneck E, Johnson PF, Smart RC. CCAAT/enhancer binding protein-beta is a mediator of keratinocyte survival and skin tumorigenesis involving oncogenic Ras signaling. Proc Natl Acad Sci USA. 2002;99:207–212. doi: 10.1073/pnas.012437299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su B, Zheng Q, Vaughan MM, Bu Y, Gelman IH. SSeCKSmetastasis-suppressing activity in MatLyLu prostate cancer cellscorrelates with VEGF inhibition. Cancer Res. 2006. [DOI] [PubMed]
- Bharti AC, Takada Y, Shishodia S, Aggarwal BB. Evidence that receptor activator of nuclear factor (NF)-kappaB ligand can suppress cell proliferation and induce apoptosis through activation of a NF-kappaB-independent and TRAF6-dependent mechanism. J Biol Chem. 2004;279:6065–6076. doi: 10.1074/jbc.M308062200. [DOI] [PubMed] [Google Scholar]
- Sum EY, Shackleton M, Hahm K, Thomas RM, O'Reilly LA, Wagner KU, Lindeman GJ, Visvader JE. Loss of the LIM domain protein Lmo4 in the mammary gland during pregnancy impedes lobuloalveolar development. Oncogene. 2005;24:4820–4828. doi: 10.1038/sj.onc.1208638. [DOI] [PubMed] [Google Scholar]
- Yamada M, Ishii N, Asao H, Murata K, Kanazawa C, Sasaki H, Sugamura K. Signal-transducing adaptor molecules STAM1 and STAM2 are required for T-cell development and survival. Mol Cell Biol. 2002;22:8648–8658. doi: 10.1128/MCB.22.24.8648-8658.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MD, Wu X, Aithmitti N, Morrison RS. Peg3/Pw1 is a mediator between p53 and Bax in DNA damage-induced neuronal death. J Biol Chem. 2002;277:23000–23007. doi: 10.1074/jbc.M201907200. [DOI] [PubMed] [Google Scholar]
- Shashidhar S, Lorente G, Nagavarapu U, Nelson A, Kuo J, Cummins J, Nikolich K, Urfer R, Foehr ED. GPR56 is a GPCR that is overexpressed in gliomas and functions in tumor cell adhesion. Oncogene. 2005;24:1673–1682. doi: 10.1038/sj.onc.1208395. [DOI] [PubMed] [Google Scholar]
- Yamada A, Kawano K, Koga M, Takamori S, Nakagawa M, Itoh K. Gene and peptide analyses of newly defined lung cancer antigens recognized by HLA-A2402-restricted tumor-specific cytotoxic T lymphocytes. Cancer Res. 2003;63:2829–2835. [PubMed] [Google Scholar]
- Soh JW, Weinstein IB. Roles of specific isoforms of protein kinase C in the transcriptional control of cyclin D1 and related genes. J Biol Chem. 2003;278:34709–34716. doi: 10.1074/jbc.M302016200. [DOI] [PubMed] [Google Scholar]
- Alekseev OM, Bencic DC, Richardson RT, Widgren EE, O'Rand MG. Overexpression of the Linker histone-binding protein tNASP affects progression through the cell cycle. J Biol Chem. 2003;278:8846–8852. doi: 10.1074/jbc.M210352200. [DOI] [PubMed] [Google Scholar]
- Varga EV, Hosohata K, Borys D, Navratilova E, Nylen A, Vanderah TW, Porreca F, Roeske WR, Yamamura HI. Antinociception depends on the presence of G protein gamma2-subunits in brain. Eur J Pharmacol. 2005;508:93–98. doi: 10.1016/j.ejphar.2004.11.062. [DOI] [PubMed] [Google Scholar]
- Gelman IH, Tombler E, Vargas J., Jr A role for SSeCKS, a major protein kinase C substrate with tumor suppressor activity, in cytoskeletal architecture, formation of migratory processes, and cell migration during embryogenesis. Histochemical Journal. 2000;32:13–26. doi: 10.1023/a:1003950027529. [DOI] [PubMed] [Google Scholar]
- Xu H, Dembski M, Yang Q, Yang D, Moriarty A, Tayber O, Chen H, Kapeller R, Tartaglia LA. Dual specificity mitogen-activated protein (MAP) kinase phosphatase-4 plays a potential role in insulin resistance. J Biol Chem. 2003;278:30187–30192. doi: 10.1074/jbc.M302010200. [DOI] [PubMed] [Google Scholar]
- Furusawa M, Taira T, Iguchi-Ariga SM, Ariga H. AMY-1 interacts with S-AKAP84 and AKAP95 in the cytoplasm and the nucleus, respectively, and inhibits cAMP-dependent protein kinase activity by preventing binding of its catalytic subunit to A-kinase-anchoring protein (AKAP) complex. J Biol Chem. 2002;277:50885–50892. doi: 10.1074/jbc.M206387200. [DOI] [PubMed] [Google Scholar]
- Nagata Y, Oda M, Nakata H, Shozaki Y, Kozasa T, Todokoro K. A novel regulator of G-protein signaling bearing GAP activity for Galphai and Galphaq in megakaryocytes. Blood. 2001;97:3051–3060. doi: 10.1182/blood.v97.10.3051. [DOI] [PubMed] [Google Scholar]
- Ram PT, Iyengar R. G protein coupled receptor signaling through the Src and Stat3 pathway: role in proliferation and transformation. Oncogene. 2001;20:1601–1606. doi: 10.1038/sj.onc.1204186. [DOI] [PubMed] [Google Scholar]
- Liu T, Bohlken A, Kuljaca S, Lee M, Nguyen T, Smith S, Cheung B, Norris MD, Haber M, Holloway AJ, Bowtell DD, Marshall GM. The retinoid anticancer signal: mechanisms of target gene regulation. Br J Cancer. 2005;93:310–318. doi: 10.1038/sj.bjc.6602700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueki K, Kondo T, Kahn CR. Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol Cell Biol. 2004;24:5434–5446. doi: 10.1128/MCB.24.12.5434-5446.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber A, Hengge UR, Bardenheuer W, Tischoff I, Sommerer F, Markwarth A, Dietz A, Wittekind C, Tannapfel A. SOCS-3 is frequently methylated in head and neck squamous cell carcinoma and its precursor lesions and causes growth inhibition. Oncogene. 2005;24:6699–6708. doi: 10.1038/sj.onc.1208818. [DOI] [PubMed] [Google Scholar]
- Raccurt M, Tam SP, Lau P, Mertani HC, Lambert A, Garcia-Caballero T, Li H, Brown RJ, McGuckin MA, Morel G, Waters MJ. Suppressor of cytokine signalling gene expression is elevated in breast carcinoma. Br J Cancer. 2003;89:524–532. doi: 10.1038/sj.bjc.6601115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Candia P, Benera R, Solit DB. A role for Id proteins in mammary gland physiology and tumorigenesis. Adv Cancer Res. 2004;92:81–94. doi: 10.1016/S0065-230X(04)92004-0. [DOI] [PubMed] [Google Scholar]
- Stighall M, Manetopoulos C, Axelson H, Landberg G. High ID2 protein expression correlates with a favourable prognosis in patients with primary breast cancer and reduces cellular invasiveness of breast cancer cells. Int J Cancer. 2005;115:403–411. doi: 10.1002/ijc.20875. [DOI] [PubMed] [Google Scholar]
- Kienast J, Berdel WE. c-maf in multiple myeloma: an oncogene enhancing tumor-stroma interactions. Cancer Cell. 2004;5:109–110. doi: 10.1016/s1535-6108(04)00030-3. [DOI] [PubMed] [Google Scholar]
- Provot S, Pouponnot C, Lecoq O, Calothy G, Felder-Schmittbuhl MP. Characterization of a novel quiescence responsive element downregulated by v-Src in the promoter of the neuroretina specific QR1 gene. Oncogene. 2000;19:4736–4745. doi: 10.1038/sj.onc.1203837. [DOI] [PubMed] [Google Scholar]
- Bernardin-Fried F, Kummalue T, Leijen S, Collector MI, Ravid K, Friedman AD. AML1/RUNX1 increases during G1 to S cell cycle progression independent of cytokine-dependent phosphorylation and induces cyclin D3 gene expression. J Biol Chem. 2004;279:15678–15687. doi: 10.1074/jbc.M310023200. [DOI] [PubMed] [Google Scholar]
- Yanagida M, Osato M, Yamashita N, Liqun H, Jacob B, Wu F, Cao X, Nakamura T, Yokomizo T, Takahashi S, Yamamoto M, Shigesada K, Ito Y. Increased dosage of Runx1/AML1 acts as a positive modulator of myeloid leukemogenesis in BXH2 mice. Oncogene. 2005;24:4477–4485. doi: 10.1038/sj.onc.1208675. [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Horiuchi S, Adachi Y, Taniguchi H, Nosho K, Min Y, Imai K. Expression of ets-related transcriptional factor E1AF is associated with tumor progression and over-expression of matrilysin in human gastric cancer. Carcinogenesis. 2004;25:325–332. doi: 10.1093/carcin/bgh011. [DOI] [PubMed] [Google Scholar]
- ST I, Nie Z, Stewart A, Najdovska M, Hall NE, He H, Randazzo PA, Lock P. ARAP3 is transiently tyrosine phosphorylated in cells attaching to fibronectin and inhibits cell spreading in a RhoGAP-dependent manner. J Cell Sci. 2004;117:6071–6084. doi: 10.1242/jcs.01526. [DOI] [PubMed] [Google Scholar]
- Umetani N, Takeuchi H, Fujimoto A, Shinozaki M, Bilchik AJ, Hoon DS. Epigenetic inactivation of ID4 in colorectal carcinomas correlates with poor differentiation and unfavorable prognosis. Clin Cancer Res. 2004;10:7475–7483. doi: 10.1158/1078-0432.CCR-04-0689. [DOI] [PubMed] [Google Scholar]
- Chan AS, Tsui WY, Chen X, Chu KM, Chan TL, Chan AS, Li R, So S, Yuen ST, Leung SY. Downregulation of ID4 by promoter hypermethylation in gastric adenocarcinoma. Oncogene. 2003;22:6946–6953. doi: 10.1038/sj.onc.1206799. [DOI] [PubMed] [Google Scholar]
- Takenaka K, Ogawa E, Oyanagi H, Wada H, Tanaka F. Carbonyl reductase expression and its clinical significance in non-small-cell lung cancer. Cancer Epidemiol Biomarkers Prev. 2005;14:1972–1975. doi: 10.1158/1055-9965.EPI-05-0060. [DOI] [PubMed] [Google Scholar]
- Manda R, Kohno T, Matsuno Y, Takenoshita S, Kuwano H, Yokota J. Identification of genes (SPON2 and C20orf2) differentially expressed between cancerous and noncancerous lung cells by mRNA differential display. Genomics. 1999;61:5–14. doi: 10.1006/geno.1999.5939. [DOI] [PubMed] [Google Scholar]
- Ikehara M, Oshita F, Sekiyama A, Hamanaka N, Saito H, Yamada K, Noda K, Kameda Y, Miyagi Y. Genome-wide cDNA microarray screening to correlate gene expression profile with survival in patients with advanced lung cancer. Oncol Rep. 2004;11:1041–1044. [PubMed] [Google Scholar]
- Vairapandi M, Balliet AG, Hoffman B, Liebermann DA. GADD45b and GADD45g are cdc2/cyclinB1 kinase inhibitors with a role in S and G2/M cell cycle checkpoints induced by genotoxic stress. J Cell Physiol. 2002;192:327–338. doi: 10.1002/jcp.10140. [DOI] [PubMed] [Google Scholar]
- Zhang X, Ma L, Enkemann SA, Pledger WJ. Role of Gadd45alpha in the density-dependent G1 arrest induced by p27(Kip1) Oncogene. 2003;22:4166–4174. doi: 10.1038/sj.onc.1206599. [DOI] [PubMed] [Google Scholar]
- Yang J, Kiefer S, Rauchman M. Characterization of the gene encoding mouse retinoblastoma binding protein-7, a component of chromatin-remodeling complexes. Genomics. 2002;80:407–415. doi: 10.1006/geno.2002.6844. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Carroll M, Papa FR, Hochstrasser M, D'Andrea AD. DUB-1, a deubiquitinating enzyme with growth-suppressing activity. Proc Natl Acad Sci USA. 1996;93:3275–3279. doi: 10.1073/pnas.93.8.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin I, Kim S, Song H, Kim HR, Moon A. H-Ras-specific activation of Rac-MKK3/6-p38 pathway: its critical role in invasion and migration of breast epithelial cells. J Biol Chem. 2005;280:14675–14683. doi: 10.1074/jbc.M411625200. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Hollmen J, Raty R, Aalto Y, Nagy B, Elonen E, Kere J, Mannila H, Franssila K, Knuutila S. Investigatory and analytical approaches to differential gene expression profiling in mantle cell lymphoma. Br J Haematol. 2002;119:905–915. doi: 10.1046/j.1365-2141.2002.03931.x. [DOI] [PubMed] [Google Scholar]
- Tsujimoto K, Ono T, Sato M, Nishida T, Oguma T, Tadakuma T. Regulation of the expression of caspase-9 by the transcription factor activator protein-4 in glucocorticoid-induced apoptosis. J Biol Chem. 2005;280:27638–27644. doi: 10.1074/jbc.M501304200. [DOI] [PubMed] [Google Scholar]
- Jiang BH, Agani F, Passaniti A, Semenza GL. V-SRC induces expression of hypoxia-inducible factor 1 (HIF-1) and transcription of genes encoding vascular endothelial growth factor and enolase 1: involvement of HIF-1 in tumor progression. Cancer Res. 1997;57:5328–5335. [PubMed] [Google Scholar]
- Gleadle JM, Ratcliffe PJ. Induction of hypoxia-inducible factor-1, erythropoietin, vascular endothelial growth factor, and glucose transporter-1 by hypoxia: evidence against a regulatory role for Src kinase. Blood. 1997;89:503–509. [PubMed] [Google Scholar]
- Gray MJ, Zhang J, Ellis LM, Semenza GL, Evans DB, Watowich SS, Gallick GE. HIF-1alpha, STAT3, CBP/p300 and Ref-1/APE are components of a transcriptional complex that regulates Src-dependent hypoxia-induced expression of VEGF in pancreatic and prostate carcinomas. Oncogene. 2005;24:3110–3120. doi: 10.1038/sj.onc.1208513. [DOI] [PubMed] [Google Scholar]
- Karni R, Dor Y, Keshet E, Meyuhas O, Levitzki A. Activated pp60c-Src leads to elevated hypoxia-inducible factor (HIF)-1alpha expression under normoxia. J Biol Chem. 2002;277:42919–42925. doi: 10.1074/jbc.M206141200. [DOI] [PubMed] [Google Scholar]
- Shoshani T, Faerman A, Mett I, Zelin E, Tenne T, Gorodin S, Moshel Y, Elbaz S, Budanov A, Chajut A, Kalinski H, Kamer I, Rozen A, Mor O, Keshet E, Leshkowitz D, Einat P, Skaliter R, Feinstein E. Identification of a novel hypoxia-inducible factor 1-responsive gene, RTP801, involved in apoptosis. Mol Cell Biol. 2002;22:2283–2293. doi: 10.1128/MCB.22.7.2283-2293.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XY, Li Y, Manjili MH, Repasky EA, Pardoll DM, Subjeck JR. Hsp110 over-expression increases the immunogenicity of the murine CT26 colon tumor. Cancer Immunol Immunother. 2002;51:311–319. doi: 10.1007/s00262-002-0287-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khokhlatchev A, Rabizadeh S, Xavier R, Nedwidek M, Chen T, Zhang XF, Seed B, Avruch J. Identification of a novel Ras-regulated proapoptotic pathway. Curr Biol. 2002;12:253–265. doi: 10.1016/s0960-9822(02)00683-8. [DOI] [PubMed] [Google Scholar]
- Zhang SQ, Yang W, Kontaridis MI, Bivona TG, Wen G, Araki T, Luo J, Thompson JA, Schraven BL, Philips MR, Neel BG. Shp2 regulates SRC family kinase activity and Ras/Erk activation by controlling Csk recruitment. Mol Cell. 2004;13:341–355. doi: 10.1016/s1097-2765(04)00050-4. [DOI] [PubMed] [Google Scholar]
- Hakak Y, Hsu YS, Martin GS. Shp-2 mediates v-Src-induced morphological changes and activation of the anti-apoptotic protein kinase Akt. Oncogene. 2000;19:3164–3171. doi: 10.1038/sj.onc.1203655. [DOI] [PubMed] [Google Scholar]
- Weernink PA, Meletiadis K, Hommeltenberg S, Hinz M, Ishihara H, Schmidt M, Jakobs KH. Activation of type I phosphatidylinositol 4-phosphate 5-kinase isoforms by the Rho GTPases, RhoA, Rac1, and Cdc42. J Biol Chem. 2004;279:7840–7849. doi: 10.1074/jbc.M312737200. [DOI] [PubMed] [Google Scholar]
- Yen A, Lin DM, Lamkin TJ, Varvayanis S. retinoic acid, bromodeoxyuridine, and the Delta 205 mutant polyoma virus middle T antigen regulate expression levels of a common ensemble of proteins associated with early stages of inducing HL-60 leukemic cell differentiation. In Vitro Cell Dev Biol Anim. 2004;40:216–241. doi: 10.1290/1543-706X(2004)40<216:RABATM>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- He HY, Fang WG, Zheng J, You JF, Heng WJ, Li Y. [Mechanism of the mitogen-activated protein kinase phosphatase-5 regulating the growth and invasion of a human prostate cancer cell line] Zhonghua Yi Xue Za Zhi. 2003;83:1812–1817. [PubMed] [Google Scholar]
- Kettunen E, Anttila S, Seppanen JK, Karjalainen A, Edgren H, Lindstrom I, Salovaara R, Nissen AM, Salo J, Mattson K, Hollmen J, Knuutila S, Wikman H. Differentially expressed genes innonsmall cell lung cancer: expression profiling of cancer-related genes in squamous cell lung cancer. Cancer Genet Cytogenet. 2004;149:98–106. doi: 10.1016/S0165-4608(03)00300-5. [DOI] [PubMed] [Google Scholar]
- Karan D, Lin FC, Bryan M, Ringel J, Moniaux N, Lin MF, Batra SK. Expression of ADAMs (a disintegrin and metalloproteases) and TIMP-3 (tissue inhibitor of metalloproteinase-3) in human prostatic adenocarcinomas. Int J Oncol. 2003;23:1365–1371. [PubMed] [Google Scholar]
- Nakahara T, Tominaga K, Koseki T, Yamamoto M, Yamato K, Fukuda J, Nishihara T. Growth/differentiation factor-5 induces growth arrest and apoptosis in mouse B lineage cells with modulation by Smad. Cell Signal. 2003;15:181–187. doi: 10.1016/s0898-6568(02)00088-8. [DOI] [PubMed] [Google Scholar]
- Borlak J, Meier T, Halter R, Spanel R, Spanel-Borowski K. Epidermal growth factor-induced hepatocellular carcinoma: gene expression profiles in precursor lesions, early stage and solitary tumours. Oncogene. 2005;24:1809–1819. doi: 10.1038/sj.onc.1208196. [DOI] [PubMed] [Google Scholar]
- Ciccarelli C, Philipson L, Sorrentino V. Regulation of expression of growth arrest-specific genes in mouse fibroblasts. Mol Cell Biol. 1990;10:1525–1529. doi: 10.1128/mcb.10.4.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermani A, Hess J, De Servi B, Medunjanin S, Grobholz R, Trojan L, Angel P, Mayer D. Calcium-binding proteins S100A8 and S100A9 as novel diagnostic markers in human prostate cancer. Clin Cancer Res. 2005;11:5146–5152. doi: 10.1158/1078-0432.CCR-05-0352. [DOI] [PubMed] [Google Scholar]
- Nagaraja GM, Othman M, Fox BP, Alsaber R, Pellegrino CM, Zeng Y, Khanna R, Tamburini P, Swaroop A, Kandpal RP. Gene expression signatures and biomarkers of noninvasive and invasive breast cancer cells: comprehensive profiles by representational difference analysis, microarrays and proteomics. Oncogene. 2006;25:2328–2338. doi: 10.1038/sj.onc.1209265. [DOI] [PubMed] [Google Scholar]