

Abstract
Bacterial surface capsular polysaccharides (CPS) that are similar in carbohydrate sequence may differ markedly in immunogenicity and antigenicity. The structural origin of these phenomena is poorly understood. Such a case is presented by the Gram-positive bacteria Streptococcus agalactiae (Group B Streptococcus; GBS) type III (GBSIII) and Streptococcus pneumoniae (Pn) type 14 (Pn14), which share closely related CPS sequences. Nevertheless, antibodies (Abs) against GBSIII rarely cross-react with the CPS from Pn14. To establish the origin for the variation in CPS antigenicity, models for the immune complexes of CPS fragments from GBSIII and Pn14, with the variable fragment (Fv) of a GBS-specific mAb (mAb 1B1), are presented. The complexes are generated through a combination of comparative Ab modeling and automated ligand docking, followed by explicitly solvated 10-ns molecular dynamics simulations. The relationship between carbohydrate sequence and antigenicity is further quantified through the computation of interaction energies using the Molecular Mechanics–Generalized Born Surface Area (MM-GBSA) method, augmented by conformational entropy estimates. Despite the electrostatic differences between Pn14 and GBSIII CPS, analysis indicates that entropic penalties are primarily responsible for the loss of affinity of the highly flexible Pn14 CPS for mAb 1B1. The similarity of the solution conformation of the relatively rigid GBSIII CPS with that in the immune complex characterizes the previously undescribed 3D structure of the conformational epitope. The analysis provides a comprehensive interpretation for a large body of biochemical and immunological data related to Ab recognition of bacterial polysaccharides and should be applicable to other Ab–carbohydrate interactions.
Keywords: amber, capsular polysaccharide, glycam, molecular dynamics, Group B Streptococcus
Streptococcus agalactiae [Group B Streptococcus (GBS)] and Streptococcus pneumoniae (Pn) are responsible for the majority of life-threatening cases of septicemia, meningitis, and pneumonia in neonates (1, 2). Gram-positive bacteria, such as GBS and Pn, are classified into serotypes according to the unique carbohydrate sequence of the bacterial surface capsular polysaccharide (CPS) and protein antigens. Serotypes vary in antigenicity, immunogenicity, virulence, and geographical distribution (3). Quantification of the structural and dynamic properties responsible for the affinity and specificity of antigenic oligosaccharide–antibody (Ab) interactions is a crucial step in furthering the understanding of the immune response to bacterial and fungal pathogens. In GBS, the CPS is a high-molecular-weight polymer composed of varying sequences of β-d-galactopyranose (β-d-Galp), β-d-glucopyranose (β-d-Glcp), β-d-N-acetylglucosamine (β-d-GlcpNAc), α-N-acetylneuraminic acid (α-Neu5Ac), and sometimes l-rhamnopyranose. The glyceryl side chain of the Neu5Ac residues may also be O-acetylated (4). In all GBS strains identified to date, the Neu5Ac residues occur in the terminal position on the side-chain branches of the polymeric repeat unit of the CPS. They play an important role in defining the antigenicity and immunogenicity of the CPS (5). Variations within the CPS sequence result in the nine known GBS serotypes: Ia, Ib, and II–VIII; among these GBS type III (GBSIII) is the most virulent and prevalent in North America (6). Despite exhibiting similar carbohydrate sequences, the GBS polysaccharides (PS) are type-specific and immunologically distinct, lacking cross-reactivity (6).
In contrast to GBS, the CPS sequences and linkages in Pn are highly heterogeneous. The antigenicity of the pneumococcal CPS is generally type-specific, but cross-reactions with GBS may occur presumably as a result of the sequential similarity of their PS (7). Notably, the CPS in both GBSIII and Pn14 share a common PS backbone and differ only in the absence of α-Neu5Ac in the side chain of Pn14. The nature of the antigenic epitope in GBSIII has been the subject of considerable study; however, the structural basis for the differing antigenicities between GBSIII and Pn14 remains unknown. Binding studies and inhibition data (8–10) indicate that the epitope of the GBSIII CPS consists of three to seven repeat units of the branched pentasaccharide [-4)-β-d-Glcp-(1–6)-β-d-GlcpNAc-[α-d-Neu5Ac-(2–3)-β-d-Galp-(1–4)-]-(1–3)-β-d-Galp-(1-]. The immunodominant core of the oligosaccharide has been identified as the -β-d-Galp-(1–4)-β-d-Glcp- sequence (8). Because of the extreme length of this epitope, a conformational dependence has been postulated (9, 10).
NMR spectroscopic and computational studies of the GBSIII CPS indicate that the PS adopts on average a helical conformation, in which the Neu5Ac residues are displayed on the surface of the helix, while the immunodominant sequence lines the helical core (11, 12). The Neu5Ac residues play a major role in CPS antigenicity and GBS virulence, and it has been proposed (9) and recently confirmed (12) that these residues exert conformational control over the CPS. Chemical removal of the Neu5Ac group (generating the equivalent of the Pn14 CPS) attenuates the antigenicity of GBSIII CPS for mAb 1B1. Providing a basis for interpreting much of this empirical serological and immunological data (9, 10, 13) requires a structural analysis of the Ab–antigen complex. To examine the relationship between CPS structure and antigenicity in GBSIII and Pn14, this work reports a computational study of large CPS fragments from GBSIII and Pn14, complexed to the Fv fragment of mAb 1B1. A computational analysis offers unique insight into the origin of the binding free energy, in terms of contributions from intermolecular interactions, as well as from solvation and entropic effects.
Despite several attempts, the antigen binding (Fab) fragment of mAb 1B1 has not been crystallized, either alone or in complex with antigen (H. J. J., unpublished data), and it is at present too large a system for structural determination by NMR methods. However, the low variability in the constant region of Ig domains, the availability of numerous crystal structures of Ab domains, and the characterization of the hypervariable loops into canonical structures facilitate the use of comparative modeling in the generation of 3D models of the antigen-binding Fab and variable Fv domains (16, 17).
Results
Structure Validation of Fv1B1.
The canonical forms for five of the six complementarity determining region (CDR) loops in Fv1B1 were readily assigned on the basis of length and the presence of key residues within, or bordering, the loop (14), as follows: CDR-L1 (class 4) and L2, L3, H1, and H2 (class 1). The carboxylate of Asp-213 in H3 forms a salt bridge with Arg-210 (Oδ1 . . . NH2 = 2.78 Å) and stabilizes an inverse γ-turn centered at Asn-211, placing this loop into the kinked structural class (18).
A Ramachandran analysis (19) of Fv1B1 indicated that only one residue (Val-56) appeared in a disallowed region of the map (Fig. 1), having ϕ and ψ angles of 60.7° and −57.7°, respectively. These values are consistent with a γ-turn, confirmed to be present in the template crystal structure [Protein Data Bank (PDB) ID code 1QFU]. Residues Trp-212 and Asp-213 also displayed uncommon ϕ, ψ angles (176°, 24.8°, and 162.5°, −87.3°, respectively) arising from the neighboring inverse γ-turn.
Fig. 1.

Aligned primary sequences of Fv1B1 and protein templates and backbone analysis of resultant comparative model. (Upper) Sequence alignments for the VL and VH domains in mAb 1B1 with the template sequences; the CDRs are in red (14, 15). (Lower) Ramachandran plot of Fv1B1 (for statistics, see Table 1).
For comparison, the 1B1 Ab sequence also was submitted to two web-based homology modeling sites for 3D structure generation. The first, wam (Web Antibody Modeling) (20) employs a combination of comparative modeling with Ab structural information; similar to the approach used by us. The second was a generic protein homology-modeling server [swiss-model (21)]. Lastly, because side-chain orientations are critical to ligand docking, the program scwrl 3.0 (22) was used to generate side-chain orientations in Fv1B1. Each resultant model was then analyzed in terms of overall fold structure and side-chain orientation by using the procheck (23) and what if (24) protein structure analysis and verification tools. Based on this analysis (Table 1), we concluded that the Fv1B1 structure was acceptable. It is notable that the binding site in Fv1B1 was predicted to be similar to that observed experimentally for an anti-Neisseria Fab that can also accommodate an unusually long segment of α-(2 → 8)-linked Neu5Ac residues (25).
Table 1.
Comparison of structural parameters for homology models of Fv1B1
| Structure | Property | Model | |||
|---|---|---|---|---|---|
| Fv1B1 | scrwl* | wam† | swiss model‡ | ||
| Overall | Fv Cα rmsd§ | 0.0 | 0.00 | 0.87 | 4.05 |
| VL Cα rmsd | 0.0 | 0.0 | 0.28 | 4.84 | |
| VH Cα rmsd | 0.0 | 0.0 | 0.74 | 2.19 | |
| FV All atom rmsd | 0.0 | 1.41 | 1.63 | 4.70 | |
| VL All atom rmsd | 0.0 | 1.21 | 1.36 | 5.47 | |
| VH All atom rmsd | 0.0 | 1.59 | 1.54 | 2.90 | |
| No. of bad contacts¶ | 4 | 26 | 1 | 0 | |
| Backbone | Core,¶ % | 86.8 | 86.8 | 83.2 | 78 |
| Allowed,¶ % | 10.5 | 10.5 | 15.3 | 18.8 | |
| Generously allowed,¶ % | 2.1 | 2.1 | 0.0 | 2.1 | |
| Disallowed,¶ % | 0.5 | 0.5 | 1.6 | 1.0 | |
| Ramachandran (Z-score)‖ | −1.467 | −1.467 | −2.518 | −1.887 | |
| Side chain | No. of bad rotamers¶ | 1 | 1 | 1 | 3 |
| χ1/χ2 (Z-score)‖ | 1.155 | 5.013 | −2.653 | 2.12 | |
Generation of the CPS–Fv Complexes.
Because of the presence of a large number of internal rotatable bonds, docking a ligand such as the CPS fragment in its entirety is inefficient and may lead to multiple spurious alignments. This problem is compounded by the number of potential orientations of the antigen in the binding site. However, division of the ligand into representative fragments enables the generation of a model for the complex by examining the structures of multiple docked fragments (26). Once a fragment has been docked, it can serve as an anchor point from which the remaining ligand structure may be extended (27). Computational strategies notwithstanding, docking flexible ligands remains a challenge. An anchor-first approach was selected for docking the GBSIII CPS to the Fv employing a trisaccharide fragment of the core residues [β-d-Glcp-(1–6)-β-d-GlcpNAc-(1–3)-β-d-Galp] for initial docking. The disaccharide side chain was removed to ensure that the primary mode of interaction would involve only the immunodominant region, common to both GBSIII and Pn14. Numerous docking simulations were performed by using autodock (28), each initiated with the trisaccharide randomly oriented within 4 Å of the antigen-binding groove. The 10 complexes with the best autodock score were then assessed for their ability to meet the following criteria. First, the trisaccharide was required to adopt orientations relative to the protein surface that facilitated the extension of the docked unit to a larger three-repeat fragment, by the addition of a single repeat unit to both the reducing and nonreducing termini. Second, only oligosaccharide conformations in which the glycosidic linkages could adopt conformationally acceptable values (29) were retained. Docked trisaccharides that did not provide an adequate template for extension to the larger structures due to insurmountable steric problems were frequently encountered and discarded. Lastly, each docked complex was assessed for its ability to tolerate the addition of the sialylated side chain in its solution conformation. This procedure was continued until a 25-residue fragment, corresponding to five repeat units of the intact GBSIII CPS, was successfully docked. The docking procedure led ultimately to only two low-energy complexes, which differed in the overall orientation of the antigen with respect to the Ab. In one complex, the reducing end of the CPS was oriented toward the VL domain (light to heavy threading; L → H), whereas in the other, the reducing end was oriented toward VH (H → L threading; Fig. 2). This reversal in the CPS polarity originated from opposing alignments of the initial trisaccharide, predicted during the automated docking phase.
Fig. 2.

Final structures of each of the Fv:GBSIII complexes from 1-ns MD simulations. (Upper) CPS aligned with the reducing terminus directed toward the VL domain (L → H threading). (Lower) Reversed (H → L) alignment. The solvent-accessible surfaces of the VL and VH domains are shown in light and dark blue, respectively; CPS is shown in purple with the neuraminic acid residues in orange. The CDRs are labeled and shown in red.
To begin to refine these two putative models of the Fv:CPS complex, each was subjected to fully relaxed, explicitly solvated 1-ns molecular dynamics (MD) simulations. Both complexes remained stable throughout the MD simulations with no significant distortion in the alignment of the oligosaccharide fragment in the binding groove being observed. Over the course of the 1-ns simulations, average rms deviation (rmsd) values of 2.9 and 3.3 Å were computed for the displacement of the CPS from the surface of the Fv in the L → H and H → L orientations, respectively. The hydrogen-bond (H-bond) occupancies and the average distances between the H-bonded atoms computed from the MD data indicated that both complexes made a similar total number of H-bond contacts (see Tables 3 and 4, which are published as supporting information on the PNAS web site). However, the L → H threading made significantly more contacts between the CPS and the residues in the hypervariable loops than did the reverse threading (Fig. 3). Therefore, qualitatively, the L → H threading represented a specific mode of interaction between the CPS fragment and the Ab CDR residues, whereas the H → L threading appeared to be nonspecific.
Fig. 3.

Schematic representations of the predicted H-bonding and aromatic stacking interactions. (Upper) L → H threading. (Lower) H → L threading. Monosaccharide residues are denoted (33) as follows: Glcp (blue circles), GlcpNAc (blue squares), Galp (yellow circles), and Neu5Ac (purple diamonds). H-bonds are represented by arrows, and hydrophobic stacking interactions are shown as dotted red lines.
In addition to the H-bond interactions, the L → H complex was further stabilized by hydrophobic stacking interactions between the sugar rings and aromatic side chains in the CDRs. Hydrophobic stacking of aromatic rings with glycosyl residues has been identified as a characteristic feature in many protein–carbohydrate complexes and is believed to contribute significantly to stability and specificity (30). The L → H complex exhibited four stacking interactions: Glc-IV/Tyr-37, Glc-V/Tyr-145, Gal-V/Tyr-212, and GlcNAc-V/Tyr-212; each of these residues is located in the CDR loops. The H → L threaded complex displayed only two such interactions: Glc-I//Tyr-145 and GlcNAc-II/Tyr-212.
Based on the predicted contacts between the CDRs and the antigen, each complex was consistent with the immunological observations pertaining to both the large size of the epitope (9) and the sequence associated with the immunodominant core (8). However, from an examination of the intermolecular interactions, we concluded that the H → L threading was essentially nonspecific, and further analysis, including extension of the MD simulation to 10 ns, was confined to the L → H complex. (see Movie 1, which is published as supporting information on the PNAS web site).
Fv:GBSIII CPS Complex.
The plateau in the computed interaction energies (Fig. 4) indicated that the complex had structurally converged by ≈5 ns. As noted previously for carbohydrate–lectin interactions (31, 32), the favorable electrostatic and van der Waals interaction energies, −294.4 kcal·mol−1, were largely offset by the positive solvation free energy, 211.9 kcal·mol−1 (Table 2). Despite the polar character of the antigen, van der Waals interactions contributed 43% (−126.9 kcal·mol−1) of the total molecular mechanical binding energy. Unlike electrostatic contributions, which may be nonspecific in origin, the van der Waals energies provide a direct indication of the level of shape complementarity between the antigen and the Ab.
Fig. 4.

Interaction energies between Fv1B1 and the CPS from Pn14 (solid line) and GBSIII (dashed line) as a function of simulation time. (Top) 〈ΔEMM〉. (Middle) 〈ΔGSolvation〉. (Bottom) Total interaction energy (〈ΔGGBTotal〉).
Table 2.
MM-GBSA analysis of the 10-ns MD trajectories of the Fv:CPS immune complexes
| Average energy* component | Interaction energies for the complexes |
|
|---|---|---|
| GBSIII | Desialylated GBSIII (Pn14) | |
| 〈ΔEElectrostatic〉 | −167.5 ± 20 | −156.5 ± 21 |
| 〈ΔEVDW〉 | −126.9 ± 9 | −98.9 ± 10 |
| 〈ΔEMM〉 | −294.4 ± 24 | −255.4 ± 20 |
| 〈ΔGNonpolar〉 | −16.8 ± 1 | −14.2 ± 1 |
| 〈ΔGPolar〉 | 228.8 ± 17 | 198.4 ± 16 |
| 〈ΔGSolvation〉 | 211.9 ± 17 | 184.1 ± 16 |
| 〈ΔGGBTotal〉 | −82.5 ± 11 | −71.3 ± 9 |
| 〈−TΔSRTV†〉 | 77.6 ± 18 | 68.7 ± 23 |
| 〈−TΔSC‡〉 | 0.01 ± 0.2 | 1.84 ± 0.2 |
| 〈ΔGBinding〉 | −4.9 ± 11 | −0.76 ± 10 |
*Values are in kcal·mol−1, averaged over the period (0.5–10 ns). ΔEMM is the total molecular mechanical energy from electrostatic and van der Waals contributions; ΔGSolvation is the total polar and nonpolar contributions to solvation from generalized Born analysis (IGB = 1); ΔGbind is the total binding energy.
†ΔSRTV is the rotational, translational, and vibrational entropy from normal mode analysis.
‡ΔSC is the conformational entropy associated with internal rotations.
Antigen binding led to a stiffening of the entire system, indicated by an entropic penalty to binding of 77.6 kcal·mol−1. The conformational entropy, associated with the change in the low-frequency, large-amplitude motions of the ω-angles, was predicted to be negligible for GBSIII, consistent with the observation that the free CPS rarely exhibits transitions in the backbone ω-angles (12). Thus, because the GBSIII CPS is relatively rigid in solution, and binds in essentially the solution conformation, there was no additional conformational entropic penalty upon its binding to the Fv, resulting in a favorable net binding energy of −4.9 kcal·mol−1.
Fv:Pn14 CPS complex.
An initial model for the Pn14 immune complex was generated by removal of the Neu5Ac residues from the GBSIII CPS in the L → H complex. After a 10-ns MD simulation, intermolecular contacts were characterized (see Tables 5 and 6, which are published as supporting information on the PNAS web site). The Pn14 CPS formed a stable, but weaker, complex with Fv1B1 than did GBSIII, as confirmed by consistently lower-magnitude interaction energies (Fig. 4). The lack of Neu5Ac residues in Pn14 led to a proportionate reduction in the van der Waals stabilization but had a surprisingly small (11 kcal·mol−1) effect on the net electrostatic interaction energy. The less-polar Pn14 CPS exhibited less desolvation penalty, and the sum of the enthalpic and solvation terms remained strongly favorable (−71.3 kcal·mol−1). It was not until the inclusion of entropic penalties that affinity was essentially abolished. Although the net change in configurational entropy penalty was smaller for Pn14 than for GBSIII, it correlated well with CPS molecular mass (31), as seen from the per-residue values of 3.4 kcal·mol−1·res−1 (Pn14) and 3.1 kcal·mol−1·res−1 (GBSIII). In contrast to GBSIII, there was a predicted conformational entropy penalty of ≈1.8 kcal·mol−1 for the binding of the flexible Pn14 CPS to Fv1B1. Comparable entropy terms have been predicted recently for conformational changes in oligopeptides (34).
Discussion
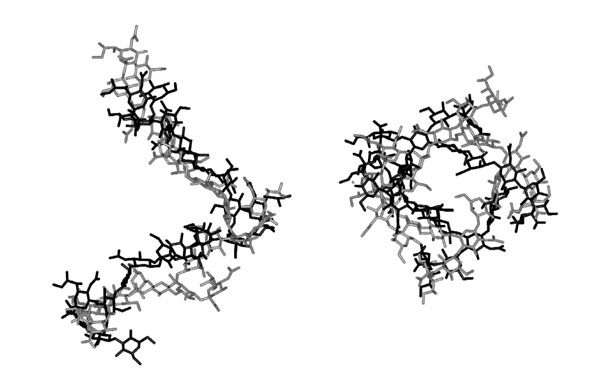
The helical conformation of the GBSIII CPS in the model immune complex is comparable with that of the free CPS in solution (12), as evidenced by the relatively low rmsd of 4.3 Å between the nonhydrogen atoms of the bound and free CPS fragments (see Fig. 5, which is published as supporting information on the PNAS web site). Further, the conformation of the immunodominant core of the CPS in the immune complex is indistinguishable from that of the free CPS. From the contacts predicted between the CDRs and the GBSIII CPS, the minimum size of a CPS fragment that would completely fill the combining site is approximately three repeat units. This finding is in accord with the observation that the minimum binding structure consists of two pentasaccharide repeat units, with structures containing from three to seven repeat units representing the optimal monovalent epitope (8, 10), and characterizes the previously undescribed 3D structure of the postulated conformational epitope (9, 10).
Analysis of the binding energies demonstrates the significance of the electrostatic and van der Waals interaction energies, the desolvation energy, and the configurational and conformational entropies. The electrostatic interaction energy was relatively insensitive to the decreased number and strength of the H-bonding interactions in the Pn14 complex (Tables 5 and 6). This result correlates with the absence of a strongly positive electrostatic potential surface in the CDRs near the Neu5Ac residues (see Fig. 6, which is published as supporting information on the PNAS web site), as well as with the lack of any direct electrostatic interactions between the CDRs and the carboxylate groups (Fig. 3).
The lack of affinity of Pn14 for Fv1B1 results primarily from entropic penalties associated with the binding of this highly flexible PS within the large groove-like combining site of Fv1B1. In the case of a smaller combining site and a more rigid epitope, for example one that recognized a single repeat unit of the CPS, this penalty would be lower. Thus, cross-reactions between some anti-GBSIII Abs and Pn14 (7) likely arise from recognition of a small linear epitope, such as that associated with the shared immunodominant region.
Materials and Methods
Generation of a 3D Model for Fv1B1.
Crystal structures and sequence homologies were identified by searching the National Center for Biotechnology Information blast database (www.ncbi.nlm.nih.gov). The Threader algorithm (D. T. Jones, University of Warwick, Coventry, U.K.) in conjunction with the Homology module in the insightii software package (Accelrys, San Diego) were used for sequence alignments and coordinate assignments from the templates to the sequence of Fv1B1 (Fig. 1). The crystal structure of a mouse IgG1 Ab (PDB ID code 1QFU) having 91% sequence identity with the light chain (VL) of Fv1B1 was selected to provide reference coordinates for the VL. VL1QFU differs from VL1B1 in only nine residues, located in the CDRs. No deletions or insertions were required for the alignment, and consequently all coordinates for the VL1B1 backbone atoms were assigned from VL1QFU. The best match for the majority of the VH1B1 sequence was found with VH1A7R from mouse monoclonal IgG D1.3, which displayed a 79% sequence identity. The H3 loop in VH1B1 is shorter than typical H3 loops (35) and alignment between VH1B1 and VH1A7R indicated the former had a four-residue deletion in the H3 region. To reduce the potential for structural errors associated with deletions and loop splicing, a reference structure with a four-residue H3 loop was identified (PDB ID code 1NSN, from mouse monoclonal IgG N10 Fab) and spliced into the PDB ID code 1A7R sequence.
Although 1NSN had only a moderate identity (59%) with 1A7R, the overall fold structures were highly similar (see Fig. 7, which is published as supporting information on the PNAS web site), with an average rmsd of 1.2 Å between the Cα atoms of the two structures. Most relevant to the loop splicing was the observation that the coordinates for the sequences on either end of the short H3 loop in 1NSN aligned closely with those in 1A7R; the rmsd of the backbone atoms of three residues on either side of the H3 loops was 0.2 Å. The need to delete and rejoin the H3 sequence in 1A7R was therefore avoided by transferring the coordinates for CDR–H3 from 1NSN. All remaining coordinates for VH1B1 were assigned from 1A7R. For nonidentical residues, preliminary coordinates were generated either by truncation or extension of the existing side chains. In the case of side-chain extensions, the replacement atoms were located initially in the fully extended conformations.
The alignment of the VH and VL domains to create the Fv domain was accomplished by superimposing the modeled domains onto the pertinent domains present in the template structure of the FV1A7R (36). To allow the side chains to better sample conformational space, the Fv model was then subjected to 2.5 ns of solvated MD simulation, under the conditions described below. The final model was generated by energy minimization.
An assessment of the quality of the resultant dynamics-refined comparative model for Fv1B1 is presented in Structure Validation of Fv1B1 in Results.
Docking Protocol.
autodock 3.0.5 (28) was used to dock the GBSIII trisaccharide fragment into the antigen binding site of Fv1B1, using a Lamarckian Genetic Algorithm (LGA). For the LGA, the initial population size of trial ligands was 50 individuals with the maximum number of generations set to 2.7 × 104. The maximum number of energy evaluations was 2.5 × 105. The number of individuals that proceeded to the next generation was 1; the probability of random change in an individual was 2% with a crossover rate of 80%. The number of generations analyzed to determine the average of the worst energy was 10. The number of iterations that used the local search algorithm was 300, and the lowest possible step size of the search was 0.01. Four consecutive successes or failures were performed before the step size was altered. The probability of performing a local search on an individual was 6%. The total number of runs for each docking trial was 10, with 50 cycles each. All results were clustered according to rmsd and ranked by total energy. The 3D grid used for docking the initial trisaccharide unit contained 613 points and was centered on the hypervariable loops. A grid-point spacing of 0.375 Å was used to systematically encompass the binding site.
MD.
All MD simulations were performed under constant pressure and temperature conditions by using the sander module of the amber 8 program (University of California, San Francisco; 2004) (37), employing the parm99 all-atom force field for proteins (38) with the glycam 2004 parameters for carbohydrates (39, 40). Histidine residues were assumed to be neutral and protonated only at the N-ε position. All other ionizable groups were treated as charged, with sodium ions added to neutralize the negative charge on the CPS where necessary. After solvation with TIP3P (41) water (8,049 and 6,898 waters for the GBSIII and Pn14–Fv complexes, respectively), the complexes were subjected to energy minimization (1,000 cycles of steepest descent and 14,000 cycles of conjugate gradient). A dielectric constant of unity was used throughout, with long-range electrostatics treated by Ewald summation. The 1–4 electrostatic and van der Waals interactions were scaled by the standard amber values (SCEE = 1.2, SCNB = 2.0). The systems were warmed to 300 K over 50 ps, with initial velocities assigned from a Boltzmann distribution at 5 K. During the 10-ns production MD simulation the temperature (300 K) and pressure (1 atm) were controlled with temperature and pressure couplings of 0.2 and 0.25 ps−1, respectively. An integration time step of 2-fs time was used. To maintain the correct relative orientations of the VL and VH domains in the Fv, the backbone atoms were restrained with a weak force constant (25 kcal·mol−·Å−1) throughout the MD simulations.
Entropy Calculations.
The vibrational, translational, and rotational contributions to the configurational entropies were derived from a normal mode analysis of the coordinates after the energies had been minimized to within a rmsd of 0.1 kcal·mol−1, employing amber 8. Each Fv:CPS complex was treated in its entirety, as were the separate CPS and the Fv domains. Snapshots were selected for normal mode analysis at 500-ps intervals. The conformational entropies were estimated from an analysis of the covariance matrix of the relevant internal coordinates (42). In the case of carbohydrates, it is particularly appropriate to focus on the conformational entropy associated with the interglycosidic torsion angles (32). Changes in the backbone ω-angles may be used to characterize the conformational differences between the GBSIII and Pn14 CPS (12); thus, these were exclusively selected for analysis.
On the basis of the contact analysis, it was concluded that only CPS repeat units IV and V interacted specifically with the CDRs, so the covariance analysis was limited to the ω-angles associated with these units, selected at 1-ps intervals from the 10-ns MD of the CPS–Fv complexes and the 50-ns MD of the free CPS (12). From the determinants of the covariance matrices for the torsion angles in the bound (σb) and free states (σf), the relative conformational entropies were derived by using Eq. 1 (42)
Molecular Mechanics Generalized Born Surface Area (MM-GBSA) Calculations.
By using the MD trajectories collected from explicitly solvated simulations of the ligand–protein complexes, the binding free energy was computed directly from the energies of the reaction components
The approach taken followed closely that reported earlier for a similar, but smaller, carbohydrate–lectin complex (31). The free energies of the components were computed by separating the energies into molecular mechanical (electrostatic and van der Waals), solvation, and entropic components
Before the analysis, the water molecules were removed from the solvated trajectories. The energy contribution from solvation was then obtained through application of the generalized Born (GB) implicit solvation model, which because of its relative speed, was suited for application to the large Fv:CPS complexes. The MM-GBSA results compare well with those employing the more rigorous Poisson–Boltzmann implicit solvent approximation (43). Based on earlier work (31), as well as on limited preliminary trials with the current system, we selected the GB parameterization of Tsui and Case (44). Although the magnitude of the computed solvation free energy is dependent on the choice of implicit solvent model, the relative values for closely related ligands are less sensitive.
Cloning and Sequencing of 1B1 VL and VH Genes.
The preparation of mAb 1B1, against GBSIII CPS, has been described elsewhere (10). Hybridoma cells producing mAb 1B1 were used to isolate messenger RNA by using the RNeasy kit (Qiagen, Valencia, CA). The RNA then was reverse-transcribed by using a cDNA-synthesis kit (Amersham Pharmacia Biotech) and primers specific to the constant regions of the VL and VH domains along with primers specific to the N terminus of the two domains. The resulting PCR products were cloned via their 3′ A overhang into pGEM-T (Promega). The subsequent ligation was transformed into Escherichia coli TG1 cells and screened via the blue/white phenotype followed by colony PCR. Several clones that passed the screening were cloned, and their DNA sequences were obtained by using a 373 automated DNA sequencer (Applied Biosystems). GenBank accession nos. are as follows: 1B1 VH, DQ519572; VL (kappa), DQ519573.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants GM55230 and RR05357.
Abbreviations
- CDR
complementarity determining region
- PS
polysaccharide
- CPS
capsular PS
- GBS
group B Streptococcus
- GBSIII
GBS type III
- Pn
Streptococcus pneumoniae
- MD
molecular dynamics
- rmsd
rms deviation
- Galp
galactopyranose
- Glcp
glucopyranose
- GlcpNAc
N-acetylglucosamine
- Neu5Ac
N-acetylneuraminic acid.
Footnotes
Conflict of interest statement: No conflicts declared.
References
- 1.Schuchat A. Lancet. 1999;353:51–56. doi: 10.1016/S0140-6736(98)07128-1. [DOI] [PubMed] [Google Scholar]
- 2.Bogaert D., Hermans P. W. M., Adrian P. V., Rümke H. C., de Groot R. Vaccine. 2004;22:2209–2220. doi: 10.1016/j.vaccine.2003.11.038. [DOI] [PubMed] [Google Scholar]
- 3.Hickman M. E., Rench M. A., Ferrieri P., Baker C. J. Pediatrics. 1999;104:203–209. doi: 10.1542/peds.104.2.203. [DOI] [PubMed] [Google Scholar]
- 4.Lewis A. L., Nizet V., Varki A. Proc. Natl. Acad. Sci. USA. 2004;101:11123–11128. doi: 10.1073/pnas.0403010101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wessels M. R. J. Appl. Microbiol. Symp. 1997;83(Suppl):20S–31S. doi: 10.1046/j.1365-2672.83.s1.3.x. [DOI] [PubMed] [Google Scholar]
- 6.Baker C. J., Barrett F. F. J. Am. Med. Assoc. 1974;230:1158–1160. [PubMed] [Google Scholar]
- 7.Guttormsen H. K., Baker C. J., Nahm M. H., Paoletti L. C., Zughaier S. M., Edwards M. S., Kasper D. L. Infect. Immun. 2002;70:1724–1738. doi: 10.1128/IAI.70.4.1724-1738.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wessels M. R., Pozsgay V., Kasper D. L., Jennings H. J. J. Biol. Chem. 1987;262:8262–8267. [PubMed] [Google Scholar]
- 9.Jennings H. J., Lugowski C., Kasper D. L. Biochemistry. 1981;20:4511–4518. doi: 10.1021/bi00519a001. [DOI] [PubMed] [Google Scholar]
- 10.Zou W., Mackenzie R., Thèrien L., Hirama T., Yang Q., Gidney M. A., Jennings H. J. J. Immunol. 1999;163:820–825. [PubMed] [Google Scholar]
- 11.Brisson J.-R., Uhrínova S., Woods R. J., van der Zwan M., Jarrell H. C., Paoletti L. C., Kasper D. L., Jennings H. J. Biochemistry. 1997;36:3278–3292. doi: 10.1021/bi961819l. [DOI] [PubMed] [Google Scholar]
- 12.Gonzalez-Outeriño J., Kadirvelraj R., Woods R. J. Carbohydr. Res. 2005;340:1007–1018. doi: 10.1016/j.carres.2004.12.034. [DOI] [PubMed] [Google Scholar]
- 13.Rubens C. E., Wessels M. R., Heggen L. M., Kasper D. L. Proc. Natl. Acad. Sci. USA. 1987;84:7208–7212. doi: 10.1073/pnas.84.20.7208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Al-Lazikani B., Lesk A. M., Chothia C. J. Mol. Biol. 1997;273:927–948. doi: 10.1006/jmbi.1997.1354. [DOI] [PubMed] [Google Scholar]
- 15.Chothia C., Gelfand I., Kister A. J. Mol. Biol. 1998;278:457–479. doi: 10.1006/jmbi.1998.1653. [DOI] [PubMed] [Google Scholar]
- 16.Padlan E. A., Kabat E. A. Methods Enzymol. 1991;203:3–21. doi: 10.1016/0076-6879(91)03003-y. [DOI] [PubMed] [Google Scholar]
- 17.Bajorath J., Novotny J. Ther. Immunol. 1995;2:95–103. [PubMed] [Google Scholar]
- 18.Shirai H., Kidera A., Nakamura H. FEBS Lett. 1999;455:188–197. doi: 10.1016/s0014-5793(99)00821-2. [DOI] [PubMed] [Google Scholar]
- 19.Ramachandran G. N., Sasisekharan V. Adv. Protein Chem. 1968;23:283–438. doi: 10.1016/s0065-3233(08)60402-7. [DOI] [PubMed] [Google Scholar]
- 20.Mandal C., Kingery B. D., Anchin J. M., Subramaniam S., Linthicum D. S. Nat. Biotechnol. 1996;14:323–328. doi: 10.1038/nbt0396-323. [DOI] [PubMed] [Google Scholar]
- 21.Peitsch M. C. Biochem. Soc. Trans. 1996;24:274–279. doi: 10.1042/bst0240274. [DOI] [PubMed] [Google Scholar]
- 22.Canutescu A. A., Shelenkov A. A., Dunbrack R. L. J. Protein Sci. 2003;12:2001–2014. doi: 10.1110/ps.03154503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- 24.Vriend G. J. Mol. Graphics. 1990;8:52–56. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- 25.Evans S. V., Sigurskjold B. W., Jennings H. J., Brisson J.-R., To R., Tse W. C., Altman E., Frosch M., Weisgerber C., Kratzin H. D., et al. Biochemistry. 1995;34:6737–6744. doi: 10.1021/bi00020a019. [DOI] [PubMed] [Google Scholar]
- 26.Wang J., Kollman P. A., Kuntz I. D. Proteins. 1999;36:1–19. [PubMed] [Google Scholar]
- 27.Leach A. R., Kuntz I. D. J. Comput. Chem. 1992;13:730–748. [Google Scholar]
- 28.Morris G. M., Goodsell D. S., Halliday R. S., Huey R., Hart W. E., Belew R. K., Olson A. J. J. Comp. Chem. 1998;19:1639–1662. [Google Scholar]
- 29.Rao V. S. R., Qasba P. K., Balaji P. V., Chandrasekaran R. Conformations of Carbohydrates. Amsterdam: Harwood Academic; 1998. [Google Scholar]
- 30.Vyas N. K. Curr. Opin. Struct. Biol. 1991;1:732–740. [Google Scholar]
- 31.Ford M. G., Weimar T., Köhli T., Woods R. J. Proteins Struct. Funct. Genet. 2003;53:229–240. doi: 10.1002/prot.10428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bryce R. A., Hillier I. H., Naismith J. H. Biophys. J. 2001;81:1373–1388. doi: 10.1016/S0006-3495(01)75793-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Varki A., Cummings R., Esko J., Freeze H., Hart G., Marth J. Essentials of Glycobiology. Woodbury, NY: Cold Spring Harbor Lab. Press; 1999. [PubMed] [Google Scholar]
- 34.Cheluvaraja S., Meirovitch H. Proc. Natl. Acad. Sci. USA. 2004;101:9241–9246. doi: 10.1073/pnas.0308201101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu T. T., Johnson G., Kabat E. A. Proteins. 1993;16:1–7. doi: 10.1002/prot.340160102. [DOI] [PubMed] [Google Scholar]
- 36.Dyekjaer J. D., Woods R. J. In: NMR Spectroscopy and Computer Modeling of Carbohydrates: Recent Advances. Vliegenthart J. F. G., Woods R. J., editors. New York: ACS; 2005. Symp. Series 930. [Google Scholar]
- 37.Case D. A., Cheatham T. E., III, Darden T. A., Gohlke H., Luo R., Merz K. M., Onufriev A., Simmerling C. L., Wang B., Woods R. J. J. Comput. Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang J., Cieplak P., Kollman P. A. J. Comp. Chem. 2000;21:1049–1074. [Google Scholar]
- 39.Woods R. J., Dwek R. A., Edge C. J., Fraser-Reid B. J. Phys. Chem. 1995;99:3832–3846. [Google Scholar]
- 40.Kirschner K. N., Woods R. J. Proc. Natl. Acad. Sci. USA. 2001;98:10541–10545. doi: 10.1073/pnas.191362798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L. J. Phys. Chem. 1983;79:926–935. [Google Scholar]
- 42.Karplus M., Kushick J. N. Macromol. 1981;14:325–332. [Google Scholar]
- 43.Kollman P. A., Massova I., Reyes C., Kuhn B., Huo S., Chong L., Lee M., Lee T., Duan Y., Wang W., et al. Acc. Chem. Res. 2000;33:889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 44.Tsui V., Case D. A. Biopolymers. 2001;56:275–291. doi: 10.1002/1097-0282(2000)56:4<275::AID-BIP10024>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}