

Abstract
Despite observations of age-dependent sexual dimorphisms in hypothalamic-pituitary-adrenal (HPA) axis activity, the role of androgens in the regulation of HPA axis activity in men has not been examined. We assessed this role by performing CRH stimulation tests in ten men (ages 18–45) during gonadal suppression with leuprolide acetate and during testosterone addition to leuprolide. CRH-stimulated cortisol levels as well as peak cortisol and greatest cortisol excursion were significantly lower (p < .05, .005, and .01, respectively) during testosterone replacement compared with the induced hypogonadal condition (leuprolide plus placebo); cortisol area under the curve was lower at a trend level (p < .1). Paradoxically, CRH-stimulated ACTH was increased significantly during testosterone replacement (p < .05). The cortisol:ACTH ratio, a measure of adrenal sensitivity, was lower during testosterone replacement (p < .1). A mixed effects regression model showed that testosterone but not estradiol or CBG significantly contributed to the variance of cortisol. These data demonstrate that testosterone regulates CRH-stimulated HPA axis activity in men, with the divergent effects on ACTH and cortisol suggesting a peripheral (adrenal) locus for the suppressive effects on cortisol. Our results further demonstrate that the enhanced stimulated HPA axis activity previously described in young men compared with young women cannot be ascribed to an activational upregulation of the axis by testosterone.
Keywords: testosterone, cortisol, HPA axis, ACTH, CRH, men
Introduction
Substantial evidence suggests that reproductive steroids modulate the response to stress. Supportive data include observations of sex differences in hypothalamic-pituitary-adrenal (HPA) axis activity/responsivity (Kitay, 1961; Brett et al, 1983; Horrocks et al, 1990; Kirschbaum et al, 1992; McCormick et al, 2002; Roelfsema et al, 1993; Greenspan et al, 1993) and demonstration of acute regulatory effects of gonadal steroids on the HPA axis in animal castration and replacement studies (Kitay, 1963; Burgess et al, 1992; Critchlow et al, 1963; Bingaman et al, 1994). As the first observations of sex differences showed increased HPA axis activity in female rodents, most attention has focused on the effects of estradiol, with little attention paid to the possible modulatory role of testosterone. In animals, several castration and androgen replacement studies suggest that androgens inhibit stress-stimulated, but not basal, corticotropin (ACTH) and cortisol concentrations (Handa et al, 1994; Papadopoulos et al, 2000), findings consonant with the many studies showing enhanced response to a variety of stressors in female compared with male rodents (LeMevel et al, 1979; Lesniewska et al, 1990). Sexual dimorphisms (largely age-dependent) in stimulated HPA axis activity have been variably reported in humans (Greenspan et al, 1993; Seeman et al, 2001), but there are virtually no studies identifying the role, if any, of androgens in the reported human dimorphisms. Indeed, in one of few studies evaluating the impact of reproductive manipulation on the HPA axis in men, estrogen was administered and observed to increase the hormonal response to psychosocial stress (Kirschbaum et al, 1996), similar to results in non-human male primates (Norman et al, 1992). Thus, the effects of testosterone on the HPA axis in human males are unknown and limited to speculations based on observed sexual dimorphisms in lower animals. To remedy this dearth of information, we investigated the stress modulatory effects of androgens by performing corticotropin-releasing hormone (CRH) stimulation tests in men under two induced hormonal conditions: leuprolide-induced suppression of the hypothalamic-pituitary-gonadal (HPG) axis, with and without testosterone replacement.
Methods
Subject Selection
Healthy male volunteers were recruited through advertisements in local newspapers. All men were between ages 18 to 45, were without current medical illness (as assessed by medical history, physical examination, laboratory tests and EKG), were medication free, and had no history of psychiatric disorders, including alcohol and substance abuse, as determined by the Structured Clinical Interview for DSM III-R or IV (SCID-IIIR or IV) (First et al, 1996). The protocol was reviewed and approved by the National Institute of Mental Health (NIMH) Institutional Review Board, and all subjects gave both written and verbal consent to study participation. Subjects were paid for their participation according to the schedule of payment issued by the National Institutes of Health Normal Volunteer Office.
Study Design
Leuprolide Study
Eleven men were recruited for the study. Ten participated in the ovine-CRH study; the eleventh individual failed to suppress testosterone levels following leuprolide acetate administration and was not included in the analysis. Of the volunteers, eight were Caucasian and two were African American. Mean age ± S.D. was 31.4 ± 7.5. Volunteers received three monthly injections of the gonadotropin-releasing hormone agonist leuprolide acetate (Lupron) (7.5 mg) following a two month screening period during which the absence of illness and mood disturbance was confirmed. Following one month of leuprolide acetate alone, men were randomly assigned to receive add-back of either 200 mg IM testosterone enanthate or sesame oil injections every two weeks for a month in a double-blind, crossover design. The ovine-CRH study was performed 1–12 days after the second injection of testosterone or placebo. CRH infusions were performed a mean (± SD) of 5.6 (± 5.6) days after the second injection of testosterone and 3.7 (± 4.0) days after the second injection of placebo.
Blood at baseline for each CRH test was analyzed for plasma levels of testosterone, estradiol and cortisol binding globulin (CBG). Additionally, 24 hour urine samples were obtained and analyzed for urinary free cortisol (UFC), the majority within one day of the infusion (i.e., most collections began one day prior to CRH, but several ranged from 2 to 5 days after CRH administration ); mean ± SD = 1.2 ± 1.7 days.
Ovine-CRH stimulation test
Subjects were instructed to fast from midnight before the test and to present to the clinic between 7 and 8 a.m. Subjects were weighed and then placed in a semi-recumbant position. An intravenous catheter was inserted in the antecubital vein and the subjects rested for 40 minutes prior to receiving 1mcg/kg ovine CRH (oCRH) by IV push. Blood samples for ACTH and cortisol were drawn at 15 and 1 minute before and 5, 15, 30, 60, 90, 120, 150 and 180 minutes after administration of oCRH. Blood samples were collected in pre-chilled tubes containing EDTA and were centrifuged and aliquoted promptly; plasma was stored at −70C until assayed.
All assays were performed by Esoterix Inc. (Calabasas Hills, CA). Individual subjects had samples from all of their tests run in the same assay.
CBG was measured directly by radioimmunoassay (RIA); the intra- and inter-assay coefficients of variation (CV) were 7.8% and 11%, respectively. Estradiol was measured by RIA after extraction with LH20 column chromatography in a modification of the procedure by Wu and Lundy (1971). The intra-assay CV was 3.2%, and the inter-assay CV was 9.2%. Testosterone was measured by RIA following extraction with column chromatography, with an intra-assay CV of 7% and an inter-assay CV of 9%.
Cortisol was measured directly by RIA with intra- and inter-assay CVs of 3.9% and 8.3%, respectively. The ACTH- immunoradiometric assay uses paired monoclonal and polyclonal antibodies, reactive respectively with the N terminal and the C terminal regions of ACTH. The intra-assay CV was 6.2%, and the inter-assay CV was 11%.
Statistics
Sample size for this repeated measures design was calculated in a power analysis performed with NCSS PASS (Kaysville, UT) and based on a delta obtained in an earlier study (Roca et al, 2003); a sample size of nine had an 80% power to detect the predicted differences as significant at an alpha of 0.05.
To determine whether the hormonal conditions were successfully created, baseline levels of testosterone and estradiol were compared with paired t-tests. Baseline measures of HPA axis function (cortisol, ACTH, CBG, UFC) also were compared across hormonal conditions with paired t-tests. Differences in CRH-stimulated ACTH and cortisol levels were assessed with analysis of variance with repeated measures (ANOVA-R), with hormonal condition (testosterone versus placebo replaced) and time (nine levels included the average of the pre-CRH values and each of the post-CRH values) as within subjects variables. As each subject had 18 data points (nine time points in each of two conditions), ANOVA-R (Systat; Port Richmond, CA) was employed as the primary statistical comparison because it takes maximal advantage of all of the data collected, unlike summary measures (e.g., area under the curve [AUC], which may not discriminate between differences in the shape of the response curve). The cortisol:ACTH ratio was used as a repeated measure to assess adrenal responsivity. Bonferroni corrected post hoc comparisons were performed when the results of the ANOVA-R were significant. Three additional summary measures of CRH-stimulated HPA axis function were compared across the two hormonal conditions with paired t tests; AUC, stimulated peak value (MAX), and difference between peak value and baseline [mean of the values 15 minutes and immediately prior to CRH] (delta MAX). (AUC was calculated by a baseline-corrected trapezoidal integration method.) Comparisons of these three measures were performed for both ACTH and cortisol. The alpha (level of significance) was set at 0.017 to adjust for type I errors associated with multiple comparisons.
Pearson product moment correlation coefficients were calculated to determine if changes in baseline levels of testosterone, estradiol, testosterone/estradiol ratio, or CBG across conditions were associated with changes in stimulated measures of ACTH and cortisol (AUC, MAX, and delta MAX). To further estimate the relative contributions of testosterone, estradiol, and CBG to the variance of CRH-stimulated cortisol and ACTH levels across hormonal conditions, a mixed effects regression analysis was performed with SAS Proc Mix (Cary, NC) with the following factors included in the predictive model: hormone condition, time, testosterone, estradiol, CBG. This procedure permits assessment of each of the in the presence of the effects of the other variables of interest.
Results
Mean plasma testosterone levels were significantly lower during leuprolide plus placebo compared with those obtained during the leuprolide plus testosterone condition (57 ng/dl vs. 783 ng/dl, (p < .001) (Table 1). Estradiol, like testosterone, was significantly higher and CBG significantly lower during testosterone replacement, as expected (p < .01, and p < 01, respectively) (Table 1). Baseline values of ACTH and cortisol did not differ across the hormonal conditions. Urinary free cortisol (UFC) did not differ between the induced hormonal conditions (although complete paired collections were obtained in only seven subjects). CRH-stimulated cortisol was significantly decreased during the testosterone-replaced condition (effect of condition, F1,9 = 7.39, p < .05) compared with the induced hypogonadal (relatively testosterone deficient) condition created by leuprolide with placebo replacement (Fig 1). Consistent with this finding, the maximum CRH-stimulated cortisol and the delta MAX were significantly lower during testosterone replacement (p < .005 and p < .01, respectively), with a trend for lower cortisol AUC (p = .057) also seen. Paradoxically, CRH-stimulated ACTH was significantly increased during testosterone replacement compared with the placebo-replaced hypogonadal condition (effect of condition, F1,9 = 5.22, p < .05) (Fig 1). While there was a trend for the ACTH max to be increased during testosterone (p = .06), no differences were seen in the delta MAX or AUC. No condition by time interactions were observed for either CRH stimulated cortisol or ACTH. Significant effects of time were observed for both cortisol and ACTH as expected. There was a trend for the cortisol/ACTH ratio, a measure of adrenal sensitivity, to be lower during testosterone replacement (F1,9 = 4.23, p = .07), largely reflecting the failure of the ratio to increase at the later time points in the testing (Fig 2). Changes in cortisol and ACTH AUC, MAX and delta MAX from the hypogonadal to testosterone replaced condition were not significantly correlated with changes across conditions in testosterone, estradiol, CBG, or the ratio of testosterone to estradiol. The correlation between changes in estradiol and AUC cortisol across conditions did reach a trend level of significance (r = −.63, p = .054). A mixed effects regression model examined the contributions of testosterone, estradiol, and CBG, alone and in combination, to the variance of stimulated cortisol and ACTH. No individual effects were seen for CBG or estradiol, and a trend was seen for a significant contribution of testosterone to the variance of cortisol (over time and across conditions). When CBG was removed from the full model (given the weakness of its contribution), the effect of testosterone surpassed significance levels (F1,131 = 3.97, p < .05). No effect of testosterone, estradiol, or CBG was seen on stimulated ACTH.
Table 1.
Effects of Leuprolide with or without testosterone add-back on basal and CRH-stimulated hormonal measures. Values are shown as mean (SD).
| Leuprolide + Placebo | Leuprolide + Testosterone | T-test | |
|---|---|---|---|
| Testosterone (ng/dl) | 57.00 (63.91) | 783.00 (413.54) | 5.50, p < .001 |
| Estradiol (pg/ml) | 0.75 (0.22) | 3.32 (1.33) | 6.08, p < .001 |
| UFC (μg/24hr) | 47.57 (17.27) | 55.14 (35.91) | 0.86, ns |
| CBG (ng/dl) | 2.93 (0.38) | 2.4 (0.52) | 3.54, p < .01 |
| ACTH baseline | 13.12 (4.27) | 15.97 (10.03) | 1.31, ns |
| ACTH AUC* | 4308.55 (3545.15) | 4426.15 (2532.89) | 0.24, ns |
| ACTH MAX* | 54.76 (31.30) | 61.08 (30.22) | 2.14, p < .1 |
| ACTH ΔMAX* | 41.64 (29.89) | 45.11 (23.70) | 1.21, ns |
| CORT baseline | 10.43 (2.95) | 10.53 (3.09) | 0.12, ns |
| CORT AUC* | 1903.18 (579.04) | 1602.20 (345.03) | 2.18, p < .1 |
| CORT MAX* | 25.75 (3.17) | 22.93 (3.24) | 3.90, p < .01 |
| CORT ΔMAX* | 15.32 (3.32) | 12.40 (1.73) | 3.48, p < .01 |
Values obtained after CRH stimulation; all others are unstimulated values ACTH values – pg/ml; cortisol values – ug/dl
Conversion factors to SI units are as follows: testosterone × 3.467 = nmol/L; estradiol × 3.4671 = pmol/L; UFC × 2.759 = nmol/d; CBG, cortisol binding globulin × 18.868 = nmol/L; ACTH × 2.2202 = pmol/L; cortisol × 27.59 = nmol/L
Fig 1.
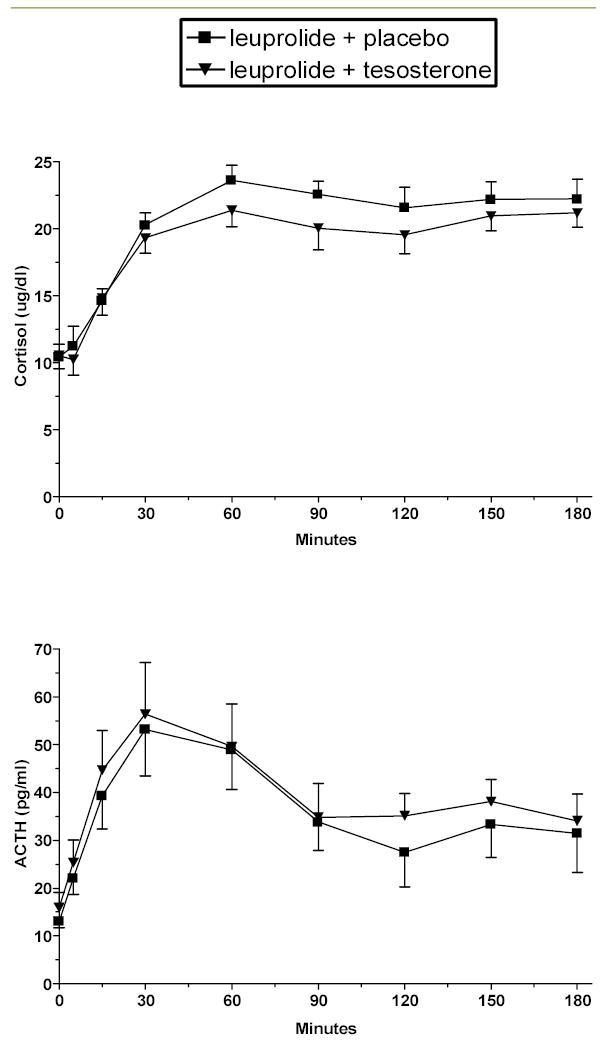
CRH-stimulated cortisol and ACTH levels (mean ± SE) during hypogonadal (leuprolide plus placebo) and testosterone replaced (leuprolide plus testosterone) conditions in men. Significantly lower stimulated cortisol levels (p < .05) and significantly higher stimulated ACTH levels (p < .05) are seen during testosterone administration.
Fig 2.
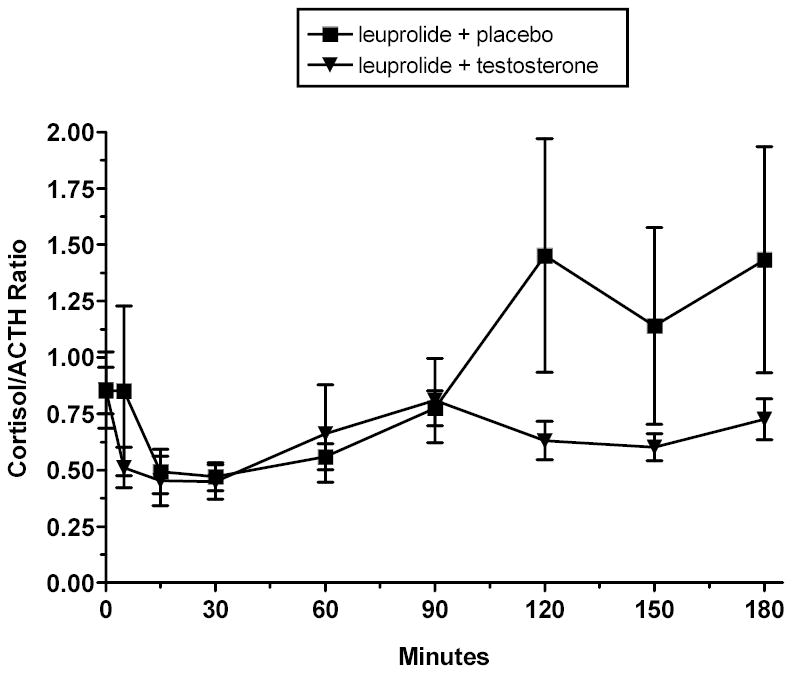
Ratio of CRH-stimulated cortisol and ACTH values (mean ± SE) during induced hypogonadal (leuprolide plus placebo) and testosterone replaced (leuprolide plus testosterone) conditions. A trend is seen for lower cortisol:ACTH ratios during the testosterone replacement condition. This trend suggests that testosterone reduces the adrenal response to ACTH.
Discussion
Prior studies suggest that young men have an enhanced cortisol (Collins et al, 1978; Forsman et al, 1982; Polefrone et al, 1987; Kirschbaum et al, 1992; Kirschbaum et al, 1995)) [but also see (Gallucci et al, 1993; Stoney et al, 1987; Heuser et al, 1994)] and ACTH (Kirschbaum et al, 1999; Kudielka et al, 2004) response to cognitive and pharmacologic stressors relative to women, presumed to represent a suppressive effect of estradiol or an augmenting effect of testosterone. In contrast, our data demonstrate that the effect of testosterone in young men is to inhibit rather than augment the cortisol response to CRH stimulation. Compared with the leuprolide-induced hypogonadal state, testosterone replacement produced a blunted cortisol response, decreased AUC, as well as decreased cortisol MAX and delta MAX. These findings are consistent with studies in rodents revealing a suppressive rather than enhancing effect of testosterone on stimulated HPA axis activity (Handa et al, 1994; Bingaman et al, 1994). A variety of central mechanisms (decreased CRH, decreased AVP, increased glucocorticoid receptor [GR] concentrations) have been postulated to underlie the suppressive effects of testosterone in rodents, with recent data suggesting that testosterone suppresses stimulated cortisol secretion through its metabolite 3 beta androstanediol, which acts through ER beta in the PVN, but not through ER alpha or the androgen receptor (Lund et al, 2004) (RJ Handa, personal communication, 4/19/2004). The surprising finding in our study of increased ACTH during testosterone replacement, however, localizes the suppressive effects on cortisol to the periphery.
A possible explanation for reduced stimulated cortisol in the face of increased ACTH is decreased adrenal sensitivity. In fact, as measured by the cortisol: ACTH ratio, adrenal sensitivity tended to decrease on testosterone + leuprolide relative to placebo + leuprolide, with the reduction primarily reflecting the two fold decrease in the ratio at time points 120–150, again suggesting diminished cortisol response to ACTH. Reductions in adrenal sensitivity have been postulated to explain similar cortisol values in young men and women despite significantly elevated ACTH levels in the young men (Roelfsema et al, 1993; Horrocks et al, 1990; Kudielka et al, 2004). The present data would suggest even greater modulation of adrenal sensitivity by testosterone, as not only is ACTH significantly higher, but cortisol is significantly lower compared with the hypogonadal state. The effect of testosterone on cortisol, however, appears to be confined to stimulated levels, as no difference in either basal cortisol or urinary free cortisol was observed across the induced hormonal conditions. Using dispersed inner adrenocortical cells, Nowak et al. (1995) showed that testosterone inhibited ACTH-stimulated but not basal corticosterone output in vitro. As testosterone reduced both pregnenolone and (particularly) post-pregnenolone steroid levels in this model, both the conversion of cholesterol to pregnenolone and the activities of several steroidogenic enzymes (3betaHSD, P450C21, and P450C11) appear to be inhibited (Stalvey, 2002). Stalvey (2002) similarly demonstrated a reduction by testosterone of 3 beta HSD activity, immunoreactivity, and m-RNA in cultured mouse adrenal cells under both basal and stimulated conditions. The restriction of the effects of testosterone in the current results to (ACTH) stimulated rather than basal conditions (Nowak et al, 1995) suggests that testosterone may interfere with ACTH signaling at the adrenal. While testosterone-related decreases in ACTH bioactivity have been observed in vitro (Coyne et al, 1969), the resultant decrease in cortisol would be expected under basal conditions, which was not observed in this study.
Downstream effects of the removal and administration of testosterone may have contributed to the results observed in this study. Testosterone replacement was associated with significant increases in estradiol (393%) and decreases in CBG (18%). While decreased CBG could account for lower stimulated cortisol values during testosterone administration, the changes in CBG across hormonal conditions were not significantly correlated with changes in the summary measures of HPA axis function (AUC, MAX, delta MAX); further, the mixed effects regression analysis demonstrated that CBG did not account for a significant portion of the variance of stimulated cortisol or ACTH when testosterone and estradiol were included in the model. The effects of estradiol on cortisol secretion are complex. In ovariectomized animals, inhibition of HPA axis responses to stressors is seen with short-term, physiological levels of estradiol replacement (Redei et al, 1994; Young et al, 2001; Dayas et al, 2000; Komesaroff et al, 1998), while higher doses and longer treatment regimens enhance HPA axis reactivity to stressors (Burgess et al, 1992; Carey et al, 1995; Viau et al, 1991). We observed a significant negative correlation between the increase in estradiol (but not testosterone) and the decrease in cortisol AUC from the hypogonadal to the testosterone replaced condition. The consequent suggestion that testosterone inhibition of stimulated cortisol is mediated by the testosterone metabolite estradiol is contradicted by several factors. First, while data are inconsistent, many in vitro and in vivo studies are convergent in showing that estradiol enhances rather than inhibits corticosteroid secretion at the level of both the adrenal (Nowak et al, 1995; Fukui et al, 1961; Kitay, 1961) and the pituitary (Kitay, 1963; Lund et al, 2004; McCormick et al, 2002; Giussani et al, 2000). Second, consistent with this possibility (and more immediately relevant) is the demonstration by Kirschbaum et al. (1996) of increased cognitive stress stimulated cortisol (and ACTH) secretion during short term estradiol administration to young men; increased mean plasma cortisol concentrations during estradiol administration were also observed in castrated male macaques (Norman et al, 1992). Finally estradiol did not account for a significant part of the variance of stimulated cortisol if testosterone was included in the regression model. Indeed, only testosterone was found to account for a significant part of the variance of stimulated cortisol secretion.
The increased stimulated ACTH secretion during testosterone replacement could reflect decreased feedback inhibition or increased corticotroph sensitivity to CRH. While both testosterone and estradiol modulate glucocorticoid receptor activity in brain regions mediating restraint of the HPA axis (Burgess et al, 1992; Carey et al, 1995; Redei et al, 1994; Peiffer et al, 1991), a more parsimonious explanation suggests that the blunted cortisol secretion during testosterone administration results in less restraint on ACTH secretion, consistent with the greatest difference in the cortisol: ACTH ratios between conditions occurring from 120–180 minutes. Similarly, a central action of testosterone or estradiol should appear as increased basal ACTH levels and would not be apparent with exogenous CRH stimulation (or would appear as blunting due to CRH receptor down regulation at the corticotroph).
As an alternative explanation, the increase in CRH-stimulated ACTH during testosterone replacement could represent the effects of increased levels and activity of vasopressin (AVP) in the paraventricular nucleus (PVN), which would augment the response to endogenous or exogenous CRH. Such an augmentation of AVP by testosterone was postulated by Rubin et al. (1999) to explain the increased ACTH response to cholinergic stimulation seen in men compared with women. Testosterone regulates AVP synthesis and receptor activity in several brain regions critical to HPA axis control, including the PVN and medial preoptic area (Viau et al, 1991; DeVries et al, 1985; Viau et al, 1996). Results from several earlier studies in animals would have predicted decreased rather than the increased ACTH response to testosterone that we observed (Viau et al, 1996; Kitay, 1963; Handa et al, 1994; Miskowiak et al, 1988). Nonetheless, the findings of Viau (1999; 1996), Patchev (1996) and others suggest that the effects of testosterone on AVP (and subsequent effects on ACTH) are complex and differ according to brain region (e.g., PVN vs. MPOA), paradigm (basal vs stress [type-dependent]- stimulated), gender and species. Despite compelling evidence in the literature for both central and peripheral regulatory effects of testosterone on the HPA axis (Handa et al, 1994; Viau et al, 1999), the changes in cortisol in our study would appear to represent a peripheral effect of testosterone (although additional effects on endogenous CRH secretion clearly cannot be ruled out).
In conclusion, we have shown that testosterone regulates CRH-stimulated HPA axis activity in men. Similar to findings in animal studies, CRH-stimulated cortisol was decreased during testosterone-replaced compared with hypogonadal conditions. The concomitant increase in ACTH suggests that the decrease in stimulated cortisol levels by testosterone or its metabolites is mediated at the level of the adrenal gland. While provocative, these results must be viewed with caution. Notwithstanding the value of the repeated measures design, the sample size is small, and the differences observed across hormonal conditions, while significant, are small in magnitude (particularly the ACTH results) and of unclear clinical significance. Nonetheless, our findings complement our earlier demonstration of the regulatory role of progesterone on the HPA axis in women (Roca et al, 2003) and suggest that, as in animals, the reproductive axis actively participates in the regulation of the stress axis and is not merely acted on by it. While the effects observed in this study are quite modest, they demonstrate that the enhanced stimulated HPA axis activity previously described in young men compared with young women cannot be ascribed to an activational upregulation of the axis by testosterone. Finally, the potential health implications of the removal of restraint on stress-stimulated cortisol secretion during the hypogonadism of aging deserve examination.
Acknowledgments
The authors gratefully acknowledge the data management assistance of Ms. Kristen Dancer.
References
- Bingaman EW, Magnuson DJ, Gray TS, Handa RJ. Androgen inhibits the increases in hypothalamic corticotropin-releasing hormone (CRH) and CRH-immunoreactivity following gonadectomy. Neuroendocrinology. 1994;59:223–234. doi: 10.1159/000126663. [DOI] [PubMed] [Google Scholar]
- Brett LP, Chong GS, Coyle S, Levine S. The pituitary-adrenal response to novel stimulation and ether stress in young adult and aged rats. Neurobiol Aging. 1983;4:133–138. doi: 10.1016/0197-4580(83)90037-4. [DOI] [PubMed] [Google Scholar]
- Burgess LH, Handa RJ. Chronic estrogen-induced alterations in adrenocorticotropin and corticosterone secretion, and glucocorticoid receptor-mediated functions in female rats. Endocrinology. 1992;131:1261–1269. doi: 10.1210/endo.131.3.1324155. [DOI] [PubMed] [Google Scholar]
- Carey MP, Deterd CH, de Koning J, Helmerhorst F, De Kloet ER. The influence of ovarian steroids on hypothalamic-pituitary-adrenal regulation in the female rat. J Endocrinol. 1995;144:311–321. doi: 10.1677/joe.0.1440311. [DOI] [PubMed] [Google Scholar]
- Collins A, Frankenhaeuser M. Stress responses in male and female engineering students. Journal of Human Stress. 1978;4:43–48. doi: 10.1080/0097840X.1978.9934986. [DOI] [PubMed] [Google Scholar]
- Coyne MD, Kitay JI. Effect of ovariectomy on pituitary secretion of ACTH. Endocrinology. 1969;85:1097–1102. doi: 10.1210/endo-85-6-1097. [DOI] [PubMed] [Google Scholar]
- Critchlow V, Liebelt RA, Bar-Sela M, Mountcastle W, Lipscomb HS. Sex difference in resting pituitary-adrenal function in the rat. Am J Physiol. 1963;205:807–815. doi: 10.1152/ajplegacy.1963.205.5.807. [DOI] [PubMed] [Google Scholar]
- Dayas CV, Xu Y, Buller KM, Day TA. Effects of chronic oestrogen replacement on stress-induced activation of hypothalamic-pituitary-adrenal axis control pathways. J Neuroendocrinol. 2000;12:784–794. doi: 10.1046/j.1365-2826.2000.00527.x. [DOI] [PubMed] [Google Scholar]
- DeVries GJ, Buijs RM, van Leeuwen FW, Caffe AR, Swaab DF. The vasopressinergic innervation of the brain in normal and castrated rats. J Comp Neurol. 1985;233 :236–254. doi: 10.1002/cne.902330206. [DOI] [PubMed] [Google Scholar]
- First MB, Spitzer RL, Gibbon M, Williams JBW. (1996) Structured clinical interview for DSM-IV axis I disorders - patient edition. Biometrics Research Department, New York State Psychiatric Institute: New York, N.Y.
- Forsman L, Lundberg U. Consistency in catecholamine and cortisol excretion in males and females. Pharmacol Biochem Behav. 1982;17:555–562. doi: 10.1016/0091-3057(82)90318-5. [DOI] [PubMed] [Google Scholar]
- Fukui S, Takeuchi K, Watanage F, Kumagai A, Yano S, Nishino K. Influences of some steroids on the corticosterone production by rat adrenal in vitro. Endocr J. 1961;8:43–49. doi: 10.1507/endocrj1954.8.43. [DOI] [PubMed] [Google Scholar]
- Gallucci WT, Baum A, Laue L, Rabin DS, Chrousos GP, Gold PW, et al. Sex differences in sensitivity of the hypothalamic-pituitary-adrenal axis. Health Psychol. 1993;12 :420–425. doi: 10.1037//0278-6133.12.5.420. [DOI] [PubMed] [Google Scholar]
- Giussani DA, Farber DM, Jenkins SL, Yen A, Winter JA, Tame JD, et al. Opposing effects of androgen and estrogen on pituitary-adrenal function in nonpregnant primates. Biol Reprod. 2000;62:1445–1451. doi: 10.1095/biolreprod62.5.1445. [DOI] [PubMed] [Google Scholar]
- Greenspan SL, Rowe JW, Maitland LA, McAloon-Dyke M, Elahi D. The pituitary-adrenal glucocorticoid response is altered by gender and disease. J Gerontol. 1993;48:M72–M77. doi: 10.1093/geronj/48.3.m72. [DOI] [PubMed] [Google Scholar]
- Handa RJ, Nunley KM, Lorens SA, Louie JP, McGivern RF, Bollnow MR. Androgen regulation of adrenocorticotropin and corticosterone secretion in the male rat following novelty and foot shock stressors. Physiol Behav. 1994;55:117–124. doi: 10.1016/0031-9384(94)90018-3. [DOI] [PubMed] [Google Scholar]
- Heuser IJ, Gotthardt U, Schweiger U, Schmider J, Lammers CH, Dettling M, et al. Age-associated changes of pituitary-adrenocortical hormone regulation in humans: importance of gender. Neurobiol Aging. 1994;15:227–231. doi: 10.1016/0197-4580(94)90117-1. [DOI] [PubMed] [Google Scholar]
- Horrocks PM, Jones AJ, Ratcliffe WA, Holder G, White A, Holder R, et al. Patterns of ACTH and cortisol pulsatility over twenty-four hours in normal males and females. Clin Endocrinol. 1990;32:127–134. doi: 10.1111/j.1365-2265.1990.tb03758.x. [DOI] [PubMed] [Google Scholar]
- Kirschbaum C, Klauer T, Filipp S-H, Hellhammer DH. Sex-specific effects of social support of cortisol and subjective responses to acute psychological stress. Psychosom Med. 1995;57:23–31. doi: 10.1097/00006842-199501000-00004. [DOI] [PubMed] [Google Scholar]
- Kirschbaum C, Kudielka BM, Gaab J, Schommer NC, Hellhammer DH. Impact of gender, menstrual cycle phase, and oral contraceptives on the activity of the hypothalamic-pituitary-adrenal axis. Psychosom Med. 1999;61:154–162. doi: 10.1097/00006842-199903000-00006. [DOI] [PubMed] [Google Scholar]
- Kirschbaum C, Schommer N, Federenko I, Gaab J, Neumann O, Oellers M, et al. Short-term estradiol treatment enhances pituitary-adrenal axis and sympathetic responses to psychosocial stress in healthy young men. J Clin Endocrinol Metab. 1996;81:3639–3643. doi: 10.1210/jcem.81.10.8855815. [DOI] [PubMed] [Google Scholar]
- Kirschbaum C, Wüst S, Hellhammer D. Consistent sex differences in cortisol responses to psychological stress. Psychosom Med. 1992;54:648–657. doi: 10.1097/00006842-199211000-00004. [DOI] [PubMed] [Google Scholar]
- Kitay JI. Enhancement of steroidogenesis by rat adrenal slices in vitro with estradiol-17-beta. Nature. 1961;192:358–359. doi: 10.1038/192358b0. [DOI] [PubMed] [Google Scholar]
- Kitay JI. Sex differences in adrenal cortical secretion in the rat. Endocrinology. 1961;68 :818–824. doi: 10.1210/endo-68-5-818. [DOI] [PubMed] [Google Scholar]
- Kitay JI. Pituitary-adrenal function in the rat after gonadectomy and gonadal hormone replacement. Endocrinology. 1963;73:253–260. doi: 10.1210/endo-73-2-253. [DOI] [PubMed] [Google Scholar]
- Komesaroff PA, Esler M, Clarke IJ, Fullerton MJ, Funder JW. Effects of estrogen and estrous cycle on glucocorticoid and catecholamine responses to stress in sheep. Am J Physiol. 1998;275:E671–E678. doi: 10.1152/ajpendo.1998.275.4.E671. [DOI] [PubMed] [Google Scholar]
- Kudielka BM, Buske-Kirschbaum A, Hellhammer DH, Kirschbaum C. HPA axis responses to laboratory psychosocial stress in healthy elderly adults, younger adults, and children: impact of age and gender. Psychoneuroendocrinology. 2004;29:83–98. doi: 10.1016/s0306-4530(02)00146-4. [DOI] [PubMed] [Google Scholar]
- LeMevel JC, Abitbol S, Beraud G, Manley J. Temporal changes in plasma adrenocorticotropin concentration after repeated neurotropic stress in male and female rats. Endocrinology. 1979;105:812–817. doi: 10.1210/endo-105-3-812. [DOI] [PubMed] [Google Scholar]
- Lesniewska B, Miskowiak B, Nowak M, Malendowicz LK. Sex differences in adrenocortical structure and function. XXVIII. The effect of ether stress on ACTH and corticosterone in intact, gonadectomized, and testosterone- or estradiol-replaced rats. Res Exp Med (Berl ) 1990;190:95–103. doi: 10.1007/pl00020011. [DOI] [PubMed] [Google Scholar]
- Lund TD, Munson DJ, Haldy ME, Handa RJ. Androgen inhibits, while oestrogen enhances, restraint-induced activation of neuropeptide neurones in the paraventricular nucleus of the hypothalamus. J Neuroendocrinol. 2004;16:272–278. doi: 10.1111/j.0953-8194.2004.01167.x. [DOI] [PubMed] [Google Scholar]
- Lund TD, Munson DJ, Handa RJ. Dihydrotestosterone may inhibit hypothalamo-pituitary-adrenal activity by acting through estrogen receptor in the male mouse. Neurosci Lett , in press. [DOI] [PubMed]
- McCormick CM, Linkroum W, Sallinen BJ, Miller NW. Peripheral and central sex steroids have differential effects on the HPA axis of male and female rats. Stress. 2002;5:235–247. doi: 10.1080/1025389021000061165. [DOI] [PubMed] [Google Scholar]
- Miskowiak B, Lesniewska B, Nowak M, Malendowicz LK. Studies on hypothalamo-pituitary corticoliberin system. V. The effects of gonadectomy and sex hormones on plasma ACTH and on the reactivity of the anterior pituitary gland to CRF. Exp Clin Endocrinol. 1988;92:1–6. doi: 10.1055/s-0029-1210773. [DOI] [PubMed] [Google Scholar]
- Norman RL, Smith CJ, Pappas JD, Hall J. Exposure to ovarian steroids elicits a female pattern of plasma cortisol levels in castrated male macaques. Steroids. 1992;57:37–43. doi: 10.1016/0039-128x(92)90094-p. [DOI] [PubMed] [Google Scholar]
- Nowak KW, Neri G, Nussdorfer GG, Malendowicz LK. Effects of sex hormones on the steroidogenic activity of dispersed adrenocortical cells of the rat adrenal cortex. Life Sci. 1995;57:833–837. doi: 10.1016/0024-3205(95)02015-b. [DOI] [PubMed] [Google Scholar]
- Papadopoulos AD, Wardlaw SL. Testosterone suppresses the response of the hypothalamic-pituitary-adrenal axis to interleukin-6. Neuroimmunomodulation. 2000;8:39–44. doi: 10.1159/000026451. [DOI] [PubMed] [Google Scholar]
- Patchev VK, Almeida OFX. Gonadal steroids exert facilitating and “buffering” effects on glucocorticoid-mediated transcriptional regulation of corticotropin-releasing hormone and corticosteroid receptor genes in rat brain. J Neurosci. 1996;16:7077–7084. doi: 10.1523/JNEUROSCI.16-21-07077.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiffer A, Lapointe B, Barden N. Hormonal regulation of type II glucocorticoid receptor messenger ribonucleic acid in rat brain. Endocrinology. 1991;129:2166–2174. doi: 10.1210/endo-129-4-2166. [DOI] [PubMed] [Google Scholar]
- Polefrone JM, Manuck SB (1987). Gender differences in cardiovascular and neuroendocrine response to stress. In: Barnett RS, Biener L, Baruch GK (eds). Gender and stress The Free Press: New York, NY. pp 13–38.
- Redei E, Li L, Halasz I, McGivern RF, Aird F. Fast glucocorticoid feedback inhibition of ACTH secretion in the ovariectomized rat: effect of chronic estrogen and progesterone. Neuroendocrinology. 1994;60:113–123. doi: 10.1159/000126741. [DOI] [PubMed] [Google Scholar]
- Roca CA, Schmidt PJ, Altemus M, Deuster P, Danaceau MA, Putnam K, et al. Differential menstrual cycle regulation of hypothalamic-pituitary-adrenal axis in women with premenstrual syndrome and controls. J Clin Endocrinol Metab. 2003;88:3057–3063. doi: 10.1210/jc.2002-021570. [DOI] [PubMed] [Google Scholar]
- Roelfsema F, vandenBerg G, Frolich M, Veldhuis JD, vanEljk A, Buurman MM, et al. Sex-dependent alteration in cortisol response to endogenous adrenocorticotropin. J Clin Endocrinol Metab. 1993;77:234–240. doi: 10.1210/jcem.77.1.8392084. [DOI] [PubMed] [Google Scholar]
- Rubin RT, Sekula LK, O’Toole S, Rhodes ME, Czambel RK. Pituitary-adrenal cortical responses to low-dose physostigmine and arginine vasopressin administration in normal women and men. Neuropsychopharmacology. 1999;20:434–446. doi: 10.1016/S0893-133X(98)00077-3. [DOI] [PubMed] [Google Scholar]
- Seeman TE, Singer B, Wilkinson CW, McEwen B. Gender differences in age-related changes in HPA axis reactivity. Psychoneuroendocrinology. 2001;26:225–240. doi: 10.1016/s0306-4530(00)00043-3. [DOI] [PubMed] [Google Scholar]
- Stalvey JRD. Inhibition of 3b-hydroxysteroid dehydrogenase-isomerase in mouse adrenal cells: a direct effect of testosterone. Steroids. 2002;67:721–731. doi: 10.1016/s0039-128x(02)00023-5. [DOI] [PubMed] [Google Scholar]
- Stoney CM, Davis MC, Matthews KA. Sex differences in physiological responses to stress and in coronary heart disease: a causal link? Psychophysiology. 1987;24:127–131. doi: 10.1111/j.1469-8986.1987.tb00264.x. [DOI] [PubMed] [Google Scholar]
- Viau V, Chu A, Soriano L, Dallman MF. Independent and overlapping effects of corticosterone and testosterone on corticotropin-releasing hormone and arginine vasopressin mRNA expression in the paraventricular nucleus of the hypothalamus and stress-induced adrenocorticotropic hormone release. J Neurosci. 1999;19:6684–6693. doi: 10.1523/JNEUROSCI.19-15-06684.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viau V, Meaney MJ. Variations in the hypothalamic-pituitary-adrenal response to stress during the estrous cycle in the rat. Endocrinology. 1991;129:2503–2511. doi: 10.1210/endo-129-5-2503. [DOI] [PubMed] [Google Scholar]
- Viau V, Meaney MJ. The inhibitory effect of testosterone on hypothalamic-pituitary-adrenal responses to stress is mediated by the medial preoptic area. J Neurosci. 1996;16:1866–1876. doi: 10.1523/JNEUROSCI.16-05-01866.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CH, Lundy LE. Radioimmunoassay of plasma estrogens. Steroids. 1971;18:91–111. doi: 10.1016/s0039-128x(71)80174-5. [DOI] [PubMed] [Google Scholar]
- Young EA, Altemus M, Parkinson V, Shastry S. Effects of estrogen antagonists and agonists on the ACTH response to restraint stress in female rats. Neuropsychopharmacology. 2001;25:881–891. doi: 10.1016/S0893-133X(01)00301-3. [DOI] [PubMed] [Google Scholar]