

Abstract
Mice in which the Jκ cluster was replaced with a VκJκ rearranged gene were studied. More than 90% of B cells from homozygous mutant mice expressed the transgenic κ chain but showed a slightly reduced level of κ transcripts compared with WT B lymphocytes. Light chain inclusion was apparent in 10% of B cells from these mice and raised 25% in hemizygous mice with a still lower expression of the knockin κ chain. Beyond the rules of clonal selection, peripheral B cells developed in such animals, with included cells being activated and differentiating into class-switched or antibody-secreting cells. The high amount of included mature B cells was associated with an increase of hybrid κ/λ immunoglobulins but not with the increased prevalence of autoantibodies. Altogether, these data suggest that light chain exclusion prevalent in normal B cells mostly results from ordered rearrangements and stochastic mechanisms but is neither tightly ensured by a stringent cell selection process nor absolutely required for normal B cell function.
Keywords: antibodies, autoimmunity, B lymphocytes, gene regulation
During B cell development, B cell receptor (BCR) assembly relies on an ordered process of rearrangements remodeling first the heavy chain (HC) locus and later on the κ and λ loci. Cells achieving expression of functional H and L chains and able to pair them can then express a unique form of functionally competent Ig at the cell surface and be selected according to the specificity of this receptor (1, 2). With very rare exceptions, B lymphocytes are thus monospecific and express a single H and L chain. This allelic and isotypic exclusion is fundamental to the establishment of the B cell repertoire and is in agreement with the clonal selection theory (3), first allowing during ontogeny the tolerization of clonal cells carrying a self-reactive BCR and later ensuring the specificity of antibody responses. Whereas this rule seems to be quite effective in the case of the HC (4, 5), it suffers remarkable exceptions at light chain (LC) loci. Indeed, several studies reported the presence of double-expressing κ/λ B cells either at very low frequencies in normal mice or in human, and more abundantly in transgenic mice (6–9) and plasmocytomas (10). Little is known about the mechanisms involved in the allelic exclusion of LC even if recent studies highlighted the importance of asymmetric demethylation of the κ locus (11, 12) and feedback inhibition of the recombination machinery after the assembly and signaling by a non-self-reactive BCR (13–16). Beyond the initial rearrangements, exclusion also has to be maintained despite the accumulation of secondary rearrangements. The latter were first reported at high frequency in LC loci of mice transgenic for an autoreactive BCR, in which receptor editing restored self-tolerance (17–19). Receptor editing also occurs in normal mice during the selection of immature B cells and may be a major pathway of tolerance induction by rescuing cells with autoreactive BCR from deletion (or anergy) provided that they express a new receptor (20, 21). Nevertheless, this editing process intrinsically bears the risk of generating B cell clones that will migrate toward peripheral lymphoid organs while carrying dual or multiple antigen receptors, one of those potentially being autoreactive (22–26). Moreover, autoreactivity may not be the only way to promote receptor editing because some non-autoreactive knockin mice also showed extensive secondary rearrangements, likely due to a low expression of the knockin gene (27, 28). It was also shown in CD19-deficient mice that a low BCR signaling could impair LC allelic exclusion (29). This finding is reminiscent of observations that HC and LC transgenes only induced allelic exclusion when expressed above a certain threshold (30). We have generated transgenic mice expressing chimeric human/mouse κ LC by inserting a rearranged human VκJκ gene upstream of the mouse Cκ while preserving recombining segment (RS) sites that could allow κ deletion through secondary rearrangements. A decrease of peripheral B cell number was observed in mutant mice together with a diminished surface BCR expression level compared with WT mice. As an effect of gene dosage, mice hemizygous and homozygous for the insertion, respectively, showed a high or a low number of B cells with inclusion, thus indicating that the stable establishment of LC exclusion strongly relies on the κ expression level. The development of cells carrying dual BCR was studied in the hemizygous mice, with a special attention to their potential ability to escape the normal negative selection and yield harmful autoantibodies. We also studied whether such cells with dual specificity were able to normally participate to immune responses and thus to undergo terminal differentiation into class-switched memory cells and plasma cells.
Results
Expression of Chimeric κ LC in Peripheral B Cells from Transgenic Mice.
Mice were obtained in which the Jκ cluster was either deleted (κD mutation) or replaced with a knockin rearranged VκJκ gene (κC mutation) (Fig. 1A). Efficient production of a chimeric LC made up of a mouse C domain and a V domain from human origin was observed both in homozygous κCH and hemizygous κCκD animals (carrying a complete deletion of the Jκ cluster on one allele and the knockin VκJκ exon on the other). Northern blotting of spleen RNA showed normal-size κ mRNAs hybridizing with a Cκ probe, with a lower abundance in heterozygous κCκD mice than in homozygous κCH mice and in κCH than in WT mice, while quantitative PCR (QPCR) experiments confirmed the decreased ratios of κ/μ transcripts in mutant animals (Fig. 1B). The decrease of κ LC transcripts was also checked by QPCR on sorted B lymphocytes from κCH and WT spleens, where the κ/μ ratio was found decreased by 30% in four independent cell sorting and QPCR experiments. Correct splicing of the V to the endogenous Cκ was confirmed by sequencing κ cDNAs.
Fig. 1.

Ig transgenic κLC expression by splenocytes from mutant animals. (A) Structure of the targeted locus. At the top is shown the WT κ locus and the 3′ probe. In the middle is shown the structure of the κC mutated locus after targeted recombination and neoR deletion. A pVH promoter and the human VκJκ exon replaced the Jκ cluster. Dashed lines mark homologous sequences used as 5′ and 3′ arms of the construct. At the bottom is shown the κD mutation after cre-deletion of all knockin elements. B, BamHI; Bs, BsmI; S, SacI. (B) Northern blot and QPCR analysis of κ mRNAs. RNA (10 μg) from spleen of WT, κCH, κC/κD, and κDH mice were analyzed on 1% agarose/0.7 M formaldehyde gels and hybridized with a Cκ probe. The 1.2-kb band corresponding to normal-size κ mRNA is indicated (arrow). κ/μ ratios were calculated by QPCR. (C) Flow cytometric analysis of κLC-expressing cells in spleen. WT, κCH, or κCκD splenocytes were stained as indicated. Numbers indicate the percentages of cells in the corresponding quadrants (from one representative of five independent experiments). (D) Flow cytometric analysis of WT, κCH, κCκD, or κDH splenocytes stained as indicated. Numbers indicate the percentages of total cells in the corresponding quadrants (from one representative of five independent experiments). (E) CD43-enriched B cells from WT and κCκD mice were analyzed in two gates corresponding to κ- and λ-only B cells for WT mice or κ-only and κ/λ B cells for κCκD mice; these two gates were then analyzed for CD23 and CD21 expression.
Surface expression level of transgenic κ LC was then studied by flow cytometry on splenocytes from hemizygous and homozygous mutant mice. In both cases, because all endogenous Jκ segments were deleted, there was no possible expression of endogenous Vκ genes. In κCH mice, ≈90% of transgenic B cells expressed the κ LC while the peripheral B cell compartment was reduced with B cells accounting for ≈25% of total splenocytes vs. 40% in WT mice and ≈15 × 106 total B220+ cells by spleen vs. 40 × 106 in WT mice (Fig. 1C; and see Table 2 and Figs. 6 and 7, which are published as supporting information on the PNAS web site). Such a reduction has been reported in mice carrying an inserted VκJκ gene (31) and may be a consequence of the restricted B cell repertoire. By flow cytometry, the BCR-associated transgenic κ chain was expressed at a lower density in κCH animals than endogenous κ LC in WT B cells. Hemizygous κCκD mice showed a more pronounced reduction in the number of peripheral B cells (≈20% of splenocytes were B cells with an absolute number of ≈10 × 106 B220+ cells by spleen) (Fig. 1C; and see Table 2 and Figs. 6 and 7) and a more pronounced reduction in the expression of membrane κ LC; the lowered BCR expression was confirmed by the decreased staining with anti-μ or anti-δ antibodies in κCκD animals (see Table 2 and Figs. 6 and 7).
High Amount of κ/λ LC-Included B Cells in Spleen and Impaired Early B Cell Development as Consequences of Low κ Transgene Expression Favoring Secondary Rearrangements.
A significant increase in λ+ B cells readily appeared in both κCH and κCκD mice, in which 10% and 25%, respectively, of B cells stained both for κ and λ LC (Fig. 1D). That these cells were real double producers was confirmed by measuring κ transcription in cells sorted by flow cytometry as the λ-positive population, where a 9-fold increase in κ transcription was observed in cells from mutant κCH mice by comparison with WT. κ/λ LC inclusion was also clearly apparent when splenocytes were analyzed by confocal microscopy, and although a number of cells simultaneously showed membrane κ and λ LC, there was no evidence of included cells only expressing membrane λ LC while κ LC would be retained intracellularly (data not shown). To characterize the phenotype of transgenic spleen B cells, IgM/IgD double-staining was carried out and showed in all animals the presence of mostly mature B cells (IgMlow/IgDhigh) (Fig. 1D Lower) together with a slight increase in transitional (T1) B cells (IgM+/IgDneg) observed among κCκD splenocytes. To specifically analyze the status of κ/λ LC-included cells, quadruple staining of magnetically enriched B cells (using CD43-coated beads) with κ, λ, CD21, and CD23 fluorescent antibodies was performed in κCκD and WT mice. A strong increase of CD23low/CD21high B cells, corresponding to marginal zone (MZ), was observed in mutant mice compared with WT (Fig. 1E). Particularly, ≈15% of κ-gated and 40% of included B cells appeared to be in the MZ compartment contrasting with the 5% of MZ B cells from WT mice. However, cells with a mature follicular phenotype (CD23+/CD21int) were also clearly present (≈40%) in mutant mice both within the κ-only and κ/λ-positive cells.
To check the effects of LC inclusion on B cell ontogeny, we studied early B cell compartments by flow cytometry with specific surface markers. The B220+/CD43+ pre-B population was slightly increased, and conversely, total B220+/IgD+ recirculating B cells were decreased by ≈2-fold (n = 5) (Table 1). Finally, B1 cells appeared to be unaffected in transgenic animals, as observed by CD5+/IgM double-staining of peritoneal or spleen B cells (data not shown).
Table 1.
B cell development in bone marrow from mutant animals
| Genotype | Total (B220+), % | CD43 (B220+), % | CD25 (B220+), % | IgM (B220+), % | IgD (B220+), % |
|---|---|---|---|---|---|
| WT | 27.1 (6.5) | 14 (2.8) | 31 (4.2) | 29.6 (4.4) | 22.5 (5.8) |
| κCκD | 22.3 (8.6) | 19.3 (2.1) | 24 (6) | 23.6 (4.6) | 11.5 (5) |
| N.S. | P < 0.05 | N.S. | N.S. | P < 0.05 |
Values in parentheses are standard deviations. Mean results from five mice. N.S., not significant.
κ/λ LC Inclusion in Activated or Terminally Differentiated B Lineage Cells.
κ/λ inclusion was checked on LPS-stimulated splenocytes from κCκD mice and various types of peripheral differentiated cells (Fig. 2A). Although anergic B cells are known to poorly proliferate under stimulation by LPS (32, 33), FACS analysis of 3-days LPS-stimulated mutant splenocytes showed a proliferation and an increased proportion of the κ/λ-stained B cells, indicating that those cells featuring LC inclusion were not anergic.
Fig. 2.

LC-inclusion analysis in LPS-stimulated splenocytes (A) and Peyer’s patch B cells (B). (A) Flow cytometric analysis of LPS-stimulated splenocytes in WT and κCκD mice. Splenocytes stimulated with LPS for 3 days were stained with anti-κ and anti-λ antibodies. (B) For Peyer’s patch B cells, triple-staining with antibodies specific for IgA, κ LC, and λ LC was performed (κ and λ LC expression was checked on IgA+-gated cells). Numbers indicate the percentages of cells in one representative of three independent experiments.
Triple-staining of cells with κ-, λ-, and IgA-specific fluorescently labeled antibodies allowed for specific analysis of LC inclusion in Peyer’s patch IgA-switched memory B cells from κCκD mice. As seen in Fig. 2B, about one-fourth of IgA+ B cells simultaneously expressed κ and λ LC, i.e., a proportion similar to that observed for IgM+ cells.
When bone marrow cells were studied for κ/λ LC inclusion by confocal microscopy, about one-fourth of plasma cells from κCκD mice appeared to simultaneously produce both LC types even if a low proportion of λ-only plasma cells were observed. Moreover, triple-staining allowed us to analyze HC in included cells and clearly demonstrated the presence of the T-dependent isotype IgG1 (Fig. 3A and B).
Fig. 3.

Confocal microscopy examination of bone marrow plasma cells. (A) Bone marrow cells from κCκD mice, stained with Cy5 anti-IgG1 antibodies (blue), Alexa 488-conjugated anti-κ antibodies (green), and Alexa 594-conjugated anti-λ antibodies (red). Arrows indicate plasma cells (high intracellular staining). The ×40 objective was used. (B) Details of WT (Upper) and κCκD (Lower) plasma cells. The ×63 objective was used. Differential interferential contrast (DIC) images are also shown with merge staining.
κ/λ-Included B Cells Are Able to Develop into Antibody-Secreting Cells.
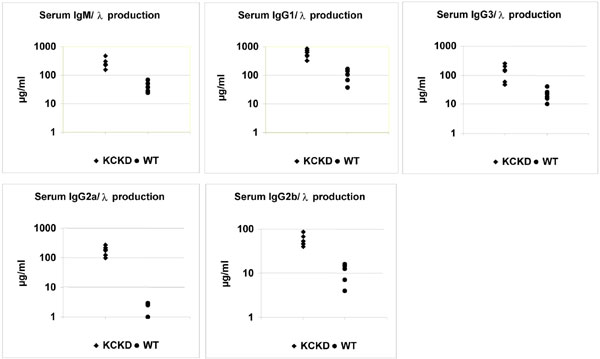
In agreement with the bone-marrow presence of plasma cells showing LC inclusion, it was expected that hybrid κ/λ Ig molecules would be present in sera of κCκD mice and would constitute one part of the λ-containing Ig. In addition, because most λ+ B cells simultaneously expressed κ LC, most λ-containing serum Ig are likely secreted by plasma cells that derive from κ/λ LC-included B lymphocytes. Crossed ELISA assays (retaining hybrid molecules on anti-λ-coated plates and revealing their binding with anti-κ or anti-HC antibodies, or reciprocally) indeed both showed higher amounts of hybrid κ/λ Ig molecules (Fig. 4) and of λ-containing Ig molecules (see Fig. 6) in κCκD than in WT mice. Because rheumatoid factors could be simultaneously detected by using this method, we checked their presence by ELISA but found no increased amount of rheumatoid factors in sera from transgenic animals compared with WT control.
Fig. 4.

Serum levels of hybrid κ/λ Ig. Serum hybrid κ/λ Ig levels were determined in κCκD and WT mice. Concentrations were relative to the optical density obtained by comparison with κDH mice (right) sera as control and were compared between transgenic κCκD mice (left) and WT controls (middle). Goat anti-mouse λ LC and goat anti-mouse κ LC were used at 2 and 1 μg/ml, respectively. The vertical axis is logarithmic. All ELISAs were performed on six different sera of each mouse strain.
High Proportion of κ/λ Ig-Producing Hybridomas.
Hybridoma supernatants from mutant hemizygous mice were analyzed by ELISA. Likely because of the reduced number of B cells in the spleen, less hybridomas (55 from two spleens) were derived from mutant than from WT mice (102 from a single spleen). Among those 55 hybridomas, 18 secreted both LC isotypes. Five of these double producers were IgG (including IgG1, IgG3, and IgG2a isotypes) whereas 13 were IgM producers, as determined by ELISA. The other 37 were conventional single-LC producers (34 κ and 3 λ) (see Table 3 and Figs. 6 and 7, which are published as supporting information on the PNAS web site). These data were confirmed by confocal microscopy with triple-staining on permeabilized hybridomas and ELISAs done on supernatants of three IgG double-producer hybridomas showing the presence of both κ/IgG and λ/IgG molecules (data not shown). No double LC producers were found among 102 hybridomas from WT mice.
Transgenic Animals Do Not Produce Elevated Levels of Autoantibodies.
Sera from two WT mice, four positive control autoimmune lpr mice, eight κCκD mice, and three κ/λ-expressing IgG hybridoma supernatants were assayed for the presence of antinuclear antibodies (ANA) (Fig. 5), antiphospholipid, and antimitochondrial antibodies. By contrast to all four lpr mice used as controls in which ANA were readily detected, no autoreactive antibodies were detected in any of the κCκD mice or in hybridoma supernatants.
Fig. 5.

Indirect immunofluorescence assays on fixed Hep2 cells. Sera from lpr (positive control), κCκD, and WT mice all diluted 1:20 were incubated with fixed Hep2 cells and secondarily stained with a FITC-conjugated anti-mouse Ig. Slides were examined on a fluorescent microscope with the ×63 objective.
Discussion
We generated mice expressing chimeric human/mouse κ LC through targeted insertion of a rearranged human VκJκ gene upstream of the mouse Cκ. By comparison with normal κ LC, the chimeric LC expression was reduced in transgenic animals, especially in hemizygous mice carrying both a knockin allele and a Jκ-deleted allele. Although homozygous and hemizygous animals exhibited a reduction in peripheral B cells, they both displayed a significant increase in proportion of λ+ B cells (≈10% and 25% of total B cells, respectively) and thus showed that λ rearrangements occurred despite the early expression of a rearranged κ gene. Most of the λ+ cells in both strains indeed underwent allelic inclusion with the suboptimally expressed transgenic κ chain. The proportion of LC-included cells was higher in hemizygous animals, likely because the diminished BCR expression in immature B cells inefficiently silenced λ LC rearrangements. Such a condition may theoretically allow a B cell clone to proliferate in response to a single exogenous BCR ligand and then to inadequately produce a second form of antibody specific for an unrelated antigen. Similarly to a situation reported for transgenic B cells with an autoreactive anti-DNA H/L combination (24), ≈40% of dual BCR-expressing cells accumulated in the splenic MZ. However, and in contrast with this previous report, more than a half of included κ/λ B cells showed a mature follicular phenotype (CD23high/CD21int) and demonstrated that such cells are not necessarily trapped in the MZ. It has also been hypothesized that dual BCR expression resulted from receptor editing of an autoreactive primary Ig HC/LC combination (22, 23, 25, 26). In such models of LC inclusion, the expression of multiple antigen receptors was thought to dilute the autoreactive BCR and permit autoreactive cells to escape deletion. Similarly, in a recent model of B cell nuclear transfer, allelic inclusion was suggested to be potentially responsible for the presence of serum autoreactive antibodies (34). A strong bias of such studies was the use of V genes chosen as autoreactive. However, because a major proportion of randomly combined VH and VL domains from unselected B cell precursors form autoreactive antibodies (mostly ANA) (35), it should be expected that any condition allowing LC inclusion (even without a known “autoimmune” transgene) would allow a high amount of random H/L autoreactive combinations. Autoreactive dual BCR B cells could thus be expected in the herein reported LC inclusion model, unless they are controlled by those very same selection processes that control or delete single BCR autoreactive B cells (36).
LC inclusion due to secondary rearrangements in this model was apparently not related to autoreactivity imposed to H/L combinations by the sole transgenic Vκ sequence but rather to its expression level, because it differed between hemizygous and homozygous animals and was inversely correlated with the level of transgenic LC expression. Weak membrane BCR expression due to a lowered expression of the knockin Vκ gene likely increased receptor editing by extending the bone marrow pre-B cell stage prominent in the hemizygous mice. In addition to the reduced diversity of the B cell repertoire, this likely accounted for the reduced size of the peripheral B cell compartment.
By contrast with previously documented transgenic mice expressing autoreactive combinations of V genes, autoreactivity was not promoted. B cells carried WT IgH loci and thus expressed diversified combinations of endogenous HC with the knockin κ LC (and/or with endogenous λ LC) and had to go through the process of negative selection accompanying B cell differentiation in bone marrow. Neither ANA, which are the hallmark of humoral autoimmunity (37), nor rheumatoid factors, antiphospholipid, or antimitochondrial antibodies were detected in LC-included mice. Despite the fact that dual BCR-expressing cells should express each form of the BCR at roughly half the normal level or below, acquisition of self-tolerance thus apparently occurred normally in both hemi- and homozygous mutant animals. Neither the potential selection of B cells by two BCR of different specificities nor their suboptimal BCR expression appeared to mask autoreactivity, “dilute” autoimmune BCR molecules, or allow the survival of cells producing harmful antibodies.
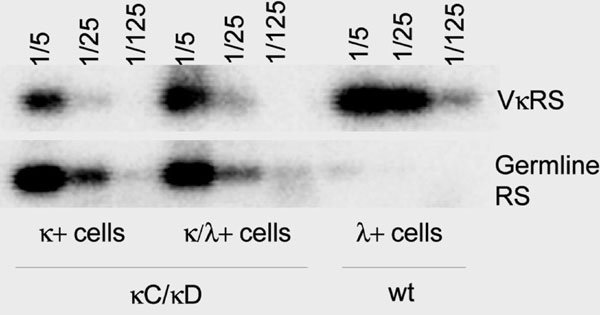
Alternatively, it could have been postulated that dual BCR-expressing cells would simply be anergic or restricted to the spleen MZ and thus never differentiate into antibody-producing cells, explaining why autoantibodies would not arise at a higher frequency in such animals. Analysis of dual BCR-expressing cells clearly shows that this is not the case and that LC inclusion is not restricted to poorly responsive peripheral resting B cells or MZ compartment. In fact, terminal differentiation of these cells clearly occurred, and there was a high frequency of LC inclusion among mature follicular B cells and IgA+ Peyer’s patch memory cells. In bone marrow, IgG-expressing plasma cells showing inclusion were also readily detected and produced T-dependent isotypes (IgG1), while hybrid κ/λ Ig molecules were abundant in the serum of hemizygous and homozygous mice. Probably because of low-level κ inactivating events by RS recombination or somatic hypermutations, rare λ-only B cells and plasma cells were observed. By contrast, λ-expressing lymphocytes were overwhelmingly κ/λ included cells and the high amounts of λ-containing Ig molecules in sera were consistent with preserved activation capacity of included B cells. Moreover, LC-included hybridomas concomitantly secreted κ/IgG and λ/IgG, showing that in most mature B cells, both LC were able to pair with the HC. In the absence of autoreactivity, included B cells could in fact be beneficial for the immune system by extending the BCR repertoire as shown for dual TCR cells (38) especially in a situation where a unique transgenic LC narrows the Ig repertoire. Because the structure of the knockin allele still permits RS-mediated Cκ deletion, it may be surprising that only rare λ-only-expressing cells emerged (see Fig. 7 and Supporting Text, which are published as supporting information on the PNAS web site). Accordingly, RS deletions were detected at a much lower level in λ-expressing cells from mutant than from WT animals, suggesting that these deletions do not provide any selective advantage over κ/λ simultaneous expression. If this assumption is correct, it implies that also during differentiation and editing of wt B cells, λ+/κ− cells mostly arise when κ genes are nonfunctional or deleted and have no clear selective advantage over κ/λ-included cells. A similar observation was made in a model of B cell nuclear transfer despite the presence of a demonstrated autoreactive H/L combination (34). Altogether, these observations confirm that RS deletions are not a prerequisite for λ LC expression but simply prolong RAG expression in B cell precursors by down-modulating BCR expression, thus further allowing diversification of the LC repertoire. In a model where BCR expression is decreased by other means (in this study through suboptimal membrane expression of a knockin κ gene), immature B cells proceed to λ rearrangement even in the absence of RS deletion.
This study reports completely functional dual BCR (LC-included) B lymphocytes that are neither harmful and autoreactive nor anergic, but successfully differentiate until the memory and plasma cell stages. We conclude that only intrinsic properties of the transgenic VκJκ account for the partial blockade of B cell development at the pre-B to B cell transition in hemizygous mice where the transgenic LC is expressed at a lower level than in homozygous mice or than normal κ LC. Weak expression of LC (and consequently of the BCR) in differentiating B cells appears insufficient to down-regulate RAG expression and thus permits ongoing LC editing (27, 28). That only a very low proportion of LC included-cells are present in normal mice may result mostly from stochastic reasons: because functional rearrangements are rare and slow events, they will most often occur only once at each Ig chain locus, then rapidly silencing RAG expression. When in competition with normal cells expressing diversified single BCR, it is also possible that dual BCR cells are less efficiently selected by foreign antigens, and that even if not harmful, they will only rarely show up.
Materials and Methods
Mice.
A 12.7-kb BamHI genomic fragment of the mouse JκCκ cluster was used to generate the targeting construct. A 2.2-kb BsmI/SacI fragment containing Jκ segments was replaced with a “κ-neo” cassette including a lox-P-flanked neomycin-resistance gene (NeoR), a rearranged human VκJκ, and a third lox-P site downstream of the VκJκ. The human Vκ1Jκ1 sequence (GenBank accession no. M87478) was cloned from a patient with LC myeloma and without any autoimmune disease. It was somatically mutated and carried seven codon changes by comparison with germ line. All three flanking lox-P sites were in the same orientation, allowing cre-mediated deletion of either the NeoR gene alone (κC locus) or both the NeoR gene and the VκJκ gene (κ“del” or κD locus) (Fig. 1A).
RNA Analysis.
RNA (10 μg) prepared with TriPure (Roche Applied Science) from spleen of WT, homozygous κC (κCH), heterozygous κC/κD, and homozygous κD (κDH) mice was analyzed on 1% agarose/0.7 M formaldehyde gels, transferred onto nylon sheets, and hybridized with a mouse κ probe corresponding to the 1.6-kb AflII Cκ genomic fragment.
RNA was treated with DNase I and reverse-transcribed into cDNA. QPCRs were performed in duplicate by using SYBR Green PCR Master Mix and analysis on an ABI Prism 7000 system (Applied Biosystems). Analysis of fluorescence allowed determination of a threshold cycle (Ct). Relative amounts of PCR products between samples were determined based on the Ct values. Primers used for QPCR were as follows: Cκ forward TGAGCAGTTAACATCTGGAGGTG; Cκ reverse AAGTTGTTCAAGAAGCACACGACT; Cμ forward (CH1-CH2) GGAGGCAAAAACAAAGATCTGC; and Cμ reverse (CH1-CH2) CATCTCTGCGACAGCTGGAAT.
For cloning and sequencing the cDNA corresponding to the chimeric κ transcript, PCR was carried out by using a human leader VκI primer (5′-AAGTCGACATGGACATGAGGGTGCC-3′) and a murine Cκ reverse primer (5′-GCACCTCCAGATGTTAACTGC-3′).
Flow Cytometry and Confocal Microscopy.
Antibodies were from Southern Biotechnology Associates. Cells from spleen and bone marrow were washed in RPMI medium supplemented with 10% FCS/2% rabbit serum (to avoid Fc receptor staining) and stained with appropriate antibodies (rat anti-mouse B220-SPRD, CD19-PE, CD43-PE, CD25-PE, CD5-PE, CD21-PE, CD23-PC7, IgM-FITC, IgD-PE, IgA-PE, and goat anti-mouse λ LC-biotin). Biotin-conjugated anti-λ antibodies were revealed by Spectral red-conjugated streptavidin. For some experiments, anti-κ or anti-λ antibodies were labeled with Alexa 488, 594, or 633 (Molecular Probes). Fluorescence analysis and cell sorting were carried out on a FACSVantage flow cytometer calibrated every day by using fluorescent beads (Becton Dickinson).
For confocal microscopy, bone marrow cells and hybridomas were stained with Alexa 488-conjugated goat anti-mouse κ LC, Alexa 594-conjugated goat anti-mouse λ LC, and biotin-conjugated goat anti-mouse IgG (γ HC-specific) or IgG1, followed by Cy5-conjugated streptavidin. Cells were allowed to adhere to poly(l-lysine)-treated glass slides for 30 min at 37°C. After fixation with a 4% paraformaldehyde/PBS solution, cells were permeabilized for 30 min at 20°C with PBS, 0.2% gelatin, and 0.1% saponin, washed in PBS, and incubated for 45 min at 37°C with the appropriate conjugated antibodies. Slides were examined by using an LSM 510 (Zeiss) confocal microscope and the ×40 or ×63 objective. When necessary, B cell enrichment over 90% purity (B220+ cells) was performed by using CD43-coated magnetic beads (Miltenyi Biotec).
Spleen Cell Cultures and Generation of Hybridomas.
Splenocytes were cultured for 3 days at 5 × 105 cells per ml in RPMI medium supplemented with 10% FCS and 20 μg/ml LPS, and then submitted to FACS analysis. Hybridomas were obtained from hemizygous mutant mice splenocytes as described in ref. 39.
ELISA Assays.
Antibodies were from Southern Biotechnology Associates. The various IgH classes containing λ LC and the presence of κ/λ hybrid Ig were estimated. Plates were coated with 2 μg/ml goat anti-mouse λ LC antibodies. After blocking with 1% BSA/PBS buffer and washing, successive dilutions of assayed sera, hybridoma supernatants, or standards for each Ig class were incubated for 2 h at 37°C. Bound Ig were revealed with the relevant alkaline phosphatase-conjugated goat anti-mouse Ig class (IgM, IgG1, IgG2a, IgG2b, or IgG3) or goat anti-mouse κ LC (1 μg/ml) added (1.5 h at 37°C). Standards for ELISAs quantifying the various λ LC-associated IgH classes were sera from homozygous κD mice containing only λ-associated Ig and precisely quantified for their amounts of each Ig class as described in ref. 40. For rheumatoid factors (IgM antibodies specific for IgG), a similar ELISA was used with goat anti-mouse IgG-coated plates; bound rheumatoid factors were revealed with an alkaline phosphatase-labeled goat anti-mouse IgM.
Autoantibody Testing.
Hep2 cells seeded at 0.4 × 106 cells per ml on microscope slides were grown overnight in 15% fetal calf serum RPMI medium. Slides were fixed in 50% ethanol/50% acetone. For ANA determinations, sera were diluted 1:20 and 1:80 and incubated for 1 h on Hep2 slides. After washing, slides were further incubated for 1 h with a FITC-conjugated anti-mouse Ig (DAKO). Sera from κCκD and κCH mice were quoted by immunofluorescence microscopy for the presence of ANA, by comparison with sera from WT mice and control positive sera from lpr mice. The presence of antiphospholipid or antimitochondrial antibodies was also checked in sera from κCκD and κCH mice by using ELISA with Varelisa assays (Pharmacia).
Supplementary Material
Acknowledgments
We thank Sylvie Desforges, Nadine Cogné, Marc Lebert, and Chantal Jayat-Vignoles for help and advice. This work was supported by grants from Ligue Nationale Contre le Cancer and Conseil Régional du Limousin.
Abbreviations
- ANA
antinuclear antibodies
- BCR
B cell receptor
- HC
heavy chain
- LC
light chain
- MZ
marginal zone
- QPCR
quantitative PCR
- RS
recombining segment.
Footnotes
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Rajewsky K. Nature. 1996;381:751–758. doi: 10.1038/381751a0. [DOI] [PubMed] [Google Scholar]
- 2.Meffre E., Casellas R., Nussenzweig M. C. Nat. Immunol. 2000;1:379–385. doi: 10.1038/80816. [DOI] [PubMed] [Google Scholar]
- 3.Burnet F. M. The Clonal Selection Theory of Acquired Immunity. Cambridge, U.K.: Cambridge Univ. Press; 1959. [Google Scholar]
- 4.Nussenzweig M. C., Shaw A. C., Sinn E., Danner D. B., Holmes K. L., Morse H. C., III, Leder P. Science. 1987;236:816–819. doi: 10.1126/science.3107126. [DOI] [PubMed] [Google Scholar]
- 5.Manz J., Denis K., Witte O., Brinster R., Storb U. J. Exp. Med. 1988;168:1363–1381. doi: 10.1084/jem.168.4.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Storb U., Engler P., Manz J., Gollahon K., Denis K., Lo D., Brinster R. Ann. N.Y. Acad. Sci. 1988;546:51–56. doi: 10.1111/j.1749-6632.1988.tb21618.x. [DOI] [PubMed] [Google Scholar]
- 7.Giachino C., Padovan E., Lanzavecchia A. J. Exp. Med. 1995;181:1245–1250. doi: 10.1084/jem.181.3.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barreto V., Cumano A. J. Immunol. 2000;164:893–899. doi: 10.4049/jimmunol.164.2.893. [DOI] [PubMed] [Google Scholar]
- 9.Pauza M. E., Rehmann J. A., LeBien T. W. J. Exp. Med. 1993;178:139–149. doi: 10.1084/jem.178.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diaw L., Siwarski D., DuBois W., Jones G., Huppi K. Mol. Immunol. 2000;37:775–781. doi: 10.1016/s0161-5890(00)00100-0. [DOI] [PubMed] [Google Scholar]
- 11.Mostoslavsky R., Singh N., Kirillov A., Pelanda R., Cedar H., Chess A., Bergman Y. Genes Dev. 1998;12:1801–1811. doi: 10.1101/gad.12.12.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldmit M., Schlissel M., Cedar H., Bergman Y. EMBO J. 2002;21:5255–5261. doi: 10.1093/emboj/cdf518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hertz M., Kouskoff V., Nakamura T., Nemazee D. Nature. 1998;394:292–295. doi: 10.1038/28419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Torres R. M., Flaswinkel H., Reth M., Rajewsky K. Science. 1996;272:1804–1808. doi: 10.1126/science.272.5269.1804. [DOI] [PubMed] [Google Scholar]
- 15.Yu W., Nagaoka H., Jankovic M., Misulovin Z., Suh H., Rolink A., Melchers F., Meffre E., Nussenzweig M. C. Nature. 1999;400:682–687. doi: 10.1038/23287. [DOI] [PubMed] [Google Scholar]
- 16.Monroe R. J., Seidl K. J., Gaertner F., Han S., Chen F., Sekiguchi J., Wang J., Ferrini R., Davidson L., Kelsoe G., Alt F. W. Immunity. 1999;11:201–212. doi: 10.1016/s1074-7613(00)80095-3. [DOI] [PubMed] [Google Scholar]
- 17.Radic M. Z., Erikson J., Litwin S., Weigert M. J. Exp. Med. 1993;177:1165–1173. doi: 10.1084/jem.177.4.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tiegs S. L., Russell D. M., Nemazee D. J. Exp. Med. 1993;177:1009–1020. doi: 10.1084/jem.177.4.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pelanda R., Schwers S., Sonoda E., Torres R. M., Nemazee D., Rajewsky K. Immunity. 1997;7:765–775. doi: 10.1016/s1074-7613(00)80395-7. [DOI] [PubMed] [Google Scholar]
- 20.Retter M. W., Nemazee D. J. Exp. Med. 1998;188:1231–1238. doi: 10.1084/jem.188.7.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Casellas R., Shih T. A., Kleinewietfeld M., Rakonjac J., Nemazee D., Rajewsky K., Nussenzweig M. C. Science. 2001;291:1541–1544. doi: 10.1126/science.1056600. [DOI] [PubMed] [Google Scholar]
- 22.Kenny J. J., Rezanka L. J., Lustig A., Fischer R. T., Yoder J., Marshall S., Longo D. L. J. Immunol. 2000;164:4111–4119. doi: 10.4049/jimmunol.164.8.4111. [DOI] [PubMed] [Google Scholar]
- 23.Iliev A., Spatz L., Ray S., Diamond B. J. Immunol. 1994;153:3551–3556. [PubMed] [Google Scholar]
- 24.Li Y., Li H., Weigert M. J. Exp. Med. 2002;195:181–188. doi: 10.1084/jem.20011453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rice J. S., Newman J., Wang C., Michael D. J., Diamond B. Proc. Natl. Acad. Sci. USA. 2005;102:1608–1613. doi: 10.1073/pnas.0409217102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu S., Velez M. G., Humann J., Rowland S., Conrad F. J., Halverson R., Torres R. M., Pelanda R. J. Immunol. 2005;175:5067–5076. doi: 10.4049/jimmunol.175.8.5067. [DOI] [PubMed] [Google Scholar]
- 27.Kouskoff V., Lacaud G., Pape K., Retter M., Nemazee D. Proc. Natl. Acad. Sci. USA. 2000;97:7435–7439. doi: 10.1073/pnas.130182597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Braun U., Rajewsky K., Pelanda R. Proc. Natl. Acad. Sci. USA. 2000;97:7429–7434. doi: 10.1073/pnas.050578497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shivtiel S., Leider N., Sadeh O., Kraiem Z., Melamed D. J. Immunol. 2002;168:5596–5604. doi: 10.4049/jimmunol.168.11.5596. [DOI] [PubMed] [Google Scholar]
- 30.Storb U., Ritchie K. A., O’Brien R., Arp B., Brinster R. Immunol. Rev. 1986;89:85–102. doi: 10.1111/j.1600-065x.1986.tb01474.x. [DOI] [PubMed] [Google Scholar]
- 31.Pelanda R., Schaal S., Torres R. M., Rajewsky K. Immunity. 1996;5:229–239. doi: 10.1016/s1074-7613(00)80318-0. [DOI] [PubMed] [Google Scholar]
- 32.Borrero M., Clarke S. H. J. Immunol. 2002;168:13–21. doi: 10.4049/jimmunol.168.1.13. [DOI] [PubMed] [Google Scholar]
- 33.Noorchashm H., Bui A., Li H. L., Eaton A., Mandik-Nayak L., Sokol C., Potts K. M., Pure E., Erikson J. Int. Immunol. 1999;11:765–776. doi: 10.1093/intimm/11.5.765. [DOI] [PubMed] [Google Scholar]
- 34.Gerdes T., Wabl M. Nat. Immunol. 2005;5:1282–1287. doi: 10.1038/ni1133. [DOI] [PubMed] [Google Scholar]
- 35.Wardemann H., Yurasov S., Schaefer A., Young J. W., Meffre E., Nussenzweig M. C. Science. 2003;301:1374–1377. doi: 10.1126/science.1086907. [DOI] [PubMed] [Google Scholar]
- 36.Fukuyama H., Nimmerjahn S., Ravetch J. V. Nat. Immunol. 2005;6:99–106. doi: 10.1038/ni1151. [DOI] [PubMed] [Google Scholar]
- 37.Leslie D., Lipsky P., Notkins A. L. J. Clin. Invest. 2001;108:1417–1422. doi: 10.1172/JCI14452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xin H., Janeway A. C., Jr., Levine M., Robinson E., Preston-Hurlburt P., Viret C., Bottomly K. Nat. Immunol. 2002;3:127–134. doi: 10.1038/ni751. [DOI] [PubMed] [Google Scholar]
- 39.Rengers J. U., Touchard G., Decourt C., Deret S., Michel H., Cogne M. Blood. 2000;95:3467–3472. [PubMed] [Google Scholar]
- 40.Pinaud E., Khamlichi A. A., Le Morvan C., Drouet M., Nalesso V., Le Bert M., Cogne M. Immunity. 2001;15:187–199. doi: 10.1016/s1074-7613(01)00181-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}