

Abstract
Purpose: Deamidation of lens crystallins and specific deamidation sites have been suggested to be associated with aging and cataracts. However, these studies have been hindered by the lack of suitable quantitative methods of measurement of protein deamidation. We demonstrate herein a method to quantitatively measure deamidation of proteins and peptides without prior sample preparation or separation in order to directly compare the amidated and deamidated forms. We have tested the hypothesis that the 19 mDa mass defect that distinguishes deamidated peptides and proteins from the ordinary natural isotopic species can be utilized for quantitative measurement of their rate and extent of deamidation. The measurement technique used was ion cyclotron resonance Fourier transform mass spectrometry (FTMS), alone with no prior sample preparation or separation. The amidated and deamidated species were recombinantly expressed human eye lens βB2-crystallins and the peptides GlyIleAsnAlaGly and GlyAsnAsnAsnGly. FTMS measurements of lens proteins from a 1-month-old human donor were also carried out.
Methods: Wild type and mutant human eye lens βB2-crystallins with Gln162 replaced by Glu162 were produced in bacteria, and GlyIleAsnAlaGly and GlyAsnAsnAsnGly were synthesized by Merrifield solid-phase peptide synthesis. The peptides were deamidated in pH 7.4, 37.00 °C, 0.15 M Tris-HCl aqueous solution for 18 successive time intervals before analysis. Mutant and wildtype βB2-crystallin solutions at various compositional percentages were mixed and analyzed. The peptides were introduced by electrospray ionization and immediately analyzed in the ion cyclotron resonance (ICR) Fourier transform mass analyzer. Two mass defect analysis procedures were demonstrated for the proteins. In the first, βB2-crystallin was introduced into the mass spectrometer by electrospray ionization and the +29 isotopic group was selectively introduced into the ICR mass analyzer, where 14 residue and 18 residue laser-induced fragments were separated and the extent of deamidation determined by mass defect analysis. In the second, βB2-crystallin was introduced into the mass spectrometer by electrospray ionization and the entire sample was fragmented by collision ionization before introduction into the ICR mass analyzer, where 14 residue fragments were separated and the extent of deamidation determined by mass defect analysis.
Results: The βB2-crystallin mass spectra showed a good quantitative dependence upon extent of deamidation. Direct injection by electrospray ionization followed by ion selection and laser fragmentation or by collision fragmentation produced fragments of amidated and deamidated βB2-crystallin that were appropriate for FTMS quantitative analysis. The two peptides exhibited the expected four deamidation rate curves with acceptable precision.
Conclusions: Mass defect FTMS quantitative analysis of protein deamidation, as reported for the first time herein and illustrated with βB2-crystallin, should prove quite useful. This procedure omits gel separation, chromatography, enzymatic digestion, derivatization, and other procedures that currently add cost and time while degrading quantitative comparison of the amidated and deamidated forms. Mass defect FTMS is also well suited to quantitative deamidation rate studies of peptides. The substantial potential significance of this technique is evident, as example, for lens crystallins where it makes possible quantitative studies of age and disease-dependent deamidation that have heretofore been very difficult. This technique should allow convenient and reliable identification and quantitative measurement of specific deamidation sites that may play a role in aging and cataracts.
The residues of glutamine and asparagine, Gln and Asn, in peptides and proteins are uniquely unstable under physiological conditions. With genetically determined in vivo deamidation half-times ranging from a few hours to more than 100 years, these residues nonenzymatically deamidate into L and D glutaminyl and asparaginyl residues, Glu and Asp, and isoGlu and isoAsp in which the side chain is incorporated into the peptide backbone. The rates of these reactions depend upon peptide and protein structure. At physiological pH, deamidation results in the introduction of a negative charge at the deamidating residue and some isomerization [1].
While peptides and proteins undergo many other postsynthetic changes such as oxidation, phosphorylation, glycosylation, cleavage, and racemization, deamidation is the most prevalent [1]. This is especially true of lens crystallins [2,3]. Each Asn or Gln residue in a protein is a miniature molecular clock that changes protein charge and structure as a function of time. Moreover, deamidation rates are determined by primary, secondary, tertiary, and quaternary structure and require no other reactant species and no enzymatic catalyst. There is no report of any enzyme that catalyzes the deamidation of Asn in the internal sequence of a peptide or protein. It is hypothesized that deamidation serves as a ubiquitous molecular clock for the regulation of biological processes [4-8]. Nonenzymatic deamidation-mediated biological processes have been reported for cytochrome c [9-11], aldolase [12,13], triosephosphate isomerase [14,15], and Bcl-xL [16,17].
Gln deamidates, on average, about 50 times more slowly than Asn, therefore most experimental observations have been of Asn. Eye lens crystallins, however, have very long in vivo lifetimes, so substantial amounts of Gln deamidation are observed [3,18-21]. Therefore, crystallins are excellent proteins in which to study deamidation of Gln. While crystallin deamidation appears to be more prevalent in eye lenses with cataracts [22-24], only a few such specific deamidation sites have been discovered. This is partly due to the difficulty of measuring deamidation quantitatively. Levels of deamidation at specific sites in the adult normal lens are estimated to be approximately 5-20% [21]. Accumulation of low levels of deamidation at multiple sites may lead to cataract formation. Accurate measurement is essential in determining associations between deamidation and function or disease.
Measurements of deamidation in proteins usually involve separations by chromatography, isoelectric focusing, electrophoresis, chemical derivatives, enzymatic digests, or mass spectrometric differentiation of peptide mixtures. These procedures often result in differential losses or responses of the amidated and deamidated species, which spoils quantitation, and they are generally labor-intensive and time-consuming. While extensive quantitative peptide deamidation rate measurements have been made by direct injection mass spectrometry [1,25-27], no similar procedure has been available for proteins.
Deamidation shifts the peptide or protein mass one nominal Dalton higher in the mass spectrum. Therefore, the first deamidated species occupies the same mass position as the first isotopic species, which usually contains a single 2H, 13C, 15N, 17O, or 33S. In ordinary mass spectrometry, the deamidated and isotopic species are not resolved. Similar unresolved deamidated species occur for the higher mass isotopic components.
In studies of single deamidations of small peptides, the relative areas of the isotopic peaks can be estimated and successfully subtracted. This permits peptide deamidation rate measurements that have accuracies of 1% or less [1,25,26]. For proteins, this is much more difficult because there are many more isotopic peaks, and each peak represents more molecular species. Also, for peptides and proteins with more than one deamidating residue, deconvolution and assignment of the resulting deamidation rate measurements to specific residues is difficult.
The masses of the isotopic species and the deamidated species are not, however, identical. Differences in nuclear binding energies lead to “mass defects”. The relevant isotopic mass defects are 12C-13C +3.35, 14N-15N −2.96, 16O-17O +4.22, 16O-18O +4.25, 1H-2H +6.28, 32S-33S −0.61, 32S-34S −4.20, and 32S-36S −4.99 mDa. Thus, with the usual isotopic abundances in peptides, the isotopic envelope has an average mass defect of about +3 mDa.
Deamidation, conversely, has a mass defect of −15.99 mDa. Therefore, the deamidated species occur with a net mass defect of about −19 mDa relative to the isotopic envelope. This allows separation of the deamidated species and the isotopic species in mass spectrometers of sufficient resolution. The 19 mDa defect has been qualitatively demonstrated in a 14 residue peptide and a 16 residue peptide by means of a 4.7 Tesla electrospray Fourier transform mass spectrometer [28].
In the research reported here, we demonstrate that direct injection mass defect FTMS can be used to measure the deamidation rate of a peptide with three deamidating amides (GlyAsnAsnAsnGly) and a peptide with one deamidating amide (GlyIleAsnAlaGly). This demonstration is then extended to show that direct injection mass defect FTMS can be used to quantitatively measure the percentage of deamidation at residue Gln/Glu162 in lens βB2-crystallin, a 204 residue protein.
Alternatively, electron capture dissociation FTMS can be used to detect the isoaspartyl products of Asn deamidation [29], and this, too, may be developed into a quantitative procedure. This technique is valuable because the isoAsp products of deamidation have biological significance and also reflect structural information about the deamidating amide. IsoAsp can also arise from Asp residues through isomerization. The relative percentages of Asp and isoAsp produced during Asn deamidation depend strongly on peptide and protein structure near the Asn. For example, the isoAsp:Asp ratio from deamidation of Asn67 in ribonuclease A varies from 3:1 to 0.3:1 as a function of the structure surrounding the amide [30]. Following deamidation, isoAsp and Asp can equilibrate through an imide mechanism. At equilibrium, the isoAsp:Asp ratio in intact RNAase A is 1:2 rather than 3:1 as is initially observed [31]. The relative biological importances of isoAsp formation compared to negative charge formation during Asn deamidation and the expected parallel products during Gln deamidation are unknown.
In view of the structural variability in isoAsp/Asp ratios produced during deamidation and equilibration and the additional isoAsp source from Asp isomerization, overall quantitative extent and rate of deamidation is probably better measured, where possible, by mass defect FTMS rather than isoAsp FTMS, except where isoAsp values are experimentally required.
Although nonenzymatic deamidation is the most prevalent and probably most biologically significant postsynthetic change in proteins, it is studied less frequently than other modifications because it is relatively difficult to measure. Deamidation measurements are especially difficult for eye lens crystallins because, during their long residence in vivo, additional modifications such as oxidation, truncation, racemization, and aggregation increase heterogeneity. Deamidation is a particularly important phenomenon in the lens where deamidation increases with age and the accumulation of multiple sites of deamidation may significantly destabilize lens proteins, contributing to cataracts. Direct injection fragmentation mass defect FTMS offers a powerful, high resolution solution to this problem. The purpose of these experiments is to demonstrate the feasibility of this technique.
METHODS
Preparation of βB2-crystallin: The 204 residue protein βB2-crystallin and the site-specific mutant βB2-crystallin in which Gln162 had been changed to Glu162 were synthesized in bacteria as previously described [32]. The expression plasmid pET3a, βB2 was kindly provided by Drs. Nicolette Lubsen (University of Nijmegen) and Orval Bateman (University of London). Wild type βB2Gln162 and mutant βB2Glu162 were purified from E. coli protein by anion-exchange chromatography to concentrations of 1.82 mg/ml and 1.50 mg/ml, respectively, in 0.3 M ammonium sulfate, 0.001 M EDTA, 0.002 M DTT, and 0.1 M pH 7.0 sodium phosphate buffer. Mixtures of these two preparations were prepared by combination and dilution in 49/49/2 H2O/methanol/acetic acid to give a concentration of 0.0075 mg/ml for the sum of βB2Gln162 and βB2Glu162.
Preparation of pentapeptides: GlyIleAsnAlaGly and GlyAsnAsnAsnGly were synthesized by Merrifield solid-phase peptide synthesis and purified as previously described [25,26]. The two peptides were incubated at 37.00 °C in pH 7.4, 0.15 M Tris-HCl for 102 days and 85 days, respectively. Each of 18 100 μl incubated samples was removed from 37.00 °C and frozen at −80 °C after a specified time interval. After all 18 samples had been frozen, each sample was diluted to 2×10−6 M with H2O. These 2×10−6 M solutions were frozen at −80 °C until further diluted 3 fold in 49/49/2 H2O/methanol/acetic acid for GlyIleAsnAlaGly and 10 fold for GlyAsnAsnAsnGly before FTMS analysis.
Fourier transform mass spectrometry (FTMS) protocols: In the first procedure, the βB2-crystallins were measured in an IonSpec QFT-9 FTMS 9.4 Tesla mass spectrometer manufactured by IonSpec, Lake Forest, CA. The acidified samples were introduced into a Z-type electrospray source at a flow rate of 5 μl/min. The m/z 803 protein ion with charge state +29 was isolated in the second quadrupole and accumulated in the third quadrupole before injection into the ion cyclotron resonance (ICR) cell. Inside the ICR cell, the +29 ion was irradiated with a 25 W continuous wave CO2 laser at 20% of full intensity, which produced several fragments including a +2 charge 18 residue fragment at m/z 1051.5 and a +2 charge 14 residue fragment at m/z 795.9. ProGlyTyrArgGlyLeuGln162 TyrLeuLeuGluLysGlyAspTyrLysAspSer mass 2101.0 and ProGlyTyrArgGlyLeuGln162 TyrLeuLeuGluLysGlyAsp mass 1589.8 were the corresponding peptides. The corresponding Glu162 fragments were also observed at 0.5 nominal m/z higher in the spectra. These species were measured with an average resolving power of about 200,000, which was sufficient to resolve the mass defect peaks.
In the second procedure, the βB2-crystallins were measured in a Bruker Daltonics Apex IV 70e FTMS 7.0 Tesla mass spectrometer manufactured by Bruker Daltonics, Inc., Billerica, MA. The samples were introduced into a Bruker electrospray source. The entire sample was fragmented in a low pressure atmospheric collision cell before being introduced into the ICR cell. The +2 charge 14 residue fragment at m/z 795.9 corresponding to peptide ProGlyTyrArgGlyLeuGln162 TyrLeuLeuGluLysGlyAsp mass 1589.8 and the analagous Glu162 fragment were analyzed. These measurements were made with an average resolving power of about 200,000, which was sufficient to resolve the mass defect peaks.
GlyAsnAsnAsnGly was analyzed in the IonSpec QFT-9 FTMS 9.4 Tesla mass spectrometer, but without fragmentation. GlyIleAsnAlaGly was similarly analyzed in a Bruker Daltonics Apex IV 70e FTMS 7.0 Tesla mass spectrometer without fragmentation.
RESULTS
Rate of deamidation of GlyIleAsnAlaGly: Figure 1 shows the mass spectrum at the 30 day deamidation point of GlyIleAsnAlaGly. Simple peak heights from two FTMS runs were averaged to provide the amidated and deamidated values for the rate of deamidation plot shown in Figure 2. The first-order rate curve shown in Figure 2 is linear throughout the entire deamidation rate experiment, thus demonstrating the utility of this technique. Peak height values for the first isotope peak of the amidated peptide and for the corresponding deamidated peptide, resolved by mass defect from the second isotope peak, were used for Figure 2.
Figure 1.
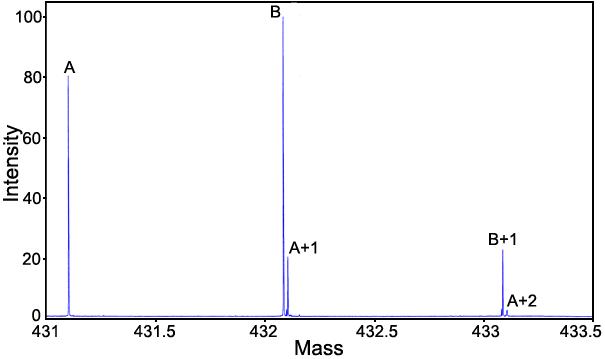
Mass spectrum of the 30 day deamidation sample of GlyIleAsnAlaGly. The peptide was incubated in 37.00 °C, pH 7.4, 0.15 M Tris-HCl buffer for 54 days. It was diluted 100 fold in H2O and then further diluted 3 fold with 49/49/2 H2O/methanol/acetic acid before being introducted by electrospray into the Fourier transform mass spectrometer. Peak A is the first isotopic peak. Peak B is the deamidation product from deamidation of the first isotopic peak. Peak A+1 of the doublet is the second isotopic peak.
Figure 2.
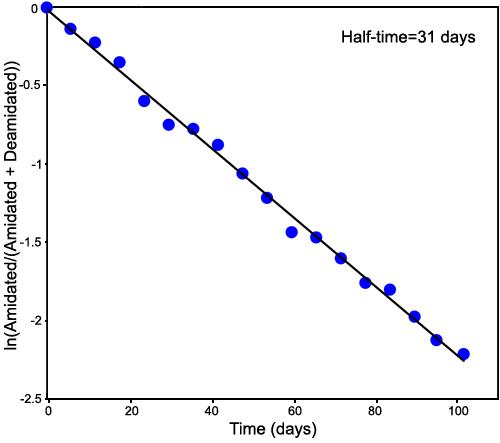
Deamidation of GlyIleAsnAlaGly. First-order rate of deamidation plot of the deamidation of GlyIleAsnAlaGly in 37.00 °C, pH 7.4, 0.15 M Tris-HCl buffer as determined by direct injection mass defect Fourier transform mass spectrometer. The plot shows the expected linearity over the entire 102 days of the experiment. The deamidation half-time is 31 days.
Rate of deamidation of GlyAsnAsnAsnGly: Figure 3 shows the mass spectrum of the 5 day and 85 day samples in the 18-point first-order deamidation rate curve for GlyAsnAsnAsnGly. Figure 4, Figure 5, and Figure 6 show “first-order” rate curves for the first, second, and third deamidations of this triple amide peptide.
Figure 3.
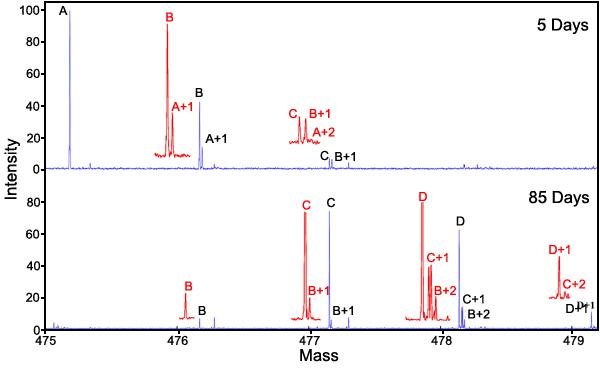
Mass spectrum of the deamidation samples at 5 and 85 days of GlyAsnAsnAsnGly. The peptide was incubated in 37.00 °C, pH 7.4, 0.15 M Tris-HCl buffer. It was diluted 100 fold in H2O and then further diluted 3 fold with 49/49/2 H2O/methanol/acetic acid before being introducted by electrospray into the Fourier transform mass spectrometer. Peak A is the first isotopic peak. Peak B is the deamidation product from deamidation of the first isotopic peak. Peak A+1 of the doublet is the second isotopic peak. Peak C is the second deamidation peak of the first isotopic peak, peak B+1 is the first deamidation peak of the second isotopic peak, and peak A+2 is the third isotopic peak. The quadruplet shows the third deamidation peak of the first isotopic peak, the second deamidation product of the second isotope peak, the first deamidation product of the third isotope peak, and the fourth isotopic peak. Only one mass defect peak is observed for the fifth isotopic peak quintuplet because the amounts of deamidated products were low. Insets are magnified 2.5x.
Figure 4.
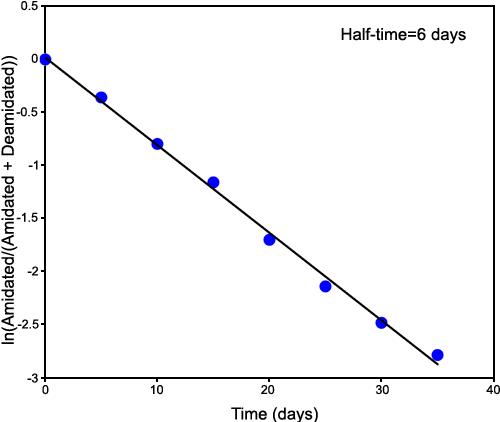
First deamidation of GlyAsnAsnAsnGly. The first-order deamidation rate curve for single deamidation of GlyAsnAsnAsnGly, using deamidation of the first isotopic cluster. This figure is ln[A/(A+B+C+D)] versus time. The measured deamidation half-time is six days. The quantitative linearity and zero deamidation intercept of the plot demonstrate that this deamidation has been accurately and separately measured by the mass defect ICR FTMS technique.
Figure 5.
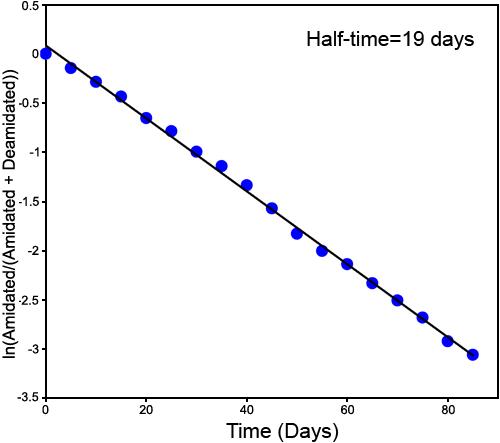
Second deamidation of GlyAsnAsnAsnGly. The first-order deamidation rate curve for the second deamidation of GlyAsnAsnAsnGly, using deamidation of the first singly deamidated peak. This figure is ln[B/(B+C+D)] versus time. The measured deamidation half-time is 19 days. The quantitative linearity and zero deamidation intercept of the plot demonstrate that this deamidation has been accurately and separately measured by the mass defect ICR FTMS technique.
Figure 6.
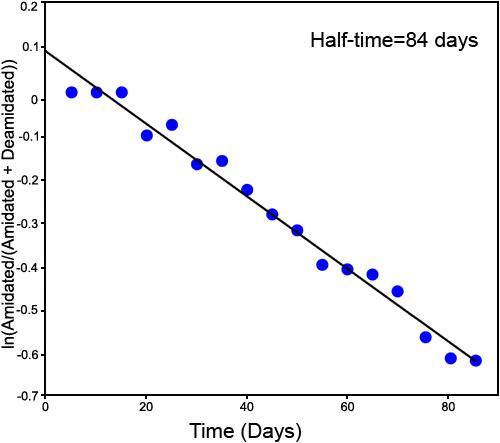
Third deamidation of GlyAsnAsnAsnGly. The first-order deamidation rate curve for the third deamidation of GlyAsnAsnAsnGly, using deamidation of the second doubly deamidated peak. This figure is ln[C/(C+D)] versus time. The measured deamidation half-time is 84 days. The linearity and deamidation intercept of the plot demonstrates that this deamidation has been separately measured with moderate accuracy by the mass defect ICR FTMS technique. Heterogeneity arising from earlier deamidations and the longer deamidation half-time as compared with the sampling interval have diminished this accuracy as compared with that of the first two deamidations.
Assuming, within experimental error, the apparent linearity of these first-order curves for the duration of the experiment, Figure 4, Figure 5, and Figure 6 provide direct measurement of the deamidation indexes of the peptide. By definition [1], the deamidation coefficient is given by
and the deamidation index is given by
This experiment gives 0.06, 0.19, and 0.84 for deamidation indexes ID1, ID2, and ID3. So, the overall deamidation half-times for 1, 2, and 3 deamidations of GlyAsnAsnAsnGly are 6, 19, and 84 days, respectively.
Figure 3 illustrates the peak labeling. A, A+1, and A+2 are the first three isotopic envelopes. Shifted -19 mDa to lower mass from A+1, A+2 and A+3 are singly deamidated peaks B, B+1, and B+2; shifted -38 mDa to lower mass are C, C+1, and C+2; and shifted -57 mDa to lower mass are D and D+1. Figure 4 plots ln[A/(A+B+C+D)], Figure 5 plots ln[B/(B+C+D)], and Figure 6 plots ln[C/(C+D)]; all are plotted against time. Peak areas were computed and used for this analysis.
While the GlyAsnAsnAsnGly deamidation indexes are easy to calculate, the individual deamidations lead to many deamidation coefficients. Ignoring D isomers, the first deamidation involves six deamidation reactions and six singly deamidated products, the L-Asp and isoAsp products at residues 2, 3, and 4 [1]. The second deamidation involves six singly deamidated species deamidating to nine doubly deamidated products by means of twelve different deamidation reactions. The third deamidation involves nine doubly deamidated species deamidating to eight triply deamidated products by means of eight deamidation reactions. The rates of these 26 deamidation reactions cannot be separately measured by this experiment. The overall deamidation indexes and corresponding rates are, however, easily determined.
In crystallins and other proteins where multiple deamidations occur, the use of FTMS of fragments containing single amides will avoid the complication of multiple amides on one fragment. There are, however, instances where the amides appear so close in the protein sequence that the method shown here for GlyAsnAsnAsnGly will be useful. For example, in βB1-crystallin, there are Gln residues at sequence positions 222, 224, and 226, and in βB2-crystallin, there are Gln residues at positions 182 and 184.
While excellent first order rate curves have been obtained for GlyIleAsnAlaGly by means of direct injection low resolution mass spectrometry [25], wherein the unresolved isotopic contributions were subtracted by computation, attempts to resolve all three deamidations in low resolution rate measurements of GlyAsnAsnAsnGly by means of computation have proved much more difficult (Robinson NE and Robinson AB, unpublished observations). Figure 3, Figure 4, Figure 5, and Figure 6 show that direct injection mass defect FTMS provides a solution to this problem.
Extent of deamidation of βB2-crystallin: Figure 7 shows the mass spectra of the mass defect separations of the 18 residue crystallin fragments produced by laser fragmentation in an IonSpec QFT-9 FTMS 9.4 Tesla mass spectrometer for a 70% deamidated mixture of wild type Gln162 βB2-crystallin and mutant Glu162 βB2-crystallin.
Figure 7.
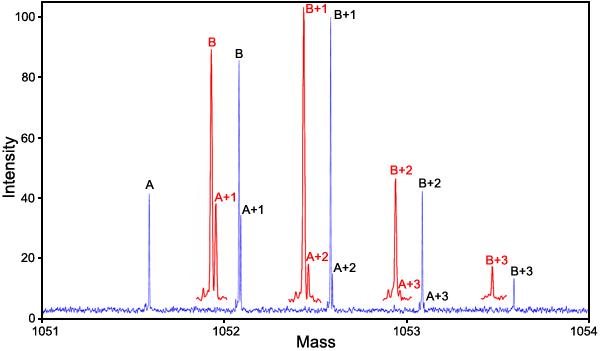
Mass spectrum of the Glu162 mutant and Gln162 wild type βB2-crystallin. Mass spectrum of the 70% Glu162 mutant and 30% Gln162 wild type of human eye lens βB2-crystallin. This mixture was diluted to 0.0075 mg/ml total protein with 49/49/2 H2O/methanol/acetic acid. This acidified sample was introduced into a Z-type electrospray source at a flow rate of 5 μl/min. The 803 Da protein ion with charge state +29 was isolated in the second quadrupole and accumulated in the third quadrupole before being injected into the ion cyclotron resonance cell. In the analyzer cell, the +29 ion was irradiated with a 25 W continuous wave CO2 laser at 20% of full intensity, which produced several fragments including the 18 residue and 14 residue fragments that contained sequence position Q/E162 and were used for quantitative analysis. The +2 charge, m/z=1051.5, 18 residue PGYRGLQ/E162YLLEKGDYKDS fragment is illustrated here. This analysis was carried out in an IonSpec QFT-9 Fourier transform mass spectrometer with ten 20 s Fourier transform scans. Red insets are magnified horizontally 2.5x.
Figure 8 shows the expected and measured deamidation percentages for laser fragmentation mass defect measurements using the first procedure for both the 14 and 18 residue fragments. These values exhibit a sigmoidal character that is thought to arise from nonlinearity in quantitative response of ion detection in the ICR cell. Ions present in lower quantities tend to be detected with lower efficiencies. In quantitative use, this effect can be corrected with a standard curve measured for the instrument and instrument settings in use. Alternatively, a correction can be made as is illustrated in Figure 9.
Figure 8.
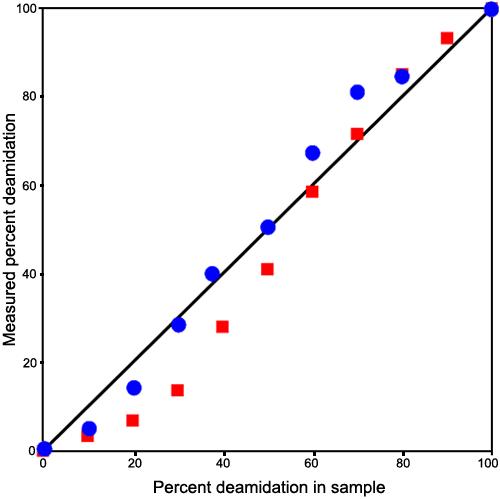
Plot of the expected and measured deamidation percentages for laser fragmentation. Plot of the expected and measured deamidation percentages for laser fragmentation, mass defect measurements using the first procedure. The red squares identify the 18 residue PGYRGLQ/E162YLLEKGDYKDS fragments and the blue circles identify the 14 residue PGYRGLQ/E162YLLEKGD fragments. These values exhibit a sigmoidal character that arises from nonlinearity in quantitative response of ion detection in the ion cyclotron resonance cell. Ions present in lower quantities are detected with lower efficiencies. In quantitative use, this effect can be corrected with a standard curve measured for the instrument and settings used. Alternatively, a correction can be made as is illustrated in Figure 10.
Figure 9.
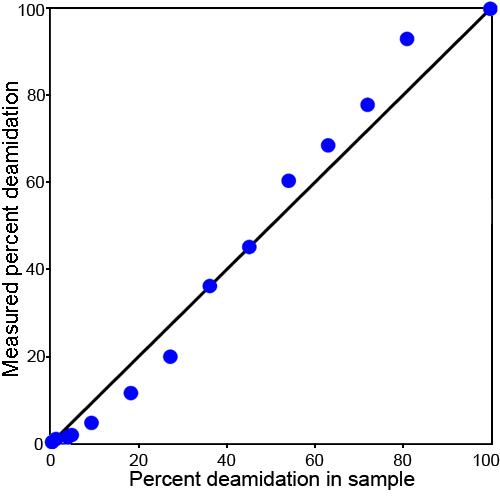
Plot of the expected and measured deamidation percentages for collision fragmentation. Plot of the expected and measured deamidation percentages for collision fragmentation, mass defect measurements using the second procedure. The blue circles identify the 14 residue PGYRGLQ/E162YLLEKGD fragment. These measurements also show the sigmoidal characteristic seen in Figure 8.
Figure 9 shows the expected and measured deamidation percentages for collision fragmentation, mass defect measurements using the second procedure and again showing the sigmoidal characteristic seen in Figure 8. In Figure 10, a correction for the nonlinearity observed in Figure 9 has been applied. This correction was calculated by means of the isotopic peak ratios of the measured isotopic peaks in these same spectra. As illustrated in Figure 7, the mass defect peaks are resolved from the isotopic peaks. Since the theoretical ratios of the isotopic peaks are known and these peaks vary in magnitude, a correction curve for ion abundance can be calculated from them. This provides an internal standard with which to correct the nonlinear ion response.
Figure 10.
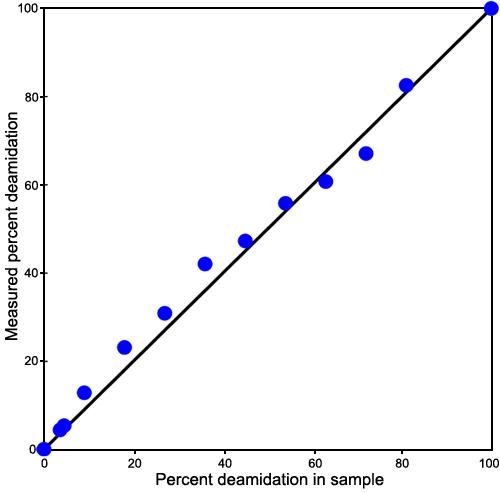
Detection-efficiency-corrected plot of the expected and measured deamidation percentages for collision fragmentation. Plot of the expected and measured deamidation percentages for collision fragmentation, mass defect measurements using the second procedure and with an applied correction for the nonlinearity observed in Figure 9. This correction was calculated by means of the isotopic peak ratios of the measured isotopic peaks in these same spectra. As illustrated in Figure 7, the mass defect peaks are resolved from the isotopic peaks. Since the theoretical ratios of the isotopic peaks are known and these peaks vary in magnitude, a correction curve for ion abundance can be calculated from them. A plot is made of the ratio of actual isotopic abundances to the observed isotopic abundances as a function of peak sizes. This provides a correction factor by means of an internal standard with which to correct the nonlinear ion response.
In principle, the deamidation of any one isotopic species is sufficient to determine deamidation, but isotopic redundancy improves measurement accuracy. In Figure 8, mass defect pairs A and B, A+1 and B+1, and A+2 and B+2 were calculated and averaged to obtain the percentages of deamidation in the m/z 1051.5 fragment, and mass defect pairs A and B and A+1 and B+1 were used to obtain percentages for the m/z 795.9 fragment. Mass defect pairs A and B, A+1 and B+1, and A+2 and B+2 were used for the m/z 795.9 fragment in Figure 9 and Figure 10.
While the actual application of this mass defect method to the solution of deamidation problems in biological lens research is beyond the scope of this report, the assertion that these methods can be applied to biological samples without prior sample preparation is illustrated in Figure 11. Figure 11 shows the ICR FTMS spectrum of the soluble fraction of an eye lens from a 1-month-old human donor obtained from the Lions Eye Bank of Oregon. The lens was simply homogenized in 1.0 ml of 0.02 M phosphate buffer at pH 7.0 with 0.001 M EDTA, centrifuged to remove the insoluble protein fraction, and desalted by repeated H2O centrifugal washing over a 3,000 Dalton filter. It was then diluted with 49/49/2 H2O/methanol/acetic acid before introduction into the mass spectrometer. Electronanospray was used for injection in order to conserve sample. The separations shown were carried out in the ICR FTMS cell.
Figure 11.
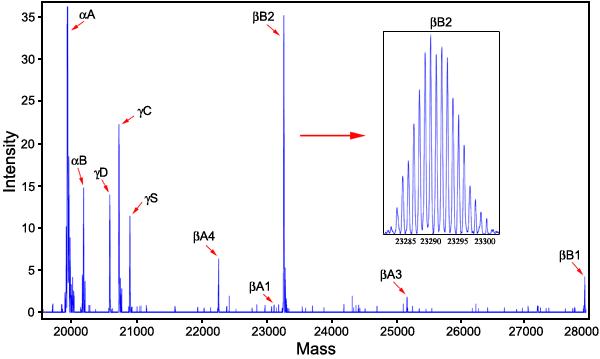
The ion cyclotron resonance Fourier transform mass spectrometry spectrum of the soluble fraction of an eye lens from a 1-month-old human subject. The lens was homogenized in 1.0 ml of 0.02 M phosphate buffer at pH 7.0 with 0.001 M EDTA, centrifuged to remove the insoluble protein fraction, and desalted by repeated H2O centrifugal washing over a 3,000 Dalton filter. It was then diluted with 49/49/2 H2O/methanol/acetic acid before being introduced into the mass spectrometer. The separations shown were carried out in the ICR FTMS cell. The labeled crystallin fractions are the ten most prevalent proteins known to exist in the young human lens [33].
For purposes of illustration, the various charge state isotopic distributions of all protein components detected in this 1-month-old human lens sample have been combined and plotted at their average natural isotopic masses. The 10 crystallins known to be present in highest quantities in newborn human lenses are all clearly resolved and easily available for mass defect analysis without further purification.
As humans age, many postsynthetic changes occur in these lens proteins. As the protein mixture becomes more complex with age, some prior purification may become necessary. Simple chromatography of whole proteins, especially at an acid pH, prior to electrospray injection would still preserve the quantitative features of this mass defect technique.
The most important quantitative features of the ICR FTMS analytical procedure illustrated here are that the whole proteins are injected into the mass spectrometer before fragmentation and that the deamidated molecules give rise to resolved peaks that can be independently observed and measured. Injection of whole proteins, especially at an acid pH, assures that artifactual differential changes in the ratios of amidated and deamidated fractions do not occur, as is often the case, for example, with enzymatic digestion followed by wet chemistry separations and mass spectrometric injection. The detection of a separately resolved mass defect component permits detection of very low percentage amounts of deamidation as compared with methods that depend upon isotopic envelopes and therefore subtracted differences between large components.
DISCUSSION
Research on the deamidation of peptides and proteins has proceeded relatively slowly, largely because the usual wet chemistry and mass spectroscopic methods for measuring peptide and protein deamidation are slow, costly, and often only semiquantitative [1]. This has been overcome for single deamidations of relatively small peptides by direct injection low resolution mass spectrometry followed by computational removal of isotopic effects [25]. This method is more difficult for long peptides, peptides with multiple amides, and proteins, especially where low levels of deamidation are present.
Eye lens protein deamidation has posed an especially difficult problem because of the long crystallin in vivo lifetimes, which provide time for the accumulation of many other postsynthetic modifications that further complicate deamidation measurements.
The experiments performed herein demonstrate that direct electrospray injection and fragmentation in a Fourier transform ion cyclotron resonance mass spectrometer provides an excellent solution for this problem. The peptide deamidation rate measurements show good quantitative results with as many as three simultaneously deamidating amides in the same peptide. This is especially valuable in lens crystallins where several structurally important potential deamidation sites occur in clusters.
The βB2-crystallin measurements show substantial utility for direct protein measurements. It is especially noteworthy that this method can be used even if the crystallin has undergone other modifications such as end-section chain cleavages. A wide range of laser-induced peptide fragments is available, so a peptide fragment for FTMS that does not also contain another postsynthetic change is likely to be found. Current enzymatic mass spectrometric methods are restricted to a small variety of fragments and, since fragmentation occurs outside of the mass spectrometer, are subject to inaccuracies due to differential enzymatic digestion, purification, and ionization.
Current methods of measurement of crystallin deamidation involve gel separations, chromatography, derivatizations, enzymatic digestions, electrospray ionization of peptides, and other procedures that often cause differential losses or responses of amidated and deamidated species, thereby spoiling quantitation. This new mass defect FTMS technique wherein the whole proteins are not fragmented until they have been injected into the mass spectrometer avoids these complications and is also much more convenient.
We conclude that FTMS as herein described is an excellent technique for the quantitative measurement of deamidation of crystallins in particular and peptides and proteins in general.
ACKNOWLEDGEMENTS
We thank IonSpec Corporation and Bruker Corporation for the use of their FTMS instruments, National Institutes of Health grant EY 12239 to KJL, and grants to the Oregon Institute of Science and Medicine by the Kinsman Foundation and the Morse Foundation for support of this work.
REFERENCES
- 1.Robinson NE, Robinson AB. Althouse Press; Cave Junction (OR): 2004. Molecular clocks: deamidation of asparaginyl and glutaminyl residues in peptides and proteins. [Google Scholar]
- 2.Lampi KJ, Ma Z, Hanson SR, Azuma M, Shih M, Shearer TR, Smith DL, Smith JB, David LL. Age-related changes in human lens crystallins identified by two-dimensional electrophoresis and mass spectrometry. Exp Eye Res. 1998;67:31–43. doi: 10.1006/exer.1998.0481. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Z, Smith DL, Smith JB. Human beta-crystallins modified by backbone cleavage, deamidation and oxidation are prone to associate. Exp Eye Res. 2003;77:259–72. doi: 10.1016/s0014-4835(03)00159-3. [DOI] [PubMed] [Google Scholar]
- 4.Robinson AB, McKerrow JH, Cary P. Controlled deamidation of peptides and proteins: an experimental hazard and a possible biological timer. Proc Natl Acad Sci U S A. 1970;66:753–7. doi: 10.1073/pnas.66.3.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robinson AB, Rudd CJ. Deamidation of glutaminyl and asparaginyl residues in peptides and proteins. Curr Top Cell Regul. 1974;8:247–95. doi: 10.1016/b978-0-12-152808-9.50013-4. [DOI] [PubMed] [Google Scholar]
- 6.Robinson NE, Robinson AB. Deamidation of human proteins. Proc Natl Acad Sci U S A. 2001;98:12409–13. doi: 10.1073/pnas.221463198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robinson NE. Protein deamidation. Proc Natl Acad Sci U S A. 2002;99:5283–8. doi: 10.1073/pnas.082102799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robinson NE, Robinson AB. Amide molecular clocks in drosophila proteins: potential regulators of aging and other processes. Mech Ageing Dev. 2004;125:259–67. doi: 10.1016/j.mad.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 9.Flatmark T. Multiple molecular forms of bovine heart cytochrome c. V. A comparative study of their physicochemical properties and their reactions in biological systems. J Biol Chem. 1967;242:2454–9. [PubMed] [Google Scholar]
- 10.Flatmark T, Sletten K. Multiple forms of cytochrome c in the rat. Precursor-product relationship between the main component Cy I and the minor components Cy II and Cy 3 in vivo. J Biol Chem. 1968;243:1623–9. [PubMed] [Google Scholar]
- 11.Robinson AB, McKerrow JH, Legaz M. Sequence dependent deamidation rates for model peptides of cytochrome C. Int J Pept Protein Res. 1974;6:31–5. doi: 10.1111/j.1399-3011.1974.tb02355.x. [DOI] [PubMed] [Google Scholar]
- 12.Midelfort CF, Mehler AH. Deamidation in vivo of an asparagine residue of rabbit muscle aldolase. Proc Natl Acad Sci U S A. 1972;69:1816–9. doi: 10.1073/pnas.69.7.1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKerrow JH, Robinson AB. Primary sequence dependence of the deamidation of rabbit muscle aldolase. Science. 1974;183:85. doi: 10.1126/science.183.4120.85. [DOI] [PubMed] [Google Scholar]
- 14.Gracy RW, Lu HS, Yuan PM, Talent JM. Structural analysis of altered proteins. In: Adelman RC, Roth GS, editors. Altered Proteins and Aging. CRC Press; Boca Raton (FL): 1983. pp. 27–34. [Google Scholar]
- 15.Gracy RW, Talent JM, Zvaigzne AI. Molecular wear and tear leads to terminal marking and the unstable isoforms of aging. J Exp Zool. 1998;282:18–27. [PubMed] [Google Scholar]
- 16.Deverman BE, Cook BL, Manson SR, Niederhoff RA, Langer EM, Rosova I, Kulans LA, Fu X, Weinberg JS, Heinecke JW, Roth KA, Weintraub SJ. Bcl-xL deamidation is a critical switch in the regulation of the response to DNA damage. Cell. 2002;111:51–62. doi: 10.1016/s0092-8674(02)00972-8. Erratum in: Cell. 2003; 115:503. [DOI] [PubMed] [Google Scholar]
- 17.Weintraub SJ, Manson SR, Deverman BE. Resistance to antineoplastic therapy. The oncogenic tyrosine kinase-Bcl-x(L) axis. Cancer Cell. 2004;5:3–4. doi: 10.1016/s1535-6108(03)00338-6. [DOI] [PubMed] [Google Scholar]
- 18.Takemoto L, Boyle D. Deamidation of specific glutamine residues from alpha-A crystallin during aging of the human lens. Biochemistry. 1998;37:13681–5. doi: 10.1021/bi981542k. [DOI] [PubMed] [Google Scholar]
- 19.Lapko VN, Purkiss AG, Smith DL, Smith JB. Deamidation in human gamma S-crystallin from cataractous lenses is influenced by surface exposure. Biochemistry. 2002;41:8638–48. doi: 10.1021/bi015924t. [DOI] [PubMed] [Google Scholar]
- 20.Lampi KJ, Kim YH, Bachinger HP, Boswell BA, Lindner RA, Carver JA, Shearer TR, David LL, Kapfer DM. Decreased heat stability and increased chaperone requirement of modified human betaB1-crystallins. Mol Vis. 2002;8:359–66. [PubMed] [Google Scholar]
- 21.Harms MJ, Wilmarth PA, Kapfer DM, Steel EA, David LL, Bachinger HP, Lampi KJ. Laser light-scattering evidence for an altered association of beta B1-crystallin deamidated in the connecting peptide. Protein Sci. 2004;13:678–86. doi: 10.1110/ps.03427504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takemoto L, Boyle D. Increased deamidation of asparagine during human senile cataractogenesis. Mol Vis. 2000;6:164–8. [PubMed] [Google Scholar]
- 23.Srivastava OP, Srivastava K. Existence of deamidated alphaBcrystallin fragments in normal and cataractous human lenses. Mol Vis. 2003;9:110–8. [PubMed] [Google Scholar]
- 24.Searle BC, Dasari S, Wilmarth PA, Turner M, Reddy AP, David LL, Nagalla SR. Identification of protein modifications using MS/MS de novo sequencing and the OpenSea alignment algorithm. J Proteome Res. 2005 Mar-Apr;4:546–54. doi: 10.1021/pr049781j. [DOI] [PubMed] [Google Scholar]
- 25.Robinson NE, Robinson AB. Molecular clocks. Proc Natl Acad Sci U S A. 2001;98:944–9. doi: 10.1073/pnas.98.3.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robinson NE, Robinson AB, Merrifield RB. Mass spectrometric evaluation of synthetic peptides as primary structure models for peptide and protein deamidation. J Pept Res. 2001;57:483–93. doi: 10.1034/j.1399-3011.2001.00863.x. [DOI] [PubMed] [Google Scholar]
- 27.Robinson NE, Robinson ZW, Robinson BR, Robinson AL, Robinson JA, Robinson ML, Robinson AB. Structure-dependent nonenzymatic deamidation of glutaminyl and asparaginyl pentapeptides. J Pept Res. 2004;63:426–36. doi: 10.1111/j.1399-3011.2004.00151.x. [DOI] [PubMed] [Google Scholar]
- 28.Schmid DG, von der Mulbe FD, Fleckenstein B, Weinschenk T, Jung G. Broadband detection electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry to reveal enzymatically and chemically induced deamidation reactions within peptides. Anal Chem. 2001;73:6008–13. doi: 10.1021/ac0104274. [DOI] [PubMed] [Google Scholar]
- 29.Cournoyer JJ, Pittman JL, Ivleva VB, Fallows E, Waskell L, Costello CE, O'Connor PB. Deamidation: Differentiation of aspartyl from isoaspartyl products in peptides by electron capture dissociation. Protein Sci. 2005;14:452–63. doi: 10.1110/ps.041062905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Capasso S, Balboni G, Di Cerbo P. Effect of lysine residues on the deamidation reaction of asparagine side chains. Biopolymers. 2000;53:213–9. doi: 10.1002/(SICI)1097-0282(200002)53:2<213::AID-BIP11>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 31.Capasso S, Di Cerbo P. Kinetic and thermodynamic control of the relative yield of the deamidation of asparagine and isomerization of aspartic acid residues. J Pept Res. 2000;56:382–7. doi: 10.1034/j.1399-3011.2000.00778.x. [DOI] [PubMed] [Google Scholar]
- 32.Lampi KJ, Oxford JT, Bachinger HP, Shearer TR, David LL, Kapfer DM. Deamidation of human beta B1 alters the elongated structure of the dimer. Exp Eye Res. 2001;72:279–88. doi: 10.1006/exer.2000.0950. [DOI] [PubMed] [Google Scholar]
- 33.Lampi KJ, Ma Z, Shih M, Shearer TR, Smith JB, Smith DL, David LL. Sequence analysis of betaA3, betaB3, and betaA4 crystallins completes the identification of the major proteins in young human lens. J Biol Chem. 1997;272:2268–75. doi: 10.1074/jbc.272.4.2268. [DOI] [PubMed] [Google Scholar]