

Abstract
The defining characteristic of large-conductance Ca2+- and voltage-activated K+ channels (BKCa) is their allosteric activation by two distinct stimuli, membrane depolarization and cytosolic Ca2+ ions. In this allosteric gating, increasing cytosolic Ca2+ concentration ([Ca2+]i) shifts the depolarization required for channel opening into the physiological voltage range. In fact, according to present knowledge, elevation of [Ca2+]i to micromolar levels is the only means to activate BKCa at membrane potentials below 0 mV. We recorded BKCa-mediated currents from auditory inner hair cells (IHCs) in acutely isolated organs of Corti using the patch-clamp technique in whole-cell and excised patch configuration. In inside-out and outside-out patches, activation of BKCa channels from IHCs showed the prototypic sensitivity to increased [Ca2+]i. However, channel activation at 0 [Ca2+]i occurred at unusually negative potentials (half-maximal activation (Vh) around 0 mV), indicating that a large fraction of the channels can be activated at physiological voltages without elevated [Ca2+]i. In intact IHCs, the activation curve of BKCa currents recorded in whole-cell configuration exhibited a Vh of −42 mV together with a high voltage dependence (slope factor of 10 mV) and submillisecond onset of current. Surprisingly, this activation was independent of changes in local [Ca2+]i as shown by experiments that interfered with Ca2+ influx through voltage-gated Ca2+ (Cav) channels, release of Ca2+ from internal stores, or intracellular buffer capacity. This behaviour is not due to β-subunits of BKCa (BKβ), as genetic inactivation of the β-subunit expressed in IHCs, KCNMB1, did not affect BKCa gating. We conclude that the BKCa channel protein in IHCs may be modified in order to rapidly activate and deactivate at resting [Ca2+]i. Our results suggest that BKCa may function as a purely voltage-gated K+ channel with exceptionally rapid activation kinetics, challenging the view that both increased cytosolic Ca2+ and depolarization are generally required for activation of BKCa.
BKCa channels are key modulators of cellular excitability (Vergara et al. 1998). They are assembled from four identical α-subunits of BKCa (BKα) encoded by the Slo gene (Adelman et al. 1992) and are dually activated by membrane depolarization and increase in intracellular Ca2+ concentration ([Ca2+]i) (Marty, 1981; Pallotta et al. 1981; Latorre et al. 1982). Increasing [Ca2+]i shifts the voltage required for channel activation in the hyperpolarizing direction (Cui et al. 1997; Horrigan & Aldrich, 2002). The precise voltage range of activation and the calcium sensitivity may be affected by different mechanisms including alternative mRNA splicing of BKα (Adelman et al. 1992; Tseng-Crank et al. 1994), coassembly with β-subunits (KCNMB1–4; McManus et al. 1995; Brenner et al. 2000a) or accessory proteins (Schopperle et al. 1998; Xia et al. 1998), and post-translational modification (Reinhart et al. 1991; DiChiara & Reinhart, 1997; Schubert & Nelson, 2001). However, the exact impact of the latter on both voltage-dependence and Ca2+ sensitivity has often not been determined rigorously. Despite this variability, the activation range at resting [Ca2+]i is generally not within the physiological voltage range (see Fig. 1E; e.g. Tseng-Crank et al. 1994; Brenner et al. 2000a; Ransom et al. 2003). Thus, it is assumed that under physiological conditions, BKCa channel opening inevitably requires coincident increase in [Ca2+]i and membrane depolarization (Vergara et al. 1998).
Figure 1. IK,f is mediated by BKCa channels.

A and B, outwardly rectifying K+ currents recorded in IHCs from a BKα knock-out mouse (BKα−/−; B) and a littermate control (BKα+/+; A). Currents were recorded in whole-cell mode in response to voltage steps to potentials between −104 and +6 mV (10 mV nominal increments) from a holding potential of −84 mV. Intracellular and extracellular solution contained 10 mm 4-AP and 1 μm XE991, respectively (see Methods). Note that the fast activating IK,f was absent in the BKα−/− IHC, where only a minor residual, slowly activating K+ current component was observed. C, BK currents recorded in an outside-out patch excised from a rat IHC. Current traces were evoked by voltage steps to +16, +56 and +96 mV, from a holding potential of −64 mV. Solutions were as in (A and B). Each trace is averaged from 10 individual presentations of the voltage step. D, fast activation of IK,f in a rat IHC, recorded as in A in response to 5-ms steps incremented by nominally 10 mV. Tail-current potential was −34 mV. Monoexponential fits to the current activation (not shown) yielded a time constant (τactivation) between 0.67 ms (at −44 mV) and 0.37 ms (at +6 mV). E, steady-state activation curve determined from tail currents measured in 16 rat IHCs as in C. Currents were normalized to the saturating tail-current amplitude and plotted versus the prepulse potential corrected for errors resulting from residual series resistance (see Methods); standard error of the voltage was smaller than the symbol size. The continuous line is a fit of a first-order Boltzmann function to the averaged data, yielding values for Vh and α of −42.4 mV and 10.4 mV, respectively. For comparison of the activation voltage ranges, activation curves obtained in excised patches from BKCa-expressing Xenopus oocytes at 0 [Ca2+]i are shown (□; mean data from seven patches; error bars are smaller than symbol size). The continuous line through the recombinant data (Xenopus oocyte) is a Boltzmann fit to the averaged data, yielding values for Vh and α of 161.9 mV and 22.6 mV, respectively.
In essentially all vertebrate auditory hair cells, BKCa carries a major component of the ionic current. In non-mammalian hair cells, activation of BKCa close to the resting potential results from Ca2+ entry through voltage-gated Ca2+ channels (Cav). Both channel types generally appear to be tightly colocalized allowing for a local activation route of BKCa channels by Cav (Roberts et al. 1990; Samaranayake et al. 2004). Consequently, blocking the Ca2+ current leads to a reduction of BKCa channel activity (Hudspeth & Lewis, 1988a; Art et al. 1993). In amphibian, reptilian and avian IHCs, the functional coupling between Cav-mediated Ca2+ influx and BKCa-mediated outward currents generates resonant behaviour of the membrane potential that contributes to the tuning of hair cells to specific sound frequencies (Art & Fettiplace, 1987; Fuchs et al. 1988; Hudspeth & Lewis, 1988b).
In contrast, mammalian IHCs do not exhibit significant electrical resonance. Yet, they exhibit a rapidly activating K+ current (termed IK,f for its fast activation kinetics; Kros & Crawford, 1990) thought to be mediated by BKCa channels based on its Ca2+-dependent gating observed in excised patches (Oliver et al. 2003) and its sensitivity to iberiotoxin, charybdotoxin and TEA (Kros et al. 1998; Marcotti et al. 2004; Pyott et al. 2004; Hafidi et al. 2005). In intact IHCs, this current exhibits a voltage range of activation similar to that of the L-type Cav channel of the hair cell (Cav1.3; Platzer et al. 2000). However, it has been a puzzle since its first description that IK,f was unaffected by removal of extracellular Ca2+ (Kros & Crawford, 1990; Marcotti et al. 2004). This apparent paradox may be explained by another voltage-dependent Ca2+ source providing increased [Ca2+]i such as release from intracellular stores (Marcotti et al. 2004). To elucidate the mechanism that underlies the unusual BKCa gating in IHCs, we performed patch-clamp recordings in excised patches at defined [Ca2+]i and in whole-cell configuration while interfering with [Ca2+]i. Surprisingly BKCa channels of IHCs could be activated at negative membrane potentials of around −40 mV without any increase in [Ca2+]i. Activation at negative potentials was observed both in excised patches and in whole-cell conditions, pointing towards a direct modification of the channel protein.
Methods
Tissue preparation
Apical cochlear turns of rats and mice (19–28 days after birth) were prepared as previously described (Oliver et al. 2000). Briefly, animals were anaesthetized with isoflurane, killed by decapitation, and the cochleae were dissected. After removal of the cochlear bone, the apical cochlear turn was separated from the modiolus. Stria vascularis and tectorial membrane were stripped off. This whole-mount preparation was placed into an experimental chamber continuously perfused with standard extracellular solution containing (mm): NaCl 144, KCl 5.8, CaCl2 1.3, MgCl2 0.9, Hepes 10, Na2HPO4 0.7 and glucose 5.6; pH adjusted to 7.4 with NaOH.
Most experiments were performed with IHCs from Wistar rats (Charles River Laboratories, Sulzfeld, Germany). BKα−/− mice were kindly provided by Dr P. Ruth (Department of Pharmacology and Toxicology, Tübingen, Germany; Sausbier et al. 2004). Experiments were performed on homozygous BKα−/− and BKα+/+129svj inbred littermates (Ruttiger et al. 2004). BKβ1−/− mice had a mixed 129svj/C57BL background (Brenner et al. 2000b) and were kindly provided by Dr M. Knipper (Hearing Research Center, Tübingen, Germany). 129svj inbred mice were used as a control. All animal use was performed according to institutional guidelines at the University of Freiburg.
Electrophysiological recordings
Voltage-clamp recordings were performed with an Axopatch 200B amplifier (Axon Instruments, Union City, CA, USA) at room temperature (20–24°C). Current recordings were low-pass filtered at 10 kHz and sampled at 25–100 kHz. Electrodes were pulled from quartz glass, were coated with Sylgard for excised patch experiments, and had initial resistances of 1.2–2.5 MΩ. During whole cell measurements, series resistance was typically 2–5 MΩ. Careful series resistance compensation (90–95%) was applied during all whole-cell experiments and voltages were corrected offline for errors due to residual series resistance.
Extracellular solutions were exchanged via a thin glass capillary (diameter, ∼100 μm) placed close to the IHCs. Either standard solution or one of the following modified extracellular solutions was applied. (i) For removal of all extracellular Ca2+, Ca2+ was replaced by 1.3 mm Mg2+, and 5 mm BAPTA was added. (ii) For external solution containing Sr2+, Ca2+ was replaced by 1.3 mm Sr2+. (iii) To measure currents through Cav channels, 35 mm Na+ was replaced by an equal concentration of TEA+. (iv) Complete removal of Na+ was achieved by replacement with an equal concentration of N-methyl-d-glucamine (NMDG+). Extracellular solutions usually contained 1 μm XE991 to block KCNQ-mediated potassium currents (IK,n; Oliver et al. 2003).
For most whole-cell measurements, pipettes were filled with a KCl-based intracellular solution containing (mm): KCl 135, MgCl2 3.5, EGTA 5, Hepes 5, Na2ATP 2.5 and 4-aminopyridine (4-AP) 10; pH adjusted to 7.3 with HCl. For one set of experiments, EGTA was replaced by either 0.1 or 30 mm BAPTA (either from Fluka, Seelze, Germany or Molecular Probes, Eugene, OR, USA). An appropriate amount of KCl was added to keep the osmolarity (285 mosmol l−1) constant. The effective concentration of BAPTA in these solutions was verified by titration using a Ca2+-sensitive electrode (World Precision Instruments, Berlin, Germany). Measurement of [Ca2+] in the absence of any Ca2+ buffer revealed a contamination with 3 μm Ca2+. Thus free [Ca2+]i was calculated as 0.04, 6.0 and 0.02 nm for solutions containing 5 mm EGTA, 0.1 mm BAPTA and 30 mm BAPTA, respectively (WEBMAXC v2.22; stanford.edu/~cpatton/maxc.html). To block all K+ currents for measurement of Ca2+ or Sr2+ currents, KCl was replaced by CsCl.
For outside-out patch recordings, the standard pipette solution was used for Ca2+i-free conditions. For increased [Ca2+]i, EGTA was replaced by 2 mm lower-affinity buffer di-bromo-BAPTA (KD for Ca2+, 1.8 μm at 23°C and pH 7.3; Fluka). Appropriate amounts of Ca2+ (0.706, 1.232 and 1.700 mm CaCl2) were added to yield free [Ca2+]i of 1, 3 or 10 μm[Ca2+]i, respectively; this was checked with Ca2+-sensitive electrodes and adjusted if necessary. For outside-out recordings with 1 or 10 μm intracellular Sr2+, 5 mm EDTA was used as the buffer (KD for Sr2+, 2.6 μM) and 1.355 or 3.949 mm SrCl2 was added, respectively. As KD values for Mg2+ and Sr2+ are approximately the same, Mg2+ and ATP were omitted from these pipette solutions and replaced by equal amounts of KCl. To obtain 100 μm Sr2+, SrCl2 was added without buffering.
For inside-out patches, pipettes were filled with standard extracellular solution. After excision, patches were placed in front of an array of capillaries that allowed exchange between solutions with different free [Ca2+]i. Composition of these solutions was as follows (mm): KCl 135, MgCl21, Hepes 5 and 4-AP 10; pH adjusted to 7.3 with HCl. Free [Ca2+] was buffered with 2 mm di-bromo-BAPTA to 1, 3 and 10 μm by adding 0.706, 1.232 and 1.700 mm CaCl2, respectively. For 0 [Ca2+], the solution contained 5 mm EGTA instead of di-bromo-BAPTA. Free [Ca2+] was verified with Ca2+-sensitive electrodes.
XE991 (DuPont, Wilmington, DE, USA), isradipine (kindly provided by T. Moser, Göttingen), ryanodine (Calbiochem, Bad Soden, Germany, or Tocris, Ellisville, MO, USA), cyclopiazonic acid (CPA; Calbiochem), 2,5-di-(t-butyl)-1,4-hydroquinone (BHQ; Calbiochem) and carboxy-eosine (Molecular Probes) were prepared as stock solutions in DMSO. XE991, isradipine, ryanodine, CPA and BHQ were added to the extracellular solution and carboxy-eosine to the pipette solution yielding final DMSO concentrations of 0.1%.
Giant patch recordings from Xenopus oocytes
Harvesting of Xenopus oocytes, mRNA injection and giant patch-clamp recordings were done as previously described (Oliver et al. 2000). Briefly, mRNA coding for mouse BKα (Accession No. A48206) was injected into isolated oocytes. Giant inside-out patches were excised using patch pipettes of ∼0.3 MΩ filled with (mm): NaCl 115, KCl 5, CaCl2 1 and Hepes 10 (pH 7.2). For activation curves shown in Fig. 1E, patches were perfused with intracellular solution containing 100 mm KCl, 10 mm K2-EGTA and 10 mm Hepes (pH 7.2). For experiments with varying Ca2+ buffer (0.1 versus 30 mm BAPTA), solutions applied to the patches were identical to the respective intracellular solutions used for IHCs.
Data analysis
Data analysis and fitting was performed with IgorPro (WaveMetrics, Lake Oswego, OR, USA) on a Macintosh PowerPC. All reported voltages (i.e. at steady-state current) were corrected off-line for measured liquid junction potentials and for voltage errors arising from voltage drop across the residual series resistance.
Activation curves of BKCa were determined using tail-current voltage protocols composed of brief prepulses (usually 5 ms) to minimize activation of residual slow outward currents, followed by a fixed tail potential (see Results). Tail-current amplitudes were plotted versus the corrected prepulse potential for each cell or patch; activation curves were fitted with a first-order Boltzmann function:
where Ileak is voltage-independent leak current, Imax is the amplitude of the fully activated current at the tail-current potential, V is the prepulse voltage, Vh is the voltage at half-maximal activation and α is the slope factor. Ileak was subtracted, and currents were normalized to Imax for each experiment. All data are presented as means ±s.e.m. Activation curves displayed in the figures are averaged from normalized data of n experiments and the presented fitted curves show the fits to the averaged data. Values given in the figure legends refer to fits to the averaged data whereas data given in the text represent means ±s.e.m. of the values from individual cells or patches. Horizontal error bars for voltages are smaller than the symbol size in the activation curves. Statistical significance of differences between two sets of individual measurements was assessed with the Kolmogorov-Smirnov test that does not presuppose normal distribution of the data.
Results
Hair cell IK,f is mediated by BKCa channels
Mature mammalian IHCs display two major outwardly rectifying K+ conductances that differ in their activation kinetics. A fast component, IK,f, activates with submillisecond kinetics and a slower component, IK,s, shows strongly voltage-dependent activation kinetics with time constants of several milliseconds (Kros & Crawford, 1990). A third K+ current is activated at hyperpolarized potentials and carried by KCNQ channels (Oliver et al. 2003). The currents can be separated pharmacologically and the sensitivity of the fast component to TEA, charybdotoxin and iberiotoxin strongly suggests that it is carried by BKCa channels (Kros et al. 1998; Raybould et al. 2001; Skinner et al. 2003; Marcotti et al. 2004). However, some ambiguity about the identity of IK,f remained given its apparent independence of calcium influx (Kros & Crawford, 1990; Marcotti et al. 2004).
Figure 1A and B shows whole-cell recordings obtained in IHCs from either a BKα knock-out (BKα−/−; Sausbier et al. 2004) or a littermate wild-type mouse (BKα+/+). In both experiments, slow potassium currents were blocked with 10 mm 4-AP and 1 μm XE991 (Kros & Crawford, 1990; Oliver et al. 2003; Marcotti et al. 2004). In wild-type IHCs, depolarization elicited the fast-activating, non-inactivating outward current IK,f that was absent in cells from BKα−/− mice (n = 12). This result unequivocally identified IK,f as a BKCa-mediated current. Moreover, only a small residual outward current with slow activation kinetics remained in the BKα−/− cells in the presence of 4-AP and XE991; these recordings thus confirmed that BKCa currents can be adequately isolated with these channel blockers and short depolarizing pulses.
BKCa currents displayed rapid onset at voltages positive to −60 mV with time constants between 0.7 and 0.4 ms (Fig. 1D). Inactivation of BKCa currents was not observed (Fig. 1A). However, as IHC currents are very large, especially when not separated pharmacologically, recordings are prone to series resistance-dependent voltage errors that vary with time as the currents activate (Marcotti et al. 2004). Such voltage errors may distort the apparent current kinetics. BK currents recorded in excised patches are much smaller and do not suffer from this problem. Patch recordings (Fig. 1C) showed the same kinetic properties of BK, confirming the proper isolation of BK in the whole-cell recordings, and in particular the absence of inactivation. Whole-cell steady-state activation was determined from tail currents following 5-ms steps to potentials between −104 mV and 6 mV. Fitting these activation curves with a Boltzmann function (see Methods) yielded a mean Vh value of −41.8 ± 0.3 mV (n = 16) and a slope factor of 10.3 ± 0.3 mV (Fig. 1E), similar to the values previously published for IK,f in guinea pig (Kros & Crawford, 1990) and mouse (Oliver et al. 2003; Marcotti et al. 2004). This very negative steady-state activation obtained in whole IHCs under physiological conditions was strikingly different from channel activation determined under Ca2+-free conditions in patches excised from Xenopus oocytes that expressed the α-subunit of the IHC BKCa channels (Langer et al. 2003). These recombinant BKCa channels activated with a Vh of 162.1 ± 2.4 mV and a slope factor of 21.9 ± 3.2 mV at 0 [Ca2+]i (estimated free [Ca2+]i, 0.04 nm, see Methods; n = 7). The difference in activation of about 200 mV would be consistent with the IHC BKCa being gated by both transmembrane voltage and increased [Ca2+]i (Oliver et al. 2003; Marcotti et al. 2004).
Ca2+- and voltage-dependence of BKCa channels in excised IHC patches
The contribution of both factors was investigated by probing Ca2+- and voltage-dependence of BKCa gating in excised patches from IHCs, allowing for a precise control of [Ca2+]i. Patches from rat IHCs typically harboured many BKCa channels yielding large ensemble K+ currents (Fig. 2A). In both outside-out (Fig. 2B) and inside-out configurations (Fig. 2C), voltage-driven activation of currents was dependent on [Ca2+]i with increasing [Ca2+]i leading to a leftward shift of the activation curve. At 0 [Ca2+]i, Vh was 10.8 ± 5.9 mV and −15.2 ± 3.0 mV for inside-out (n = 11) and outside-out (n = 5) patches, respectively (Fig. 2D). Notably, these values for Vh at 0 [Ca2+]i were much more negative than those reported for both native and recombinant BKCa channels (e.g. Tseng-Crank et al. 1994; Xie & McCobb, 1998; Jones et al. 1999), but were still more positive than Vh values determined in whole IHCs (−41.8 mV; Fig. 1D). Thus, channel activation approached the whole-cell Vh value at micromolar levels of [Ca2+]i (∼3 μm) at the cytoplasmic face of the excised patches. The slope of voltage dependence (α) was essentially constant throughout all Ca2+ concentrations tested.
Figure 2. Ca2+- and voltage-dependence of BKCa channels in patches excised from rat IHCs.

A, BKCa currents recorded in outside-out patches from IHCs at the free [Ca2+]i indicated. Holding potential was −83 mV, currents were elicited by step depolarizations to potentials between −143 and +37 mV (10-mV increments). Tail potential was −3 mV for 0, 1 and 3 μm and −37 mV for 10 μm[Ca2+]i. Each trace is averaged from 20 repetitions. Current scale bar, 0.2 nA. B, steady-state activation curves determined by tail-current analysis from outside-out patch recordings as in A. Free [Ca2+]i in the pipette was as indicated, and data points show mean values of 5, 6, 4 and 11 patches for 0, 1, 3 and 10 μm[Ca2+]i, respectively. Continuous lines show Boltzmann fits to the averaged data. Vh and α were −14.1 mV and 18.1 mV (0 μm), −31.2 mV and 18.3 mV (1 μm), −64.1 mV and 15.9 mV (3 μm), −115.9 mV and 16.9 mV (10 μm[Ca2+]i), respectively. C, steady-state activation curves obtained from inside-out patches as in B. The indicated [Ca2+]i was applied to the cytoplasmic face of the patches via a multibarrel pipette, data points are mean values of 9, 9, 9 and 11 patches for 0, 1, 3 and 10 μm[Ca2+]i, respectively. Continuous lines are Boltzmann fits to the averaged data. Vh and α were −11.6 and 26.3 mV (0 μm), −0.5 mV and 27.0 mV (1 μm), −53.9 mV and 25.2 mV (3 μm) and −83.2 mV and 25.5 mV (10 μm[Ca2+]i), respectively. D, summary of the Vh as a function of [Ca2+]i for outside-out (○) and inside-out patches (•). E, activation time constants obtained from monoexponential fits to the rising phase of BKCa currents plotted against the membrane potential. Data are from the same outside-out patches analysed in B, symbols indicate values obtained with 0, 1, 3 and 10 μm[Ca2+]i as in B. F, τactivation of inside-out patches from C at the various [Ca2+]i indicated by the symbols used in C.
Next we examined the activation kinetics and their dependence on [Ca2+]i in excised patches (Fig. 2F). Activation time constants (τactivation) in inside-out patches were close to 1 ms at 0 [Ca2+]i, slightly slower at 1 μm, and faster at higher [Ca2+]i. Similar results were obtained in outside-out patches (Fig. 2E), but with a smaller effect of the lower Ca2+ concentrations on time constants, especially at positive potentials. Note that fast activation time constants equivalent to the rapid kinetics obtained in the whole-cell configuration (< 1 ms) required [Ca2+]i of between 3 and 10 μm.
In conclusion, both the Vh and kinetics of BKCa activation in excised IHC patches in the presence of micromolar [Ca2+]i are similar to the whole cell BKCa currents. However, the voltage dependence of the whole-cell currents is significantly steeper (∼10 mV per e-fold change in voltage) than that determined for currents in isolated patches (∼20 mV). As suggested previously, such voltage dependence may be due to a steeply voltage-dependent elevation of [Ca2+]i in the intact IHC (Oliver et al. 2003).
Gating of BKCa channels in whole IHCs is independent of local increase in [Ca2+]i
To test this interpretation, we next performed experiments that manipulated the intracellular and extracellular Ca2+ concentration ([Ca2+]ex) while measuring BKCa currents in the whole-cell configuration.
Removal of extracellular Ca2+ (buffered with 5 mm BAPTA) abolished the Ca2+ current of IHCs (Fig. 3A, inset) and thus voltage-dependent influx via Cav. However, neither voltage-dependent activation of BKCa nor the maximal BKCa current recorded at −4 mV were significantly altered (Fig. 3A). This is in agreement with previous reports (Kros & Crawford, 1990; Marcotti et al. 2004). Also, blocking the IHC Cav channels with isradipine (10 μm; Platzer et al. 2000; Koschak et al. 2001) had no effect on the activation curve and the amplitude of the BKCa currents (Fig. 3B). These results suggested, that voltage-dependent Ca2+ influx is not involved in BKCa gating in IHCs.
Figure 3. Ca2+ influx through Cav channels is not required for BKCa gating in IHCs.

A, upper panel shows steady-state activation curves of BKCa channels measured in whole-cell mode in rat IHCs before and after withdrawal of extracellular Ca2+ (5 mm extracellular BAPTA). Continuous lines show Boltzmann fits to the averaged data yielding Vh and α of −39.3 mV and 12.9 mV with 1.3 mm Ca2+, and −35.6 mV and 12.9 mV after withdrawal of extracellular Ca2+, respectively. Vh values were not significantly different (P = 0.45; Vh values of individual experiments were −39.0 ± 1.0 mV (1.3 mm Ca2+, n = 6) and −35.0 ± 2.0 mV (Ca2+-free, n = 6). Inset, the Ca2+-current in response to a step to −24 mV measured with a CsCl-based pipette solution was abolished by application of the Ca2+-free extracellular solution (dark grey, control; black, 0 [Ca2+]ex; light grey, wash). Lower panel shows that the absolute BKCa current amplitude was not changed by removal of extracellular Ca2+. Bars represent mean currents at −4 mV, where whole-cell activation was complete. B, upper panel show steady-state activation curves of BKCa channels measured in whole-cell mode in IHCs before and after block of Cav channels with isradipine (10 μm). Continuous lines show Boltzmann fits to the averaged data yielding Vh of −45.8 mV in the presence of isradipine (n = 7) and Vh of −45.1 mV in control conditions (n = 7). Slope factor α was 11.6 and 10.2 mV with and without isradipine, respectively. Vh values obtained from fits to individual experiments were not significantly different between both conditions (P = 0.20). Lower panel shows that current amplitude at −4 mV was not changed by application of isradipine. Currents in A and B (lower panels) were corrected for errors resulting from voltage drop across the residual series resistance (see Marcotti et al. 2004). C, BKCa currents measured in outside-out patches excised from IHCs were sensitive to [Sr2+]i. Steady-state activation curves at the [Sr2+]i indicated were obtained as in Fig. 2B and fitted with a Boltzmann function (continuous lines). At 0, 1, 10 and 100 μm[Sr2+]i, values for Vh and α of BKCa activation were −5.5 mV and 17.6 mV (n = 4), −10.2 mV and 23.6 mV (n = 3), −51.4 mV and 24.6 mV (n = 4) and −95.6 mV and 20.3 mV (n = 3), respectively. Activation curves obtained with 0, 1, 3 and 10 μm[Ca2+]i are shown for comparison (grey lines). Note that Sr2+ was about 10-fold less effective than Ca2+. D, inward currents through Cav channels measured in whole-cell mode with extracellular solutions containing either 1.3 mm Ca2+ (n = 5 IHCs) or 1.3 mm Sr2+ (n = 4). Currents were recorded upon depolarizing voltage steps from a holding potential of −84 mV. CsCl-based pipette solution was used and extracellular solution contained 35 mm TEA to block all K+ currents. E, whole-cell BKCa currents measured from the same IHC either at 1.3 mm[Ca2+]ex (left-hand panel) or 1.3 mm[Sr2+]ex (right-hand panel). Voltage protocol as in Fig. 1C. F, steady-state activation of BKCa channels determined from six experiments as in E. Lines are results of Boltzmann fits yielding values for Vh and α of −55.1 mV and 12.4 mV for 1.3 mm[Sr2+]ex, and −45.8 mV and 10.6 mV for 1.3 mm[Ca2+]ex, respectively.
This conclusion was further supported by experiments that replaced Ca2+ by the divalent Sr2+ which is able to substitute for Ca2+ in activating BKCa channels (Fig. 3C) and readily permeates the IHC Cav channels (Fig. 3D). Increasing intracellular Sr2+ concentration ([Sr2+]i) caused a leftward shift of the BKCa activation curve in excised outside-out patches (Fig. 3C), similar to the action of Ca2+. However, Sr2+ was about 10-times less effective than Ca2+, as 100 μm[Sr2+]i shifted the Vh to −95 mV, while a Vh of −109 mV was obtained with 10 μm[Ca2+]i (Fig. 3C). Equivalent results were obtained with recombinant BKCa channels expressed in Xenopus oocytes and investigated in giant inside-out patches (data not shown). In contrast to the large difference in activation potency, both divalent cations permeated equally well through the Cav channels of IHCs. Thus voltage-dependent inward currents recorded after blockade of all K+ currents were similar in amplitude in the presence of 1.3 mm extracellular Ca2+ or Sr2+ (Fig. 3D). These findings predicted that if BKCa gating in whole IHCs required the influx of a divalent cation, replacement of extracellular Ca2+ by Sr2+ should lead to a shift of the activation curve to more positive values due to the lower efficiency of the inflowing Sr2+; however, this was not observed. Instead, with Sr2+ as the permeating divalent cation, the BKCa activation curve appeared slightly shifted to the left (Fig. 3E and F). This shift may be explained by a surface charge effect, as a similar shift in peak current is apparent in the current–voltage relation of the Ca2+ current (Fig. 3D).
Together, these results indicate that voltage-dependent Ca2+influx is not involved in the gating of BKCa in IHCs and question whether BKCa channels in whole IHCs are sensitive to a rise in [Ca2+]i at all. As IHCs are equipped with efficient Ca2+ clearing mechanisms (Kennedy, 2002), it appeared difficult to achieve a well-defined Ca2+ elevation via the whole-cell pipette without an exceedingly high free [Ca2+] in the pipette. We therefore used carboxy-eosin (50 μm; Gatto & Milanick, 1993), a blocker of plasma membrane Ca2+ ATPases, to induce a global rise in [Ca2+]i. In the presence of carboxy-eosin and 5 mm EGTA in the patch pipette, the activation curve of BKCa currents steadily shifted to more negative potentials (average slope, 2.1 mV min−1), reaching a Vh of about −100 mV after 25 min (Fig. 4A). In contrast, Vh remained constant over 30 min in the absence of carboxy-eosin (Fig. 4A). When Ca2+ clearance via the patch pipette was lowered by a 10-fold reduction in the buffer concentration (to 0.5 mm EGTA), the shift of Vh was considerably accelerated to 4.6 mV min−1. Removal of extracellular Na+ further accelerated the shift to 9.2 mV min−1, most probably by inhibiting Na+–Ca2+ exchange activity (Fig. 4A).
Figure 4. BKCa channels in intact IHCs are sensitive to a global increase in [Ca2+]i.

A, activation of BKCa currents measured in whole-cell mode in IHCs under various conditions interfering with Ca2+ extrusion. Vh values were obtained from Boltzmann fits to the activation curves obtained as in Fig. 1C and plotted against the time after establishment of whole-cell access. Vh remained constant under control conditions (▵; n = 6 IHCs) but shifted to more negative potentials when 50 μm carboxy-eosin was included in the pipette solution ( 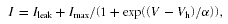 ; n1= 5). Reduction of the intracellular Ca2+ buffer EGTA to 0.5 mm (□; n = 5) and replacement of external Na+ by NMDG+ (•; n = 6) further accelerated the leftward shift. Symbols show pooled data for each condition. Cells usually deteriorated when reaching a Vh of around −100 mV, precluding the observation of the BKCa activation curve beyond this value. B, voltage protocol for measuring BKCa currents with a depolarizing prepulse to −44 mV to allow for a prolonged inward current through Cav channels prior to each 5-ms test voltage step in nominal 10-mV increments. Intervals between successive sweeps were 55 ms. C, BKCa current response to the voltage protocol in B under control conditions with 1.3 mm[Ca2+]ex. D, BKCa current response from the same cell as in C after replacement of Ca2+ by Sr2+. Note that the amplitude of the BKCa current increased during the prepulse with each voltage step. Consequently, currents during the short test voltage step were larger and were activated already at more negative potentials. The first sweep is shown in light grey; subsequent sweeps are shown in increasingly darker grey. E, BKCa current response as in C after removal of extracellular Sr2+.
; n1= 5). Reduction of the intracellular Ca2+ buffer EGTA to 0.5 mm (□; n = 5) and replacement of external Na+ by NMDG+ (•; n = 6) further accelerated the leftward shift. Symbols show pooled data for each condition. Cells usually deteriorated when reaching a Vh of around −100 mV, precluding the observation of the BKCa activation curve beyond this value. B, voltage protocol for measuring BKCa currents with a depolarizing prepulse to −44 mV to allow for a prolonged inward current through Cav channels prior to each 5-ms test voltage step in nominal 10-mV increments. Intervals between successive sweeps were 55 ms. C, BKCa current response to the voltage protocol in B under control conditions with 1.3 mm[Ca2+]ex. D, BKCa current response from the same cell as in C after replacement of Ca2+ by Sr2+. Note that the amplitude of the BKCa current increased during the prepulse with each voltage step. Consequently, currents during the short test voltage step were larger and were activated already at more negative potentials. The first sweep is shown in light grey; subsequent sweeps are shown in increasingly darker grey. E, BKCa current response as in C after removal of extracellular Sr2+.
Ca2+ pumps are selective for Ca2+ over other divalent cations (Graf et al. 1982). We therefore investigated the behaviour of BKCa channels in response to prolonged influx of Sr2+ through the Cav channels activated repetitively by 500-ms depolarizations to −44 mV (Fig. 4B). Under these conditions, BKCa currents increased over time even at −44 mV and were fully activated at this voltage after several subsequent depolarizations (Fig. 4D). In contrast, such activation was not observed with extracellular Ca2+ (Fig. 4C) or after removal of extracellular Sr2+ (Fig. 4E) indicating that a global increase in [Sr2+]i was able to activate BKCa channels in the whole-cell configuration.
These findings show that Ca2+ influx through Cav channels does not activate BKCa channels in the intact IHC, although BKCa channels are perfectly sensitive to a global increase in cytoplasmic concentration of Ca2+ (and Sr2+). It appeared therefore most likely that Cav channels are not tightly enough colocalized with BKCa to provide a sufficiently high increase in local [Ca2+]i.
Such a local increase in [Ca2+]i may, however, be provided by Ca2+ released from internal stores and thus explain the difference in activation range observed between BKCa channels in whole-cell and excised patch recordings at 0 [Ca2+]i (Marcotti et al. 2004). To test this possibility, the endoplasmic reticulum Ca2+ pump (SERCA) was blocked with a saturating concentration of the inhibitor cyclopiazonic acid (CPA; 50 μm). Inhibition of SERCA will deplete intracellular Ca2+ stores (Mason et al. 1991) and inhibit processes that depend on Ca2+ release. In the presence of CPA the IHCs were repetitively depolarized to accelerate any putative Ca2+ efflux from stores and induce their depletion. However as shown in Fig. 5A and B neither the Vh and slope of the BKCa activation curve, nor the current amplitude were altered significantly even after prolonged application of CPA. The values for Vh before and during application of CPA (10 min) were −38.6 ± 1.6 mV (n = 3) and −41.7 ± 1.1 mV (n = 7), respectively (P = 0.31). In addition, BHQ (100 μm; Mason et al. 1991), a chemically unrelated SERCA inhibitor, also failed to change the BKCa activation properties. After BHQ application (3–10 min) Vh was −41.1 ± 1.7 mV in five IHCs, which had a Vh of −37.7 ± 2.8 mV prior to BHQ application. Furthermore, the involvement of Ca2+ release was probed by blocking putative release channels. In immature IHCs, Ca2+ release via ryanodine receptors (RyRs) is involved in amplifying presynaptic Ca2+ signals (Kennedy & Meech, 2002). We therefore applied ryanodine (50 μm) which is known to block RyRs at micromolar concentrations (Meissner, 1986). Activation properties of BKCa were not changed significantly by ryanodine (5 min). Steady-state activation curves obtained with ryanodine and under control conditions yielded values for Vh and slope factor of −43.8 ± 2.8 mV and 10.3 ± 0.5 mV (ryanodine; n = 5) and −40.4 ± 1.4 mV and 10.0 ± 0.8 mV (control; n = 5), respectively. To ensure that intracellular concentrations were sufficiently high to block release channels we additionally applied ryanodine (30 μm; see Marcotti et al. 2004) through the patch pipette and measured BK activation curves 5–10 min after establishment of the whole-cell access. On average, Vh was slightly more positive in the presence of ryanodine (Vh, −33.3 ± 3.0 mV; α, 12.1 ± 0.6 mV; n = 6 IHCs) when compared to control cells from the same preparations without ryanodine (Vh, −38.9 ± 2.2 mV; α, 12.8 ± 0.5 mV; n = 6 IHCs); however, this difference was not statistically significant (P = 0.45). Current amplitudes with ryanodine (9.3 ± 1.8 nA at −14 mV) and in control IHCs (12.5 ± 1.2 nA) were also not significantly different (P = 0.45).
Figure 5. BKCa activation in IHCs does not require Ca2+ release from internal stores.

A, activation curves measured before (n = 3) and after prolonged application (10 min) of the SERCA blocker CPA to IHCs (50 μm; n = 7). Boltzmann fits (continuous lines) to the averaged data (mean values of three experiments) yielded values for Vh and α of −34.4 mV and 11.7 mV for controls, and −38.2 mV and 12.1 mV with CPA, respectively. During application of CPA, the cell was depolarized repetitively to facilitate store depletion. B, absolute BKCa current amplitudes measured at +6 mV from the same cells shown in A. The difference between control and CPA was not significant (P = 0.61). Correction for series resistance error as in Fig. 3A and B. C, activation curves measured with pipette solutions containing either 0.1 (n = 5 IHCs) or 30 mm (n = 6) fast Ca2+ chelator BAPTA. Continuous lines indicate Boltzmann fits to the averaged data, yielding values for Vh and α of −49.1 and 11.0 mV for 0.1 mm BAPTA, and −26.5 mV and 12.3 mV for 30 mm BAPTA. Grey line represents the fit to control data (5 mm EGTA) from Fig. 1D. D, inset shows that intracellular BAPTA (0.1 and 30 mm) directly shifted BKCa activation curves measured in inside-out patches at 0 [Ca2+]i to more negative or positive values, respectively, compared to 5 mm EGTA. Solutions applied to the cytoplasmic side of the patch were identical to the intracellular solutions used in C. Whole-cell activation curves for 0.1 (▪) and 30 mm BAPTA (□) from B are shown with this direct effect of BAPTA subtracted. Boltzmann fits (continuous lines) to the corrected data yielded Vh and α values of −45.6 mV and 11.0 mV, and −36.8 mV and 12.3 mV for 0.1 and 30 mm BAPTA, respectively. Grey line represents activation curve at 5 mm EGTA as in C.
We next explored the involvement of local [Ca2+]i in BKCa gating by using the fast Ca2+ buffer BAPTA at high (30 mm) and low concentrations (0.1 mm) in the whole-cell pipette. Free [Ca2+] was subnanomolar in these pipette solutions, as no Ca2+ was added. BKCa activation curves were recorded only after complete equilibration of BAPTA with the cytoplasm; that is, more than 3 min after establishing whole-cell access. The effective buffer concentrations of the intracellular solutions were verified by titration using a Ca2+-sensitive electrode and were found to match the respective BAPTA concentration within 10%. As shown in Fig. 5C, the Vh values obtained at 0.1 and 30 mm BAPTA were −47.5 ± 3.4 mV (n = 5) and −27.0 ± 1.4 mV (n = 6), respectively, which were significantly different from each other (P = 0.009), and Vh at 30 mm BAPTA significantly differed from measurements with 5 mm EGTA (P = 0.0003). In contrast to the Vh values, the slope factors of the activation curves recorded in 0.1 and 30 mm BAPTA were not significantly different from each other or from the measurement in the presence of EGTA (α, 10.7 ± 0.6 mV and 12.1 ± 0.5 mV for 0.1 and 30 mm BAPTA, respectively; P = 0.52).
As increasing BAPTA to 30 mm markedly changes the bulk ionic properties of the intracellular milieu (most monovalent anions are replaced by a much lower concentration of tetravalent ions), the BAPTA-containing solutions (without any added Ca2+) were tested for direct effects on BKCa channels in inside-out patches excised either from IHCs or BKCa-expressing oocytes. These solutions contained less than 6 nm free [Ca2+]i (see Methods), a concentration that does not activate BK channels even at very positive potentials (Cui et al. 1997). Thus, any change in BK activation observed with these solutions cannot be mediated by Ca2+. As shown in Fig. 5D (inset), BKCa activation was shifted towards depolarized potentials by 10.3 ± 1.2 mV (n = 7 IHC patches) when the intracellular solution with 5 mm EGTA was changed to the solution containing 30 mm BAPTA. At 0.1 mm BAPTA, Vh was slightly shifted in the opposite, negative, direction by −3.6 ± 1.7 mV (Fig. 5D, inset). The slope factor was not affected. Similarly, a shift in Vh of 16.7 ± 4.0 mV (n = 6) was determined for heterologously expressed BKCa upon changing from 0.1 to 30 mm BAPTA. Thus, the concentration and species of the Ca2+ buffer influenced BKCa channel activation, but not by affecting the [Ca2+]i sensed by the channels. Subtracting this ‘non-specific’ (not Ca2+-mediated) effect of BAPTA from the Vh values determined in whole IHCs with 0.1 and 30 mm BAPTA, results in corrected Vh values of −44.0 and −37.3 mV, respectively. The corrected activation curves were close to the data obtained with the slow buffer EGTA in the pipette (Fig. 5D); the corrected Vh values for 0.1 and 30 mm BAPTA were not significantly different from the data obtained with EGTA (P = 0.170 and 0.167, respectively). Thus, the rightward shift observed upon application of BAPTA to IHCs was predominantly due to a direct effect of the buffer molecule on the BKCa properties rather than resulting from the buffering of local [Ca2+]i in the vicinity of the channel.
Together, these experiments indicated that activation of IHC BKCa channels in the voltage range around −40 mV is not due to a local rise of [Ca2+]i fuelled either by release of Ca2+ from internal stores or by Ca2+ influx through Cav channels. Instead, the negative activation range of IK,f appears to be an intrinsic property of the channel protein.
Gating of BKCa channels in IHCs of KCNMB1 knock-out mice
Voltage dependence of BKCa channels is modified by auxiliary β-subunits, the KCNMB1–4 proteins (McManus et al. 1995; Brenner et al. 2000a). BKβ1 (KCNMB1) is coexpressed with the pore-forming BKα subunit in mature IHCs (Langer et al. 2003). To assess the role of BKβ1, BKCa currents were measured in IHCs from mice with a targeted deletion of KCNMB1 (BKβ1−/−; Brenner et al. 2000b).
The voltage dependence of whole-cell BKCa currents recorded from BKβ1−/− IHCs (Vh, −45.0 ± 1.4 mV), was identical to currents from wild-type control mice (Vh, −45.7 ± 2.4 mV; Fig. 6A). Likewise, neither current amplitudes (Fig. 6B) nor activation time constants of BKCa (Fig. 6C) were changed substantially by ablation of BKβ1. In outside-out patches, BKCa currents from BKβ1−/− animals were indistinguishable from controls at 0 [Ca2+]i (Vh, −0.7 ± 6.4 and 0.9 ± 10.4 mV, respectively; Fig. 6D). At 10 μm[Ca2+]i, BKCa channels from BKβ1−/− mice appeared to activate at potentials slightly more positive than those from control animals (Fig. 6D), although the difference was not statistically significant (P = 0.24). The activation kinetics of BKCa in patches were also not substantially different between wild-type and BKβ1−/− when measured at subnanomolar levels of [Ca2+]i or with 10 μm[Ca2+]i (Fig. 6E).
Figure 6. BKCa gating in IHCs is not affected by KCNMB1.

A, steady-state BKCa activation curves determined from whole-cell recordings in IHCs of wild-type (○; n = 7) and BKβ1−/− mice (▴; n = 9). Lines represent results of Boltzmann fits to the averaged data yielding values for Vh and α of −45.5 mV and 9.7 mV for BKβ1−/− IHCs and −46.0 mV and 9.7 mV for wild-type IHCs, respectively. B, BKCa current amplitudes measured at −4 mV from the same IHCs as in A (symbols as in A). Currents were corrected for the voltage drop across the residual series resistance as in Fig. 3A and B. C, the time constants of activation, obtained from monoexponential fits to the rising phase of the same current data from A and B are very similar in BKβ1−/− and control IHCs (symbols as in A). D, BKCa activation measured in outside-out patches from wild-type and BKβ1−/− mice at 0 and 10 μm free [Ca2+]i as in Fig. 2. Data are mean values of five and 14 patches for wild-type and eight and 16 patches for the BKβ1−/− IHCs. Lines represent results of a Boltzmann fit to the averaged data yielding values for Vh and α of −0.3 mV and 22.54 mV (wild-type at 0 Ca2+), 0.6 mV and 23.0 mV (BKβ1−/− at 0 Ca2+), −90.8 mV and 29.5 mV (wild-type at 10 μm 0 Ca2+) and −70.7 mV and 27.2 mV (BKβ1−/− at 10 μm Ca2+), respectively. E, activation time constants from the BKCa currents in D were essentially equal in BKβ1−/− and control patches, with only a slightly slower activation in BKβ1−/− at 0 Ca2+. Time constants were obtained from monoexponential fits to the onset of the currents.
Unaltered properties of BKCa upon ablation of KCNMB1 obtained in excised patches and in the intact cell thus show that the negative activation range of BKCa currents in the IHCs is not due to association of BKα with the BKβ1 protein.
Discussion
Identity of IK,f and properties in excised patches
Several lines of evidence previously indicated that IK,f, the fast activating K+ conductance in IHCs, is carried by BKCa channels. This included sensitivity to the specific BKCa channel blockers iberiotoxin and charybdotoxin (Kros et al. 1998; Marcotti et al. 2004; Pyott et al. 2004; Hafidi et al. 2005), and the [Ca2+]i dependence of its activation observed in excised inside-out patches (Oliver et al. 2003). Here we show that targeted deletion of the pore-forming BKCaα-subunit abolished IK,f in IHCs, which unequivocally identifies IK,f as a BKCa current. The minor residual current component observed in IHCs from BKα−/− mice demonstrates that IK,f can be appropriately isolated with 4-AP in the patch pipette to block IK,s (Kros & Crawford, 1990) and XE991 to block the KCNQ-type IK,n (Oliver et al. 2003). Notably, this finding supports the non-inactivating nature of BKCa in IHCs as suggested previously (Oliver et al. 2003; Marcotti et al. 2004) but contrasts with a report showing inactivation of IK,f currents obtained by subtraction of pharmacologically isolated current components (Pyott et al. 2004). The absence of inactivation cannot be attributed to the 4-AP in our recording solution, because 4-AP does not affect the inactivation of BK channels (Armstrong & Roberts, 2001). Marcotti et al. (2004) argued that an apparent inactivation of IK,f may result from time-varying voltage errors that easily occur with the large IHC currents in the presence of even a moderate series resistance. The present recordings in isolated patches in the absence of series resistance problems and at very positive potentials (Fig. 1C; see Oliver et al. 2003) support this view.
Recordings in patches from rat (and mouse) IHCs revealed the signature behaviours of BKCa channels: (i) channel activation with a relatively shallow voltage dependence (α, ∼20 mV); and (ii) the shift of the steady-state activation curve to hyperpolarized potentials with increasing [Ca2+]i. The most noteworthy observation obtained with patches is the very negative activation with Vh values of −15 mV and 11 mV even in the absence of free [Ca2+]i (i.e. subnanomolar levels) in outside-out and inside-out patches, respectively. This negative activation range was independent of Mg2+ which may also shift the voltage dependence of BKCa (Shi & Cui, 2001), as shown in Fig. 3C with EDTA-buffered intracellular solution. To our knowledge this is the most negative Ca2+-independent activation voltage range described so far for BKCa currents from any type of cell (see Adelman et al. 1992; Tseng-Crank et al. 1994; McManus et al. 1995; Cui et al. 1997; Vergara et al. 1998; Jones et al. 1999; Brenner et al. 2000a).
A role for Ca2+ for activation of BKCa in the intact cell?
BKCa currents recorded from intact IHCs differ from patch currents at 0 [Ca2+]i by their more negative range of activation (Vh, ∼−42 mV), faster kinetics (τactivation, 0.65 ms at room temperature; Fig. 1C; Marcotti et al. 2004), and steeper slope of the activation curve (α, ∼10 mV). These features would be most easily explained by an increase in [Ca2+]i. In particular, the steep slope factor suggested a voltage-dependent rise in [Ca2+]i between −70 and −20 mV (Oliver et al. 2003) compatible with the Cav1.3 channels of IHCs (Platzer et al. 2000) that activate at exceptionally low voltages, similar to the activation range of IK,f (Xu & Lipscombe, 2001). In fact, this led us previously to suggest that Ca2+ influx via Cav1.3 may provide the increased [Ca2+]i that could result in the particular activation properties of BKCa currents in IHCs (Oliver et al. 2003). The finding that targeted deletion of Cav1.3 abolished IK,f (Brandt et al. 2003), further supported this view.
However, the clear-cut finding that inhibition of Ca2+-influx – either by removal/substitution of external Ca2+ or by blocking Cav channels – did not result in any alteration of the activation of BKCa channels, excludes this interpretation.
Our present data confirm previous reports that IHC BKCa channels are insensitive to changes in [Ca2+]ex (Kros & Crawford, 1990; Marcotti et al. 2004). Moreover, we show that this does not indicate an intrinsic insensitivity for [Ca2+]i in the intact IHC, as BKCa channels were effectively activated by an elevation of global [Ca2+]i or [Sr2+]i when extrusion of the divalent cation was blocked (Fig. 4). The lack of direct impact of Ca2+ influx strongly suggests that spatial separation of Cav and BKCa channels precludes functional coupling. This is consistent with the available data on localization of both channel types. Recent antibody staining showed that BKCa is predominantly located in large clusters along the apical segment of the lateral membrane of IHCs (Pyott et al. 2004; Ruttiger et al. 2004; Hafidi et al. 2005). Though clear immunohistochemical data on the subcellular distribution of Cav1.3 in mature IHCs are not yet available, these channels should be located at the basal presynaptic pole of the cells. Synaptic release at these sites depends on Ca2+ influx via Cav1.3 (Moser & Beutner, 2000; Brandt et al. 2003) and depolarization increases [Ca2+]i close to the basal membrane in neonatal IHCs (Kennedy & Meech, 2002).
Alternatively, voltage dependence and onset kinetics of BKCa channels in IHCs may be set by Ca2+ released from internal stores (Marcotti et al. 2004). To address this issue we blocked Ca2+ release and Ca2+ reuptake to deplete stores and used high concentrations of the fast Ca2+ buffer BAPTA to diminish putative high local [Ca2+]i. All of these manipulations failed to significantly affect the amplitude or gating properties of BKCa currents (Fig. 5).
Some of these data stand in contrast to previous data (Marcotti et al. 2004) showing that manoeuvres thought to interfere with release of Ca2+ from internal stores shifted the activation curve to more positive potentials in mouse IHCs. A shift of the activation curve reported by Marcotti et al. (2004) for high concentrations of BAPTA was confirmed (Fig. 5), but demonstrated to result essentially from a Ca2+-independent effect of the buffer molecule. Thus, application onto excised patches from either IHCs or Xenopus oocytes showed that increasing BAPTA concentration directly shifted BKCa activation by ∼10–15 mV in the absence of Ca2+. Although a slight shift in Vh with intracellular ryanodine was in the same (depolarizing) direction as the shift reported by Marcotti et al. (2004), this effect was not significant. Extracellular application of ryanodine also did not produce a significant shift of the activation curves in our experiments. It is important to note that the shifts with store release blockers reported by Marcotti et al. (2004) were only moderate, yielding a Vh value not exceeding about −25 mV. Such an activation voltage is still more negative than observed in excised patches (Fig. 2) and is far more negative than the activation range of ‘conventional’ BK channels without elevated Ca2+ (e.g. Fig. 1E). Thus a contribution of Ca2+ released from internal stores can at most explain a minor part of the negative activation range, and a distinct mechanism must be present. This conclusion is fully consistent with the present data obtained from excised patches, a condition that excludes the contribution of store release and showed Ca2+-independent activation well below 0 mV (Fig. 2).
Moreover, it appears unlikely that store-released Ca2+ can account for the fast and negative BKCa activation in IHCs for the following reasons. First, if IK,f properties depend on Ca2+ release, this release must be strongly voltage dependent to match the slope factor of ∼10 mV that exceeds the value in patches by about 2-fold. Accordingly, the stores would have to be operated by a voltage sensor in the plasma membrane, similar to the excitation–contraction coupling in skeletal muscle, where Cav1.1 channels physically interact with and gate sarcoplasmic reticulum ryanodine receptors (RyRs) (Lamb, 2000). However, all Cav channels in IHCs seem to be located distantly from BKCa. Second, manipulations that interfere with a Ca2+ store release should decrease the voltage dependence of BKCa. Yet, the high slope factor is remarkably stable under all whole-cell conditions (Marcotti et al. 2004). Third, it is unlikely that the cascade of voltage sensor/RyR, release of Ca2+ and BKCa opening occurs in the submillisecond time range. Even in skeletal muscle, the delay between depolarization and Ca2+ release is reported to be of the order of a millisecond (Kim & Vergara, 1998), in contrast to the submillisecond kinetics of IHC BKCa.
Possible mechanisms that control BKCa gating phenotype
The apparent independence of BKCa channel activation on local [Ca2+]i is consistent with a constitutive modification of the BKCa channel protein that endows the high voltage dependence, negative activation range and rapid kinetics of channel gating. This conclusion is supported by the finding of very negative activation of BKCa in patches excised from IHCs at precisely defined [Ca2+]i.
Such a modification may either occur pre/post-translationally or result from association with an auxiliary protein. Indeed, BKCa channel activation is reported to be modulated by a set of different mechanisms, including alternative splicing (Adelman et al. 1992; Tseng-Crank et al. 1994; Xie & McCobb, 1998), association with β-subunits (McManus et al. 1995; Brenner et al. 2000a) and protein phosphorylation (Reinhart et al. 1991; Schubert & Nelson, 2001). IHCs predominantly express minimal mRNA splice variants with conventional gating properties (Langer et al. 2003), and, to a lesser extent, a splice variant with a moderately increased Ca2+ sensitivity that, when expressed heterologously, still activates at markedly more positive potentials than IHC BKCa (Xie & McCobb, 1998; Ha et al. 2000). The known accessory β-subunits are unlikely to contribute to BKCa gating in IHCs. The BKβ1 knock-out failed to alter BKCa gating, a finding consistent with the lack of an obvious auditory phenotype in BKβ1−/− mice (Ruttiger et al. 2004). BKβ2 and β4, which may be expressed transiently in the immature organ of Corti (Langer et al. 2003), either endow channels with inactivation (β2; Wallner et al. 1999) or slow the kinetics (β1,2 and 4; Brenner et al. 2000a), properties that contrast with the observed characteristics of BKCa gating in IHCs. Expression of β3 in the cochlea has not been examined, but coexpression with β3 has little effect on kinetics and voltage dependence of BKCa gating (Brenner et al. 2000a), arguing against a role of β3 for the unusual behaviour of BKCa in IHCs. Activation of BKCa channels may be affected by the redox potential (DiChiara & Reinhart, 1997); however, our preliminary experiments with redox reagents (not shown) indicate that they have no effect on BKCa channels in excised patches from IHCs.
The different activation properties seen in whole-cell versus excised patch recordings suggest that the modulation of BKCa properties is at least partially reversible. Such dynamic behaviour may also underlie the difference between inside-out and outside-out patches and a substantially more negative Vh value in inside-out patches compared to similar data from mouse IHCs (Oliver et al. 2003). BKCa channel activation in outside-out patches more closely resembled the whole-cell situation, suggesting that a post-translational ‘factor’, perhaps a protein phosphorylation event that may confer rapid kinetics and hyperpolarized activation voltage onto the IHC BKCa channels, is lost in the inside-out configuration, but is better maintained in outside-out patches. In any case, the molecular nature of such modification, or the identity of a potentially BKα-associated auxiliary protein, remains to be elucidated.
In conclusion, we have shown that in IHCs BKCa channels are able to rapidly gate at negative membrane potentials without the requirement of an increase in [Ca2+]i, as necessary for BKCa function in other native cells. Instead, BKCa channels in IHCs effectively operate as purely voltage-gated outward rectifiers, essentially equivalent to Kv-type K+ channels (e.g. Kv3.1, Kv1.1; Coetzee et al. 1999; Rudy et al. 1999), but with considerably faster activation and deactivation kinetics. Our conclusion challenges the concept that BKCa activation at physiological potentials generally requires micromolar [Ca2+]i. It will be interesting to see whether this mode of operation is also realized in other tissues, in particular neurones, where BKCa is involved in the regulation of firing patterns (e.g. Sausbier et al. 2004). It is important to note that in excised patches from IHCs the voltage dependence was shifted considerably to more depolarized potentials compared to the intact cell. Thus, recordings of BKCa currents in isolated patches do not necessarily faithfully report the voltage and Ca2+ dependence of the channels under physiological (whole-cell) conditions, and a Kv-like behaviour might easily be missed.
Acknowledgments
We are indebted to Drs Peter Ruth and Marlies Knipper (University of Tübingen) for providing the knock-out animals of BKα and BKβ and to Drs P. Jonas and J. P. Adelman for reading the manuscript. This work was supported by a grant from the Deutsche Forschungsgemeinschaft to B.F. (Fa 332/5-1) and the Graduiertenkolleg GRK 843.
References
- Adelman JP, Shen KZ, Kavanaugh MP, Warren RA, Wu YN, Lagrutta A, Bond CT, North RA. Calcium-activated potassium channels expressed from cloned complementary DNAs. Neuron. 1992;9:209–216. doi: 10.1016/0896-6273(92)90160-f. [DOI] [PubMed] [Google Scholar]
- Armstrong CE, Roberts WM. Rapidly inactivating and non-inactivating calcium-activated potassium currents in frog saccular hair cells. J Physiol. 2001;536:49–65. doi: 10.1111/j.1469-7793.2001.00049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Art JJ, Fettiplace R. Variation of membrane properties in hair cells isolated from the turtle cochlea. J Physiol. 1987;385:207–242. doi: 10.1113/jphysiol.1987.sp016492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Art JJ, Fettiplace R, Wu YC. The effects of low calcium on the voltage-dependent conductances involved in tuning of turtle hair cells. J Physiol. 1993;470:109–126. doi: 10.1113/jphysiol.1993.sp019850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt A, Striessnig J, Moser T. CaV1.3 channels are essential for development and presynaptic activity of cochlear inner hair cells. J Neurosci. 2003;23:10832–10840. doi: 10.1523/JNEUROSCI.23-34-10832.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner R, Jegla TJ, Wickenden A, Liu Y, Aldrich RW. Cloning and functional characterization of novel large conductance calcium-activated potassium channel beta subunits, hKCNMB3 and hKCNMB4. J Biol Chem. 2000a;275:6453–6461. doi: 10.1074/jbc.275.9.6453. [DOI] [PubMed] [Google Scholar]
- Brenner R, Perez GJ, Bonev AD, Eckman DM, Kosek JC, Wiler SW, Patterson AJ, Nelson MT, Aldrich RW. Vasoregulation by the beta1 subunit of the calcium-activated potassium channel. Nature. 2000b;407:870–876. doi: 10.1038/35038011. [DOI] [PubMed] [Google Scholar]
- Coetzee WA, Amarillo Y, Chiu J, Chow A, Lau D, McCormack T, et al. Molecular diversity of K+ channels. Ann N Y Acad Sci. 1999;868:233–285. doi: 10.1111/j.1749-6632.1999.tb11293.x. [DOI] [PubMed] [Google Scholar]
- Cui J, Cox DH, Aldrich RW. Intrinsic voltage dependence and Ca2+ regulation of mslo large conductance Ca-activated K+ channels. J Gen Physiol. 1997;109:647–673. doi: 10.1085/jgp.109.5.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiChiara TJ, Reinhart PH. Redox modulation of hslo Ca2+-activated K+ channels. J Neurosci. 1997;17:4942–4955. doi: 10.1523/JNEUROSCI.17-13-04942.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs PA, Nagai T, Evans MG. Electrical tuning in hair cells isolated from the chick cochlea. J Neurosci. 1988;8:2460–2467. doi: 10.1523/JNEUROSCI.08-07-02460.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatto C, Milanick MA. Inhibition of the red blood cell calcium pump by eosin and other fluorescein analogues. Am J Physiol. 1993;264:C1577–C1586. doi: 10.1152/ajpcell.1993.264.6.C1577. [DOI] [PubMed] [Google Scholar]
- Graf E, Verma AK, Gorski JP, Lopaschuk G, Niggli V, Zurini M, Carafoli E, Penniston JT. Molecular properties of calcium-pumping ATPase from human erythrocytes. Biochemistry. 1982;21:4511–4516. doi: 10.1021/bi00261a049. [DOI] [PubMed] [Google Scholar]
- Ha TS, Jeong SY, Cho SW, Jeon H, Roh GS, Choi WS, Park CS. Functional characteristics of two BKCa channel variants differentially expressed in rat brain tissues. Eur J Biochem. 2000;267:910–918. doi: 10.1046/j.1432-1327.2000.01076.x. [DOI] [PubMed] [Google Scholar]
- Hafidi A, Beurg M, Dulon D. Localization and developmental expression of BK channels in mammalian cochlear hair cells. Neuroscience. 2005;130:475–484. doi: 10.1016/j.neuroscience.2004.09.038. [DOI] [PubMed] [Google Scholar]
- Horrigan FT, Aldrich RW. Coupling between voltage sensor activation, Ca2+ binding and channel opening in large conductance (BK) potassium channels. J Gen Physiol. 2002;120:267–305. doi: 10.1085/jgp.20028605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudspeth AJ, Lewis RS. Kinetic analysis of voltage- and ion-dependent conductances in saccular hair cells of the bull-frog, Rana catesbeiana. J Physiol. 1988a;400:237–274. doi: 10.1113/jphysiol.1988.sp017119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudspeth AJ, Lewis RS. A model for electrical resonance and frequency tuning in saccular hair cells of the bull-frog, Rana catesbeiana. J Physiol. 1988b;400:275–297. doi: 10.1113/jphysiol.1988.sp017120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EM, Gray-Keller M, Fettiplace R. The role of Ca2+-activated K+ channel spliced variants in the tonotopic organization of the turtle cochlea. J Physiol. 1999;518:653–665. doi: 10.1111/j.1469-7793.1999.0653p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy HJ. Intracellular calcium regulation in inner hair cells from neonatal mice. Cell Calcium. 2002;31:127–136. doi: 10.1054/ceca.2001.0267. [DOI] [PubMed] [Google Scholar]
- Kennedy HJ, Meech RW. Fast Ca2+ signals at mouse inner hair cell synapse: a role for Ca2+-induced Ca2+ release. J Physiol. 2002;539:15–23. doi: 10.1113/jphysiol.2001.013171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim AM, Vergara JL. Fast voltage gating of Ca2+ release in frog skeletal muscle revealed by supercharging pulses. J Physiol. 1998;511:509–518. doi: 10.1111/j.1469-7793.1998.509bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koschak A, Reimer D, Huber I, Grabner M, Glossmann H, Engel J, Striessnig J. alpha 1D (Cav1.3) subunits can form L-type Ca2+ channels activating at negative voltages. J Biol Chem. 2001;276:22100–22106. doi: 10.1074/jbc.M101469200. [DOI] [PubMed] [Google Scholar]
- Kros CJ, Crawford AC. Potassium currents in inner hair cells isolated from the guinea-pig cochlea. J Physiol. 1990;421:263–291. doi: 10.1113/jphysiol.1990.sp017944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kros CJ, Ruppersberg JP, Rusch A. Expression of a potassium current in inner hair cells during development of hearing in mice. Nature. 1998;394:281–284. doi: 10.1038/28401. [DOI] [PubMed] [Google Scholar]
- Lamb GD. Excitation-contraction coupling in skeletal muscle: comparisons with cardiac muscle. Clin Exp Pharmacol Physiol. 2000;27:216–224. doi: 10.1046/j.1440-1681.2000.03224.x. [DOI] [PubMed] [Google Scholar]
- Langer P, Grunder S, Rusch A. Expression of Ca2+-activated BK channel mRNA and its splice variants in the rat cochlea. J Comp Neurol. 2003;455:198–209. doi: 10.1002/cne.10471. [DOI] [PubMed] [Google Scholar]
- Latorre R, Vergara C, Hidalgo C. Reconstitution in planar lipid bilayers of a Ca2+-dependent K+ channel from transverse tubule membranes isolated from rabbit skeletal muscle. Proc Natl Acad Sci U S A. 1982;79:805–809. doi: 10.1073/pnas.79.3.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus OB, Helms LM, Pallanck L, Ganetzky B, Swanson R, Leonard RJ. Functional role of the beta subunit of high conductance calcium-activated potassium channels. Neuron. 1995;14:645–650. doi: 10.1016/0896-6273(95)90321-6. [DOI] [PubMed] [Google Scholar]
- Marcotti W, Johnson SL, Kros CJ. Effects of intracellular stores and extracellular Ca2+ on Ca2+-activated K+ currents in mature mouse inner hair cells. J Physiol. 2004;557:613–633. doi: 10.1113/jphysiol.2003.060137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty A. Ca-dependent K channels with large unitary conductance in chromaffin cell membranes. Nature. 1981;291:497–500. doi: 10.1038/291497a0. [DOI] [PubMed] [Google Scholar]
- Mason MJ, Garcia-Rodriguez C, Grinstein S. Coupling between intracellular Ca2+ stores and the Ca2+ permeability of the plasma membrane. Comparison of the effects of thapsigargin, 2,5-di-(tert-butyl)-1,4-hydroquinone, and cyclopiazonic acid in rat thymic lymphocytes. J Biol Chem. 1991;266:20856–20862. [PubMed] [Google Scholar]
- Meissner G. Ryanodine activation and inhibition of the Ca2+ release channel of sarcoplasmic reticulum. J Biol Chem. 1986;261:6300–6306. [PubMed] [Google Scholar]
- Moser T, Beutner D. Kinetics of exocytosis and endocytosis at the cochlear inner hair cell afferent synapse of the mouse. Proc Natl Acad Sci U S A. 2000;97:883–888. doi: 10.1073/pnas.97.2.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver D, Klocker N, Schuck J, Baukrowitz T, Ruppersberg JP, Fakler B. Gating of Ca2+-activated K+ channels controls fast inhibitory synaptic transmission at auditory outer hair cells. Neuron. 2000;26:595–601. doi: 10.1016/s0896-6273(00)81197-6. [DOI] [PubMed] [Google Scholar]
- Oliver D, Knipper M, Derst C, Fakler B. Resting potential and submembrane calcium concentration of inner hair cells in the isolated mouse cochlea are set by KCNQ-type potassium channels. J Neurosci. 2003;23:2141–2149. doi: 10.1523/JNEUROSCI.23-06-02141.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallotta BS, Magleby KL, Barrett JN. Single channel recordings of Ca2+-activated K+ currents in rat muscle cell culture. Nature. 1981;293:471–474. doi: 10.1038/293471a0. [DOI] [PubMed] [Google Scholar]
- Platzer J, Engel J, Schrott-Fischer A, Stephan K, Bova S, Chen H, Zheng H, Striessnig J. Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell. 2000;102:89–97. doi: 10.1016/s0092-8674(00)00013-1. [DOI] [PubMed] [Google Scholar]
- Pyott SJ, Glowatzki E, Trimmer JS, Aldrich RW. Extrasynaptic localization of inactivating calcium-activated potassium channels in mouse inner hair cells. J Neurosci. 2004;24:9469–9474. doi: 10.1523/JNEUROSCI.3162-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransom CB, Liu X, Sontheimer H. Current transients associated with BK channels in human glioma cells. J Membr Biol. 2003;193:201–213. doi: 10.1007/s00232-003-2019-7. [DOI] [PubMed] [Google Scholar]
- Raybould NP, Jagger DJ, Housley GD. Positional analysis of guinea pig inner hair cell membrane conductances: implications for regulation of the membrane filter. J Assoc Res Otolaryngol. 2001;2:362–376. doi: 10.1007/s101620010087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhart PH, Chung S, Martin BL, Brautigan DL, Levitan IB. Modulation of calcium-activated potassium channels from rat brain by protein kinase A and phosphatase 2A. J Neurosci. 1991;11:1627–1635. doi: 10.1523/JNEUROSCI.11-06-01627.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts WM, Jacobs RA, Hudspeth AJ. Colocalization of ion channels involved in frequency selectivity and synaptic transmission at presynaptic active zones of hair cells. J Neurosci. 1990;10:3664–3684. doi: 10.1523/JNEUROSCI.10-11-03664.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy B, Chow A, Lau D, Amarillo Y, Ozaita A, Saganich M, et al. Contributions of Kv3 channels to neuronal excitability. Ann N Y Acad Sci. 1999;868:304–343. doi: 10.1111/j.1749-6632.1999.tb11295.x. [DOI] [PubMed] [Google Scholar]
- Ruttiger L, Sausbier M, Zimmermann U, Winter H, Braig C, Engel J, et al. Deletion of the Ca2+-activated potassium (BK) alpha-subunit but not the BKbeta1-subunit leads to progressive hearing loss. Proc Natl Acad Sci U S A. 2004;101:12922–12927. doi: 10.1073/pnas.0402660101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaranayake H, Saunders JC, Greene MI, Navaratnam DS. Ca2+ and K+ (BK) channels in chick hair cells are clustered and colocalized with apical-basal and tonotopic gradients. J Physiol. 2004;560:13–20. doi: 10.1113/jphysiol.2004.069856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sausbier M, Hu H, Arntz C, Feil S, Kamm S, Adelsberger H, et al. Cerebellar ataxia and Purkinje cell dysfunction caused by Ca2+-activated K+ channel deficiency. Proc Natl Acad Sci U S A. 2004;101:9474–9478. doi: 10.1073/pnas.0401702101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schopperle WM, Holmqvist MH, Zhou Y, Wang J, Wang Z, Griffith LC, Keselman I, Kusinitz F, Dagan D, Levitan IB. Slob, a novel protein that interacts with the Slowpoke calcium-dependent potassium channel. Neuron. 1998;20:565–573. doi: 10.1016/s0896-6273(00)80995-2. [DOI] [PubMed] [Google Scholar]
- Schubert R, Nelson MT. Protein kinases: tuners of the BKCa channel in smooth muscle. Trends Pharmacol Sci. 2001;22:505–512. doi: 10.1016/s0165-6147(00)01775-2. [DOI] [PubMed] [Google Scholar]
- Shi J, Cui J. Intracellular Mg2+ enhances the function of BK-type Ca2+-activated K+ channels. J Gen Physiol. 2001;118:589–606. doi: 10.1085/jgp.118.5.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner LJ, Enee V, Beurg M, Jung HH, Ryan AF, Hafidi A, Aran J-M, Dulon D. Contribution of BK Ca2+-activated K+ channels to auditory neurotransmission in the guinea pig cochlea. J Neurophysiol. 2003;90:320–332. doi: 10.1152/jn.01155.2002. [DOI] [PubMed] [Google Scholar]
- Tseng-Crank J, Foster CD, Krause JD, Mertz R, Godinot N, DiChiara TJ, Reinhart PH. Cloning, expression, and distribution of functionally distinct Ca2+-activated K+ channel isoforms from human brain. Neuron. 1994;13:1315–1330. doi: 10.1016/0896-6273(94)90418-9. [DOI] [PubMed] [Google Scholar]
- Vergara C, Latorre R, Marrion NV, Adelman JP. Calcium-activated potassium channels. Curr Opin Neurobiol. 1998;8:321–329. doi: 10.1016/s0959-4388(98)80056-1. [DOI] [PubMed] [Google Scholar]
- Wallner M, Meera P, Toro L. Molecular basis of fast inactivation in voltage and Ca2+-activated K+ channels: a transmembrane beta-subunit homolog. Proc Natl Acad Sci U S A. 1999;96:4137–4142. doi: 10.1073/pnas.96.7.4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X, Hirschberg B, Smolik S, Forte M, Adelman JP. dSLo interacting protein 1, a novel protein that interacts with large-conductance calcium-activated potassium channels. J Neurosci. 1998;18:2360–2369. doi: 10.1523/JNEUROSCI.18-07-02360.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, McCobb DP. Control of alternative splicing of potassium channels by stress hormones. Science. 1998;280:443–446. doi: 10.1126/science.280.5362.443. [DOI] [PubMed] [Google Scholar]
- Xu W, Lipscombe D. Neuronal Ca(V), 1.3alpha(1), L–type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J Neurosci. 2001;21:5944–5951. doi: 10.1523/JNEUROSCI.21-16-05944.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]