

Abstract
Design of helical super secondary structural motifs is expected to provide important scaffolds to incorporate functional sites, thus allowing the engineering of novel miniproteins with function. An α,β-dehydrophenylalanine containing 21-residue apolar peptide was designed to mimic the helical hairpin motif by using a simple geometrical design strategy. The synthetic peptide folds into the desired structure as assessed crystallographically at 1.0-Å resolution. The two helices of the helical-hairpin motif, connected by a flexible (Gly)4 linker, are docked to each other by the concerted influence of weak interactions. The folding of the peptide without binary patterning of amino acids, disulfide bonds, or metal ions is a remarkable observation. The results demonstrate that preferred interactions among the hydrophobic residues selectively discriminate their putative partners in space, leading to the unique folding of the peptide, also a hallmark of the unique folding of hydrophobic core in globular proteins. We demonstrate here the engineering of molecules by using weak interactions pointing to their possible further exploitation in the de novo design of protein super secondary structural elements.
De novo protein design endeavors to understand the frustrating complexity of protein architecture and has the ambitious goal of constructing novel molecules with predetermined structure and function(s) (1–4). Most design strategies rely on information from the rich database of solved protein structures (5, 6). Further, the observed patterning of polar/nonpolar residues and their different modes of interaction with the solvent is a major consideration in achieving compaction (1–4, 7). A conceptually distinct approach is to achieve stereochemical control over the local folding of the peptide by incorporation of conformation-restricting residues. For example, α,β-dehydrophenylalanine (ΔPhe) or α-aminoisobutyric acid residues nucleate helical conformation whereas d-Pro nucleates hairpins (8–11). The success in establishing these “folding rules” has encouraged subsequent attempts to design and construct super secondary structures, in particular, the apparently simple helical hairpin (helix–turn–helix) motif. Many attempts in which designed hydrophobic helices were connected by linker sequences containing various coded/noncoded amino acids with a tendency to break continuous helix formation, e.g., ɛ-aminocaprionic acid, l-lactic acid, Gly-Pro, d-Phe-Pro, and Gly-Dpg, etc., have failed to realize the desired folded conformation (9, 12). Therefore the stabilization of the monomeric helical hairpin motif without binary patterning of polar/nonpolar residues has proved to be challenging.
Our design strategy for stabilization of the helical hairpin motif is based on a shift of paradigm from overemphasis on the linker region (9) toward the optimization of weak interactions between helices (termed long-range interactions). We have used ΔPhe, the dehydro analogue of phenylalanine, with a double bond between Cα and Cβ atoms, which makes it a special planar residue that leads peptide sequences to assume 310-helical conformations (8), as the key residue in achieving the intended design of crystallographically characterized helical hairpin motif. This is an attempt where a conformation constraining residue is used in inducing long-range interactions to achieve compaction.
Design Strategy
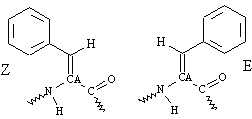
ΔPhe is an achiral planar residue (8) in which, because of the extended conjugation of the ΔPhe ring electrons with sp2-hybridized Cα and Cβ atoms, all atoms (including the hydrogens) of the residue are restricted to an approximate plane (Fig. 1A). This essentially simplifies three-dimensional complexity of the side-chain atomic positions to two dimensions. This simplified geometry and reduced rotamer complexity of ΔPhe was fully exploited in introducing long-range interactions between the two secondary structures, separated in sequence. Other properties of ΔPhe, considered as positive design elements, are its inherent preference to stabilize 310-helical structures and to adopt enantiomeric conformations (8, 13).¶ The design strategy, which incorporates ideas from the crystal structures of ΔPhe containing peptides reported from our own laboratory and by others (13–15), is geometrical and simple. Based on these ideas we speculated that in an ideal right-handed 310-helix (16) (say R) with ΔPhe at every ith and i + 3rd position (-ΔPhe1-X-X-ΔPhe4-X-X-ΔPhe7-) (Fig. 1B, marked R) all of the ΔPhe side chains protruding from the helix will stack in planes parallel to each other, with the vector perpendicular to the plane making an angle of approximately 45° to the helix axis. Inversion of the coordinates of this helix should not only result in a shape-complement left-handed helix (Fig. 1B, marked L) but also the ΔPhe side chains should appear in complementary orientation to that observed in the original helix. Further, in the 310-helical peptides, the side chains are stacked one over the other along the helical axis, creating a column of protuberant side chains at ≈120° to each other and hence forming three grooves parallel to the helix axis (Fig. 1C). The specific groove in helix R, involving the back of the ΔPhe stack with planarity around Cα of ΔPhe, provides the space for planar aromatic moiety of the another ΔPhe side chain from the shape-complement helix, which results in parallel stacking of two ΔPhe residues as shown in Fig. 1 A and B. This arrangement was expected to result in possible interaction between the two conjugated π systems (17), supported by C—H—O interactions (18, 19). This type of interdigitation may be described as a wedge into a groove arrangement as opposed to the knobs into holes in α-helices (20). Therefore, the designed model 310-helical peptide is expected to generate three such π-π stacking side-chain interactions with its shape-complement partner, forming a core with a network of weak interactions running along the helix axis (Fig. 1B). Crystal structure of a ΔPhe decapeptide¶ containing shape-complement helices (left- and right-handed) in the crystal asymmetric unit formed the basis of this geometric design strategy. Having designed two helices, which we expect to pack closely, the task left over to make a helical hairpin motif, was to connect them with a flexible linker. Model building studies with two enantiomeric helices, connected head to tail with a four-residue linker, supported the expected weak interaction-assisted association. Moreover, from the point of view of entropy, which arises because of rotamer flexibility, it is less expensive energetically to bury a residue with restricted side-chain conformation such as ΔPhe than to bury a residue with a wide variety of side-chain conformations. In the actual design of the peptide (Fig. 2) d-Ala, ΔPhe were placed at i + 1st and i + 2nd positions in the sequence (-X-X- between two ΔPhe) to stabilize the left-handed 310-helix and l-Ala, l-Leu to stabilize the right-handed helix. These two helices are connected head to tail by a highly flexible l-Ala-(Gly)4 linker, with l-Ala9 serving as a helix stop signal to the growing left-handed helix (see supplemental data for more detailed explanation and Figs. 6–9, which are published as supplemental material on the PNAS web site, www.pnas.org).
Figure 1.

Design strategy of HH21. (A) The planarity of ΔPhe (ΔF) residue and hence the possible stacking of two ΔPhe side chains belonging to two different secondary structural elements. (B) The model sequence -ΔPhe-X-X-ΔPhe-X-X-ΔPhe- and the speculated right-handed helix (marked R, colored green), a left-handed helix (marked L, colored mauve), Cβ atoms of the residues X are shown for clarity. The planar ΔPhe side chains with restricted rotamer flexibility stack one above the other in each helix, where the vector perpendicular to mean plane of all of the atoms of ΔPhe makes an angle of approximately 45° to the helix axis. The specific groove in helix R, involving the back of the ΔPhe stack acts as a host for the ΔPhe stack from the helix L, wherein the entire ith ΔPhe residue of helix R stacks on to the entire (i ± 3n, n = 0, 1, 2) ΔPhe residue of the helix L, resulting in three sets of extended phenyl embrace arrangement at the helix–helix interface. Dotted lines represent the expected C—H—O hydrogen bonds. (C) View along the helical axes, showing orientation of ΔPhe residues with respect to helical axis and their stacking. Three wedges and grooves along the helical axis can be seen (see Figs. 8 and 9 for more information).
Figure 2.

Scheme depicting the target backbone structure of the design. The model peptide -ΔPhe-X-X-ΔPhe-X-X-ΔPhe- was modified to get the desired right-handed helix and a left-handed helix. The two helices were connected by a flexible linker (Gly)4. l-Ala acts as a stop signal to the growing left-handed helix.
Experimental Section
The final designed sequence, named HH21 (Fig. 2), was synthesized by manual solid-phase peptide synthesis on Rinkamide MBHA resin by using fluorenylmethoxycarbonyl (Fmoc) chemistry. Twelve of the 21 amino acids were coupled by segment condensation of dipeptides and tripeptides, which were activated as their azalactones. Azalactone derivatives of ΔPhe peptides were prepared by dissolving Fmoc-Leu-x/Fmoc-Gly-x/Fmoc-d-Ala-ΔPhe-x (x = dlβ phenylserine) in freshly distilled acetic anhydride and adding anhydrous sodium acetate. The reaction was stirred at 25°C overnight and finally added to ice, stirred, triturated, filtered on sinter, and washed with cold water. The azalactones thus obtained were dried on blotting paper and then on P2O5 in vacuo. The purity of the azalactone peptide intermediates was checked on TLC. Fmoc-alanines (residues 9, 15, 18, and 21) and Fmoc-glycines (residues 10–13) were coupled by using carbodiimide. At the completion of assembly the amino terminal of Gly1 was acetylated by using 20% acetic anhydride in dichloromethane. HH21 was cleaved from the resin by using 95% trifluroacetic acid, 2.5% water, and 2.5% triisopropyl silane cleavage mixture. Work-up of the peptide was carried out by ether precipitation and subsequent lyophilization from diluted acetic acid. The reverse-phase-HPLC analysis (detector: 280 nm) on C18 (30% acetonitrile to 80% acetonitrile in 40 min) at a flow rate of 2.5 ml/min showed two dominant peaks (retention times: 26.6 min and 28.8 min) with mass values of 2,169 and 2,160 Da, respectively (expected 2158.5).
The HH21 (C115H121O22N22) was crystallized by controlled evaporation of the peptide solution in acetic acid at room temperature. The x-ray (wavelength 1.07 Å) data were collected at the beamline X8C, National Synchrotron Light Source, at the Brookhaven National Laboratory, New York, by using a crystal cryocooled to 100 K. The crystal to detector distance was 50 mm for high-resolution pass and 150 mm for low-resolution pass. The diffraction spots were recorded on a Quantum-4 charge-coupled device detector with 3.0° and 5.0° oscillation for each image, respectively (see Table 2, which is published as supplemental material). Data were processed by using a HKL2000 suite (21). The crystal belongs to the monoclinic space group P21 with a = 21.178, b = 13.997, c = 21.956(10) Å, β = 104.556°, Z = 2, GOF = 1.264 and dc = 1.179 gm⋅cm-3 (crystal size ≈0.20 × 0.015 × 0.010 mm.) A total of 6,557 unique reflections were used for refinement of which 5,517 reflections had |Fo|>4σ|Fo|. Structure determination and refinement was carried out by using SHELXS97 and SHELXL97 (22), respectively on |F| (residue 2) by using all of the reflections with anisotropic temperature factors for nonhydrogen atoms. During the course of refinement a few water molecules with full as well as partial occupancy were located in a difference Fourier map. Atoms having unusually high temperature factors were refined isotropically. All of the hydrogen atoms were fixed by using stereochemical criteria and were used only for structure factor calculations. The conventional R factor for reflections (5,517) with |Fo|>4σ|Fo| is 12.9%. We did not observe any significant electron density near the N terminal of the peptide corresponding to the acetyl group (three nonhydrogen atoms). It appears that during crystallization and/or x-ray data collection the peptide undergoes an unusual deacetylation reaction, leading to a five-membered ring structure, resulting from the formation of a covalent bond between NH2 of Gly1 and Cα of ΔPhe2, and with simultaneous saturation of double bond between Cα and Cβ of ΔPhe2. As a result of this unusual conversion the expected stacking between ΔPhe2 and ΔPhe20 is disturbed (Fig. 3). However, the desired folding of the peptide has been achieved despite the unexpected perturbation at the terminal region. The methyl group in the terminal NHMe was not observable presumably because of disorder at the C terminal of the peptide (Fig. 4).
Figure 3.

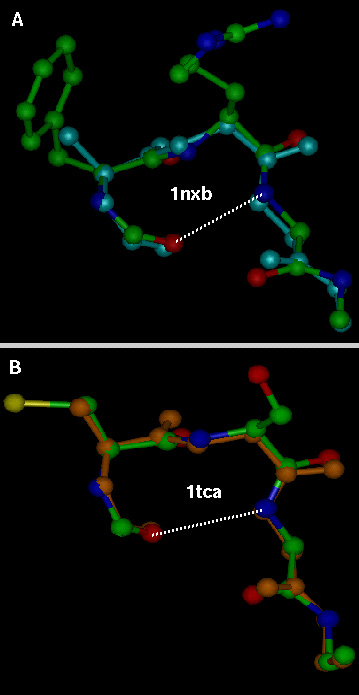
Two views of the structure of HH21. (A) ΔPhe side chains from two shape-complement helices stack one against the other resulting in a core with extended phenyl embrace arrangement. The thin dotted lines represent additional weak interactions such as C—H—O, N—H—π responsible for helix–helix association and turn stabilization and thick dotted (colored white) line represents the 4→1 N—H—O hydrogen bond at the type I β-turn region. Thin lines (green) with the corresponding values (4.62 Å, 4.96 Å) are the distances between the Cα atoms of the two ΔPhe residues stacking one above the other and belong to the two shape-complement helices. These distances highlight the extent of intercalation of the side chains belonging to two shape-complement helices. (B) Another view of the molecule.
Figure 4.

Ortep representation (32) of the peptide HH21 in stereo. The ellipsoids are drawn at 40% probability level. View of the molecule similar to that in Fig. 3A.
Results and Discussion
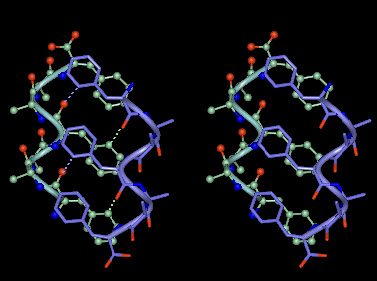
A perspective view of the crystal-state molecular structure of the peptide HH21 is illustrated in Fig. 3A. As designed, the N terminus of the molecule assumes a left-handed helical conformation, followed by a turn, and a right-handed helix at the C terminus. The angle between the two helices is approximately 166°. It is noteworthy that clear electron density can be seen for the flexible -(Gly)4 (residues 10–13)-linker (Fig. 5). The segment from Phe2 to ΔPhe8 is characterized by a left-handed 310-helical conformation, composed of four consecutive, overlapping type III′ β-bends (Fig. 3A, colored green), stabilized by appropriate 4→1 intramolecular N—H—O hydrogen bonds (see Table 3, which is published as supplemental material for more details). As expected l-Ala9, which assumes right-handed helical conformation (φ9, ψ9 = −59°, −35°), acts as a stop signal for the growing left-handed helix. The segment ΔPhe8-l-Ala9-Gly10-Gly11, assuming type I β-turn conformation (φ8, ψ8 = 50°, 31°; φ9, ψ9 = −59°, −35°; φ10, ψ10 = −97°, 9°; φ11, ψ11= 109°, −168°), causes chain-reversal (Fig. 3A, colored purple). Although the β-turn classification (23) is based on the dihedral angles at residues i + 1 and i + 2 position, it should be noted that the Gly11 at i + 3 position of the β-turn with φ11, ψ11 = +109°, −168°, appears to be crucial for the change of direction of the continuing helix. The role of weak interactions (C—H—O, N—H—π) (18, 19, 24) in stabilizing the turn region can be clearly seen in this structure (Fig. 3A, Table 1).
Figure 5.

Clear electron density for the flexible Ala-(Gly)4 linker region. (2Fo − Fc) map contoured at 2.5-σ level.
Table 1.
Intramolecular hydrogen* bonding network† observed at the helix–helix interior and at the turn region of the structure of HH21 (Fig. 3A)
| Donor D | Acceptor A | Distance (Å) D–A | Distance (Å) H–A | Angle (°) D—H–A | Type |
|---|---|---|---|---|---|
| C2B | O17 | 3.441 | 2.39 | 163 | SB/L/CHO |
| C5B | O14 | 3.507 | 2.52 | 152 | SB/L/CHO |
| C5D2 | O14 | 3.446 | 2.43 | 156 | SB/L/CHO |
| C14B | O5 | 3.422 | 2.42 | 154 | SB/L/CHO |
| C14D2 | O5 | 3.473 | 2.47 | 153 | SB/L/CHO |
| C17B | O2 | 3.535 | 2.57 | 147 | SB/L/CHO |
| C17D2 | O2 | 3.466 | 2.52 | 146 | SB/L/CHO |
| C8B | O11 | 3.258 | 2.30 | 147 | SB/T/CHO |
| N10 | C14R‡ | 3.611 | 2.67 | 152 | BS/T/NHπ |
| N11 | O8 | 3.014 | 2.01 | 164 | BB/T/NHO |
SB/L/CHO: Side chain to backbone lateral (interhelix) hydrogen bond (C—H—O). BB/T/NHO: Backbone to backbone turn stabilizing hydrogen bond (N—H—O). SB/T/CHO: Side chain to backbone turn stabilizing hydrogen bond (C—H—O). BS/T/NHπ: Backbone to aromatic ring center of the side chain interaction (N—H—π) at the turn region.
Helix stabilizing 4 → 1(N—H—O) hydrogen bonds are not shown (see Table 3).
C14R is the pseudo atom at the ring-center of the side chain of the residue ΔPhe14.
The segment from Gly12 to Ala21 folds into a right-handed 310-helix, containing seven consecutive, overlapping type III β-bends (Fig. 3A, colored mauve), stabilized by appropriate 4→1 intramolecular N—H—O hydrogen bonds. Interestingly, Gly12 and Gly13 assume helical values (φ12, ψ12 = −64°, −18° and φ13, ψ13 = −61°, −21°) and are involved in the initiation of the right-handed helix. As designed, the ΔPhe side chains stack parallel to each other; the ΔPhe8 stacks against ΔPhe14 and ΔPhe5 stacks against ΔPhe17. Further, as expected from our design model, specific C—H—O interactions between the side chains of one helix to the backbone of the other are also maintained (Fig. 3A, Table 1). HH21 is hydrated at its C and N termini with water molecules forming a hydration slab between the peptide molecules, packed in a continuous array.
Based on experimental (25) and theoretical (26) studies involving the “minimum protein folding alphabet” and “five-residue solution” hypotheses it has been proposed that for a structured protein, two hydrophobic and two polar plus a flexible Gly residues are essential for the sequence to achieve native-like folding. The implication may be that the presence of polar residue and binary patterning is important to achieve compaction. The apolar peptide HH21, which assumes a folded structure, sets a standard for the number/type of residues needed to form a folded structure, suggesting that although the five-residue solution may be relevant for solubility of a given peptide in water, it may not be so for its folding per se. Therefore in HH21 the possibility of replacement of some of the apolar noncore residues with polar ones to introduce water solubility would be an added positive design element and can be expected to result in a more stable association of the helices.
In the present structure as per the design strategy, only six ΔPhe residues (of 21 residues) are mainly responsible for the helix–helix association and stabilization. Further φ, ψ values of the residues in the linker region except Gly11 fall within the allowed region of Ramachandran's map for non-Gly residues. A search for loop regions in protein crystal structures (using the program spasm) (27) considering only the backbone atoms of the residues in the segment -Ala9 to Gly12 suggests that various residues can be accommodated in such a loop (see Figs. 10 and 11, which are published as supplemental material). Based on these considerations it is tempting to suggest that HH21 is a suitable template for mutational experiments at the noncore region and could serve as a host structure to accommodate functional sites.
The present crystal structure is remarkable because it reports a designed helix–turn–helix super secondary motif and the design strategy allows one to address the intricate problem of the involvement of weak interactions (C—H—O, π-π stacking, N—H—π) in molecular compaction and stability. The present structure highlights the important role that weak interactions can play in the association of secondary structures and hence in the design strategy. The helix–helix interior with a network of weak interactions, acting in a concerted manner, appears to be strong enough to destabilize all other possible associations of helices, maintaining a large energy gap, thus helping the molecule achieve a unique compact structure.
Conclusions
The Meccano (or Lego) set approach (11, 28) using conformation-constraining residues, which was very successful in designing secondary structural motifs, here achieves marvelous compact helical-hairpin super secondary structure. The novelty of our design strategy is that we have used conformation constraining amino acid (ΔPhe) for both restricting the backbone conformation and inducing long-range interactions. It is the concerted effect of weak interactions (29) that brings about the folding, a fact that may be profitably used in the engineering of novel molecules with desired properties and functions. The results described here assume considerable significance, because the broader aim of de novo design is not only to synthetically mimic the structures of natural proteins but also to design new proteins with novel structural arrangements and functions. We feel that these results should encourage peptide designers in pursuing the ambitious goal of de novo design of enormous pool of new shapes, new functions, and new materials (4).
Supplementary Material
Acknowledgments
We are grateful to Dr. K. R. Rajashankar for collecting data and critical review and Dr. Joel Berendson of Los Alamos National Laboratory for providing beam time in X8C beamline. U.A.R. thanks Mr. Anil K. Padyana for his encouragement and discussions on weak interactions in the course of design. We thank Dr. Nagasuma Chandra for critical reading. The financial support from the Department of Science and Technology (DST), India is acknowledged. We thank the Department of Biotechnology, India for access to facilities at Bioinformatics and interactive graphics facility, IISc, Bangalore. We are extremely grateful to the academy member and the referees for very thorough reading of the manuscript and helpful suggestions.
Abbreviations
- ΔPhe
α,β−dehydrophenylalanine
- Fmoc
fluorenylmethoxycarbonyl
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: Crystallographic data (excluding structure factors) for the structure in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC-153089. Copies of the data can be obtained, free of charge, on application to Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, United Kingdom, (fax: +44 1223 336033 or E-mail: deposit@ccdc.cam.ac.uk).
Ramagopal, U. A., Ramakumar, S. & Chauhan, V. S. (1999) Acta Crystallogr. A55 Supplement, 376 (abstr.).
References
- 1.DeGrado W F, Summa C M, Pavone V, Nastri F, Lombardi A. Annu Rev Biochem. 1999;68:779–819. doi: 10.1146/annurev.biochem.68.1.779. [DOI] [PubMed] [Google Scholar]
- 2.Richardson J S, Richardson D C, Tweedy N B, Gernet K M, Quinn T P, Hecht M H, Erickson B W, Yan Y, McClain R D, Donlan M E, Surles M C. Biophys J. 1992;63:1186–1209. [PMC free article] [PubMed] [Google Scholar]
- 3.Imperiali B, Ottesen J J. J Peptide Res. 1999;54:177–184. doi: 10.1034/j.1399-3011.1999.00121.x. [DOI] [PubMed] [Google Scholar]
- 4.Baltzer T. Top Curr Chem. 1999;202:39–76. [Google Scholar]
- 5.Fezoui Y, Weaver D L, Osterhout J J. Proc Natl Acad Sci USA. 1994;91:3675–3679. doi: 10.1073/pnas.91.9.3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kortamme T, Ramirez-Alvarado M, Serrano L. Science. 1998;281:253–256. doi: 10.1126/science.281.5374.253. [DOI] [PubMed] [Google Scholar]
- 7.Walsh S T R, Cheng H, Bryson J W, Roder H, DeGrado W F. Proc Natl Acad Sci USA. 1999;96:5486–5491. doi: 10.1073/pnas.96.10.5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jain R M, Chauhan V S. Biopolymers. 1996;40:105–119. doi: 10.1002/(sici)1097-0282(1996)40:1<105::aid-bip5>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 9.Balaram P. J Peptide Res. 1999;54:195–199. doi: 10.1034/j.1399-3011.1999.00119.x. [DOI] [PubMed] [Google Scholar]
- 10.Karle I L, Awasthi S K, Balaram P. Proc Natl Acad Sci USA. 1996;93:8189–8193. doi: 10.1073/pnas.93.16.8189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karle I L, Das C, Balaram P. Proc Natl Acad Sci USA. 2000;97:3034–3037. doi: 10.1073/pnas.070042697. . (First Published March 21, 2000; 10.1073/pnas.070042697) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karle I L, Flippen-Anderson J L, Sukumar M, Uma K, Balaram P. J Am Chem Soc. 1991;113:3952–3956. [Google Scholar]
- 13.Tuzi A, Ciajolo M R, Guarino G, Temussi P A, Fissi A, Pironi O. Biopolymers. 1993;33:1111–1121. doi: 10.1002/bip.360330713. [DOI] [PubMed] [Google Scholar]
- 14.Rajashankar K R, Ramakumar S, Chauhan V S. J Am Chem Soc. 1992;144:9225–9226. [Google Scholar]
- 15.Rajashankar K R, Ramakumar S, Jain R M, Chauhan V S. J Am Chem Soc. 1995;117:11773–11779. [Google Scholar]
- 16.Toniolo C, Benedetti E. Trends Biochem Sci. 1991;16:350–353. doi: 10.1016/0968-0004(91)90142-i. [DOI] [PubMed] [Google Scholar]
- 17.Burley S K, Petsko G A. Science. 1985;229:23–28. doi: 10.1126/science.3892686. [DOI] [PubMed] [Google Scholar]
- 18.Desiraju G R. Acc Chem Res. 1996;29:441–449. doi: 10.1021/ar950135n. [DOI] [PubMed] [Google Scholar]
- 19.Gu Y, Kar T, Scheiner S. J Am Chem Soc. 1999;121:9411–9422. [Google Scholar]
- 20.Crick F H C. Acta Crystallogr. 1953;6:689–697. [Google Scholar]
- 21.Otwinowski Z, Minor V. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 22.Sheldrick G M. Program for Crystal Structure Solution and Refinement. Gottingen, Germany: University of Gottingen; 1997. [Google Scholar]
- 23.Wilmot C M, Thornton J M. J Mol Biol. 1988;203:221–232. doi: 10.1016/0022-2836(88)90103-9. [DOI] [PubMed] [Google Scholar]
- 24.Steiner T. Acta Crystallogr D. 1998;54:584–588. doi: 10.1107/s090744499701500x. [DOI] [PubMed] [Google Scholar]
- 25.Riddle D S, Santiago J V, Bray-Hall S T, Doshi N, Grantcharova V P, Yi Q, Baker D. Nat Struct Biol. 1997;4:805–809. doi: 10.1038/nsb1097-805. [DOI] [PubMed] [Google Scholar]
- 26.Wang J, Wang W. Nat Struct Biol. 1999;6:1033–1038. doi: 10.1038/14918. [DOI] [PubMed] [Google Scholar]
- 27.Kleywegt G J. J Mol Biol. 1999;285:1887–1897. doi: 10.1006/jmbi.1998.2393. [DOI] [PubMed] [Google Scholar]
- 28.Balaram P. Pure Appl Chem. 1992;64:1061–1066. [Google Scholar]
- 29.Desiraju G R, Steiner T. The Weak Hydrogen Bonds: International Union of Crystallography Monographs on Crystallography. Oxford: Oxford Univ. Press; 1999. [Google Scholar]
- 30.Jeffrey G A, Lewis L. Carbohydr Res. 1978;60:179–184. [Google Scholar]
- 31.Taylor R, Kennard O. Acta Crystallogr B. 1983;39:133–138. [Google Scholar]
- 32.Johnson C K. ortep: A Fortran Thermal-Ellipsoid Plot Program for Crystal Structure Illustrations, ORNL Report 5138. Oak Ridge, TN: Oak Ridge National Laboratory; 1976. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}