

Abstract
Viral terminases play essential roles as components of molecular motors that package viral DNA into capsids. Previous results indicated that the putative terminase subunits of herpes simplex virus 1 (HSV-1) encoded by UL15 and UL28 (designated pUL15 and pUL28, respectively) coimmunoprecipitate with the UL33 protein from lysates of infected cells. All three proteins are among six required for HSV-1 DNA packaging but dispensable for assembly of immature capsids. The current results show that in both infected- and uninfected-cell lysates, pUL28 coimmunoprecipitates with either pUL33 or pUL15, whereas pUL15 and pUL33 do not coimmunoprecipitate unless pUL28 is present. The UL28 protein was sufficient to stabilize pUL33 from proteasomal degradation in an engineered cell line and was necessary to stabilize pUL33 in infected cells, whereas pUL15 had no such effects. The presence of pUL33 was dispensable for the pUL15/pUL28 interaction in lysates of both infected and uninfected cells but augmented the tendency for pUL15 and pUL28 to coimmunoprecipitate. These data suggest that pUL28 and pUL33 interact directly and that pUL15 interacts directly with pUL28 but only indirectly with pUL33. It is logical to propose that the indirect interaction of pUL15 and pUL33 is mediated through the interaction of both proteins with pUL28. The data also suggest that one function of pUL33 is to optimize the pUL15/pUL28 interaction.
Late in infection with all herpesviruses, capsids lacking DNA and viral concatameric DNA accumulate in infected-cell nuclei. By analogy to double-stranded DNA bacteriophages, it is presumed that capsid assembly culminates when a viral terminase cleaves the concatameric DNA into genomic lengths and hydrolyzes ATP to drive the DNA through a unique structure within the capsid, termed the portal vertex. In the case of herpes simplex virus (HSV), the portal vertex is likely composed of a dodecameric ring of the UL6 protein (pUL6) (16, 21).
Terminases consist of at least two subunits in all viral systems studied to date (7). Although obtaining direct evidence for the identity of the terminase subunits in herpesviruses has been hampered by the lack of an in vitro packaging system, several lines of indirect evidence have implicated the products of UL15 and UL28 (pUL15 and pUL28, respectively) as terminase components as follows: (i) pUL15 and pUL28 interact in vitro and in vivo with one another and in vitro with the portal protein pUL6 (1, 6, 11, 12, 22), (ii) pUL28 has been shown to bind DNA sequences necessary for formation of genomic ends (2), (iii) pUL15 contains a highly conserved Walker box motif that is essential for HSV DNA packaging and resembles motifs maintained in the ATPase domains of some bacteriophage terminases (9, 15, 23), and (iv) it is likely that the terminase functions are conserved, inasmuch as the homologs of pUL15 and pUL28 in human cytomegalovirus (hCMV), encoded by UL89 and UL56, respectively, which also interact, have been shown to form a complex with the hCMV portal protein and are required for DNA packaging (10, 13).
The approximately 19,000-Mr protein encoded by herpes simplex virus 1 (HSV-1) UL33 has also been shown to interact with pUL15 and pUL28 by immunoprecipitation from lysates of HSV-infected cells (6). Although its exact function is not known, pUL33, like pUL15 and pUL28, is required for DNA cleavage and packaging (3, 8). Thus, engineered mutations in any of these genes can cause empty capsids lacking DNA to accumulate in infected cells (3, 4, 20). Small amounts of pUL33, pUL15, and pUL28 have also been shown to associate with HSV-1 capsids, suggesting that they maintain their interaction during packaging (5, 18, 24).
Because it would provide information about the HSV terminase, one goal of the present work was to characterize the roles of the individual proteins in the formation of the pUL15/pUL33/pUL28 complex.
MATERIALS AND METHODS
Cells and virus.
Vero cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% newborn calf serum, 100 U of penicillin per ml, and 100 μg of streptomycin per ml (growth medium). A UL15 deletion virus was propagated on a cell line designated clone 17 as previously described (4). A previously described cell line designated D4 was used to propagate the UL33 deletion virus (17). Clone 17 and D4 cell lines were maintained in growth medium supplemented with 500 μg G418 per ml. HSV-1(F) virus and the UL15, UL28, and UL33 null viruses have been described previously (4, 8, 20). The CV28 cell line described herein was used to propagate the previously described UL28 deletion virus (20).
Antibodies.
Polyclonal rabbit antisera recognizing the first 35 amino acids of pUL15 (designated UL15N), the C terminus of pUL15, and the entire UL28- and UL33-encoded proteins have been described previously (6, 17, 19). Actin antibody was purchased from Santa Cruz Biotechnology.
Plasmids.
Plasmid pJB125 contained the UL15 cDNA in vector pCDNA3 (Invitrogen), whereas plasmid pJB112 contained the UL28 coding sequence cloned into this vector. To construct a shuttle plasmid (designated pJB401) to transfer UL28 sequences into a defined site within CV1 cells, pJB112 was cleaved with BamHI and EcoRV, and a 2.36-kb DNA fragment containing the entire UL28 open reading frame was isolated and cloned into pCDNA5/FRT vector (Invitrogen) at the BamHI and EcoRV sites. This vector contains the hygromycin resistance gene with an Flp recombinase targeting site (FRT site) embedded in the 5′ coding sequence. To construct pJB433, a PCR amplicon from HSV-1(F) DNA containing the entire UL33 coding sequence was cloned into pCDNA3 at the HindIII and EcoRI sites. pJB433 was digested with HindIII and EcoRI, and the UL33 coding sequences were gel purified and cloned into pCDNA5/FRT at HindIII and EcoRI sites. The resultant plasmid was designated pJB481. The genotype of each plasmid was confirmed by DNA sequencing.
Transfections for transient expression.
Ninety-five-percent-confluent cells were transfected with the plasmids indicated in Results by use of Lipofectamine 2000 according to the manufacturer's protocol (Invitrogen). Cells were harvested at 24 h posttransfection and subjected to either immunoprecipitation or immunoblot analysis as described below.
Construction and maintenance of novel UL33- and UL28-expressing cell lines.
Complementing cell lines were constructed by using the Flp-In-CV-1 system (Invitrogen) according to the manufacturer's protocol. Briefly, either pJB481 or pJB401 (see above) was cotransfected with a plasmid (pOG44) containing Flp recombinase under the control of a constitutive hCMV promoter/enhancer into an engineered cell line (Flp-CV1). This cell line was derived from CV1 cells (a derivative of Vero cells) and contains an Flp target sequence (FRT) at a single locus that also bears a lacZ gene fused to a gene encoding zeocin resistance. Transcription of the fused gene was driven by the simian virus 40 early promoter. The Flp recombination event was expected to cause insertion of the pCDNA5/FRT construct into the cellular genome at the integrated FRT site. Insertion of the pCDNA5/FRT construct at this site was expected to bring the simian virus 40 promoter and the ATG initiation codon in frame with the hygromycin resistance gene, with concomitant inactivation of the lacZ-Zeor fusion gene.
After recombination, cells resistant to hygromycin were selected by growth in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 200 μg/ml hygromycin B. Once hygromycin-resistant foci were identified, the cells were trypsinized and pooled. Monolayers of the entire population of cells containing either UL33 or UL28 were screened for the ability to complement the growth of the UL33 or UL28 null mutants, respectively. All tested cell populations were able to complement the replication of the corresponding viral null mutants (not shown), and the cells were designated CV33 and CV28, respectively. CV28 and CV33 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% newborn calf serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and 200 μg/ml hygromycin B.
Immunoprecipitation and immunoblotting.
Cells were washed with cold phosphate-buffered saline (PBS) and resuspended in radioimmunoprecipitation assay buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1% NP-40, 0.25% sodium deoxycholate, 1 mM EDTA, 2 mM phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, 5 μg/ml leupeptin, 10 μg/ml pepstatin, 10 mM NaF, 0.1 mM Na3VO4). After incubation on ice for 30 min without sonication, the lysates (800 μl from 8.8 × 106 cells) were clarified at 14,000 rpm for 15 min at 4°C in a microcentrifuge. The supernatants of all lysates were precleared by reaction with preimmune rabbit serum and 30 μl of a 50% slurry of Gammabind G-Sepharose beads (Amersham Pharmacia Biotech) for 2 h at 4°C with constant rotation. After the beads were pelleted by centrifugation, the supernatants were incubated with rabbit antibodies directed against pUL15, pUL28, or pUL33 for 2 h at 4°C. Thirty microliters of a 50% slurry of Gammabind G-Sepharose beads was then added. The mixture was incubated overnight at 4°C with constant rotation. The beads were washed four times with excess radioimmunoprecipitation assay buffer, and immune complexes were eluted in loading buffer (62.5 mM Tris, pH 6.8, 2% sodium dodecyl sulfate [SDS], 5% β-mercaptoethanol, 12.5% glycerol) and boiled for 10 min. The immunoprecipitated material was electrophoretically separated on 12% SDS-polyacrylamide gels, and proteins were transferred electrically to nitrocellulose. In some experiments, a portion of the lysates was denatured in loading buffer, electrophoretically separated, and transferred to nitrocellulose.
Nitrocellulose sheets were washed twice in PBS and blocked overnight in PBS supplemented with 10% nonfat dry milk (Carnation). Primary rabbit polyclonal antibodies directed against the C terminus of pUL15 or pUL28 were diluted 1:1,000 in PBS supplemented with 2% bovine serum albumin, whereas anti-pUL33 rabbit polyclonal antibody was diluted 1:400, as previously described (17). Actin-specific antibody was diluted 1:200 according to the manufacturer's protocol. The diluted antibodies were reacted with the blocked nitrocellulose sheets for 2 h at room temperature and washed, and horseradish peroxidase-conjugated anti-rabbit immunoglobulin G diluted 1:5,000 in PBS plus 2% bovine serum albumin was added for 2 h at room temperature. The bound immunoglobulins were revealed by enhanced chemiluminescence (Amersham Pharmacia Biotech). Where applicable, the image intensities of bands on immunoblots were quantified with a Molecular Dynamics PhosphorImager before exposure to radiographic film.
To strip and reprobe the immunoblots, developed blots were incubated in a buffer containing 62.5 mM Tris-HCl (pH 6.8), 2% SDS, and 100 mM β-mercaptoethanol at 50°C for 30 min as described in the ECL manual (Amersham), followed by immunoblotting as described above.
RESULTS
pUL33 interacts directly with pUL28 and indirectly with pUL15.
To investigate the roles of individual proteins in complex formation, cells were mock infected or were infected with wild-type HSV-1(F) or viral deletion mutants lacking UL15, UL28, or UL33. Cells were then lysed, and the pUL15, pUL33, and pUL28 proteins were separately immunoprecipitated from the lysates with appropriate antibodies. Whether pUL33 was present in immunoprecipitated material was then determined by immunoblotting. The results indicated that pUL33 was immunoprecipitated with the pUL33-specific antibody from lysates of cells infected with wild-type HSV-1(F) (not shown) and the UL28 and UL15 deletion viruses (Fig. 1, lanes 4 and 6). This indicated that the absence of pUL15 or pUL28 did not preclude expression of pUL33 within infected cells. On the other hand, levels of pUL33 were consistently lower in immunoprecipitations from cells infected with the UL28 and UL15 deletion viruses than in immunoprecipitations from cells infected with wild-type virus (not shown).
FIG. 1.

Immunoblot probed with anti-pUL33 polyclonal antibody. Vero cells were mock infected (Mock, lane 1) or infected with UL15 null (Δ15, lanes 3 and 4), UL28 null (Δ28, lanes 5 and 6), UL33 null (Δ33, lane 2) or wild-type HSV-1(F) (F, lane 7) virus at a multiplicity of infection of 5 PFU/cell. At 18 h p.i., lysates of the cells were subjected to immunoprecipitations with anti-pUL15N (α15N, lanes 5 and 7), anti-pUL28 (α28, lanes 2 and 3), or anti-pUL33 (α33, lanes 1, 4, and 6) polyclonal antibody. The immunoprecipitates were electrophoretically separated on a 12% SDS-polyacrylamide gel, transferred onto a nitrocellulose membrane, and probed with anti-pUL33 polyclonal antibody. Bound immunoglobulin was revealed by enhanced chemiluminescence. Virus and Ab, respectively, indicate the infecting virus and the antibody used for immunoprecipitation for that particular experiment.
As expected, pUL33 was not immunoprecipitated from lysates of mock- or UL33 deletion virus-infected cells (Fig. 1, lanes 1 and 2). Reaction of HSV-1(F)-infected-cell lysates with antibody directed against the N terminus of pUL15 caused coimmunoprecipitation of pUL33 (Fig. 1, lane 7). Similarly, the pUL28-specific antiserum caused coimmunoprecipitation of pUL33 from lysates of cells infected with the UL15 deletion virus (Fig. 1, lane 3), indicating that UL15 was not necessary for the interaction between pUL33 and pUL28. Surprisingly, however, pUL33 was not coimmunoprecipitated with pUL15 N-terminal-specific antibody from lysates of cells infected with the UL28 deletion mutant (Fig. 1, lane 5), despite the presence of ample pUL33 within the lysate (Fig. 1, lane 6). These data indicate that pUL28 is necessary for pUL33 to interact with pUL15 in infected cells and suggest that the pUL15/pUL33 interaction is indirect and normally mediated through pUL28.
The presence of pUL33 enhances the pUL15/pUL28 interaction.
Cells were mock infected or were infected with wild-type HSV-1(F) or viral deletion mutants lacking UL15, UL28, or UL33. Lysates were prepared, clarified, and reacted with antibodies against pUL15N, pUL28, or pUL33, and the presence of pUL15 in the immunoprecipitations was monitored by immunoblotting. The anti-C-terminal pUL15 antiserum was used for immunoblotting throughout this study due to its high sensitivity and specificity in this assay. The UL15 protein was readily immunoprecipitated with the pUL15N-specific antibody from lysates of cells infected with wild-type virus (not shown) and the UL28 and UL33 deletion viruses (Fig. 2, lanes 4 and 5) but was not immunoprecipitated from lysates of mock- or UL15 deletion mutant-infected cells (Fig. 2, lanes 1 and 2). Reaction of HSV-1(F)-infected-cell lysates with pUL33-specific antibody caused coimmunoprecipitation of pUL15 (Fig. 2, lane 7), whereas pUL15/pUL33 coimmunoprecipitation did not occur in lysates of cells infected with the UL28 deletion virus (Fig. 2, lane 3). These observations were consistent with previous data (Fig. 1) demonstrating that pUL28 was necessary for the pUL15/pUL33 interaction. Importantly, antibody against pUL28 reacted with lysates of cells infected with the UL33 deletion virus immunoprecipitated only a portion of the pUL15 that was immunoprecipitated with its cognate antibody (compare Fig. 2, lanes 5 and 6). These data indicate that while pUL33 is ultimately dispensable for the pUL15/pUL28 interaction, it also acts in some way to enhance the interaction in infected-cell lysates.
FIG. 2.

Immunoblot probed with anti-pUL15 antibody. Vero cells were mock infected (Mock, lane 1) or infected with UL15 null (Δ15, lane 2), UL28 null (Δ28, lanes 3 and 4), UL33 null (Δ33, lanes 5 and 6), or HSV-1(F) (F, lane 7) virus at a multiplicity of infection of 5 PFU/cell. At 18 h p.i., the cells were lysed and subjected to immunoprecipitation with antibodies against the N terminus of pUL15 (α15N, lanes 1, 4, and 5), pUL28 (α28, lane 6), or pUL33 (α33, lanes 2, 3, and 7). The immunoprecipitates were probed for the presence of pUL15 by immunoblotting with an antibody directed against the C terminus of pUL15. Bound immunoglobulin was revealed by enhanced chemiluminescence. Virus and Ab indicate the infecting virus and the immunoprecipitating antibody, respectively.
To confirm some of the above results, lysates of mock-infected cells or cells infected with HSV-1(F) or the UL15, UL33, or UL28 deletion virus were reacted with antibodies to these proteins, and the immunoprecipitated material was probed for the presence of pUL28 by immunoblotting. The UL28 protein was not immunoprecipitated from lysates of mock-infected cells or cells infected with the UL28 deletion virus (Fig. 3, lanes 1 and 2). In contrast, pUL28 was readily immunoprecipitated from lysates of cells infected with wild-type virus (not shown) and deletion viruses lacking either UL15 or UL33 (Fig. 3, lanes 3 and 6). Of interest was the observation that despite the presence of pUL28 in the lysates of cells infected with the UL33 deletion mutant, antibody to the pUL15 N terminus coimmunoprecipitated only barely detectable levels of pUL28 (Fig. 3, lane 5). This observation further indicated that the pUL15/pUL28 interaction was significantly augmented by the presence of pUL33. Moreover, antibody directed against pUL33 coimmunoprecipitated pUL28 from lysates of cells infected with the UL15 deletion mutant (Fig. 3, lane 4), indicating that the absence of pUL15 did not preclude an interaction between pUL33 and pUL28.
FIG. 3.

Immunoblot probed with anti-pUL28 antibody. Vero cells were mock infected (Mock, lane 1) or infected with UL15 null (Δ15, lanes 3 and 4), UL28 null (Δ28, lane 2), UL33 null (Δ33, lanes 5 and 6), or HSV-1(F) (F, lane 7) virus. At 18 h p.i., immunoprecipitations were performed with antibodies against the N terminus of pUL15 (α15N, lanes 5 and 7), pUL28 (α28, lanes 1, 3, and 6), or pUL33 (α33, lanes 2 and 4). The immunoprecipitates were denatured, separated on denaturing polyacrylamide gels, and transferred onto a nitrocellulose membrane, followed by immunoblotting with pUL28-specific polyclonal antibody. Virus and Ab indicate the infecting virus and the immunoprecipitating antibody, respectively.
pUL28 is necessary and sufficient to mediate the pUL15/pUL33 interaction in mammalian cells.
To determine if pUL28 was sufficient to mediate the interaction between pUL33 and pUL15, a cell line expressing pUL33 was constructed as described in Materials and Methods. This cell line (CV33) was able to support replication of the UL33 deletion mutant (data not shown). CV33 cells were transfected with plasmids expressing UL15, UL28, or both. Twenty-four hours later, lysates were prepared and either electrophoretically separated or immunoprecipitated with pUL33-specific antibody, followed by electrophoretic separation on denaturing polyacrylamide gels. In both cases, separated material was transferred to nitrocellulose and analyzed for the presence of pUL15 and pUL28 by immunoblotting. Transfection of plasmids containing UL15 and UL28 into CV33 cells caused production of detectable levels of the UL15- and UL28-encoded proteins (Fig. 4, lanes 2 to 4). The pUL33-specific antibody readily coimmunoprecipitated pUL28 from lysates of CV33 cells expressing pUL28 or coexpressing pUL28 and pUL15 (Fig. 4A, lanes 6 and 8), indicating that pUL28 and pUL33 can interact in the absence of other HSV proteins. In contrast, the pUL33-specific antibody coimmunoprecipitated pUL15 only from lysates of CV33 cells expressing both pUL28 and pUL15 (Fig. 4B, lane 8), whereas pUL15 was not coimmunoprecipitated by the pUL33-specific antibody when pUL15 was expressed in the absence of pUL28 (Fig. 4B, lane 7). These data therefore indicate that pUL28 is both necessary and sufficient to mediate the interaction between pUL15 and pUL33.
FIG. 4.

Immunoblots of CV33 cells (CV1 cells expressing pUL 33) transiently expressing pUL28 and/or pUL15. CV33 cells were mock transfected (Mock) or transfected with plasmids expressing the genes indicated above each lane (28, pUL28; 15, pUL15; 15+28, pUL15 and pUL28). Lysates were prepared 24 h later, and lysates (lanes 1 to 4) or immunoprecipitations from these lysates obtained using pUL33-specific antibody (lanes 5 to 8) were electrophoretically separated, transferred to nitrocellulose, and probed with the pUL28-specific antisera (A) or antisera against the C terminus of pUL15 (B).
In the reciprocal reaction, lysates of CV33 cells that were mock transfected or transfected with plasmids expressing either UL15 or UL28 or both were subjected to immunoprecipitation with antibodies directed against either the N terminus of pUL15 (Fig. 5, lanes 3 and 4) or pUL28 (Fig. 5, lanes 1 and 2). The presence of pUL33 and pUL15 in the immunoprecipitated material was then determined by immunoblotting. The UL33-encoded protein was coimmunoprecipitated with the pUL28-specific antibody when pUL28 was expressed in the absence of pUL15 (Fig. 5A, lane 2). In contrast, and despite the presence of ample amounts of pUL15 in UL15N antibody-immunoprecipitated material (Fig. 5B, lane 3), pUL33 was coimmunoprecipitated with the pUL15N-specific antibody only when pUL28 was coexpressed with pUL15 (Fig. 5A, lane 4). These data further support the conclusion that pUL28 is not only necessary for the pUL33/pUL15 interaction but also sufficient to mediate the interaction in the absence of other HSV proteins.
FIG. 5.
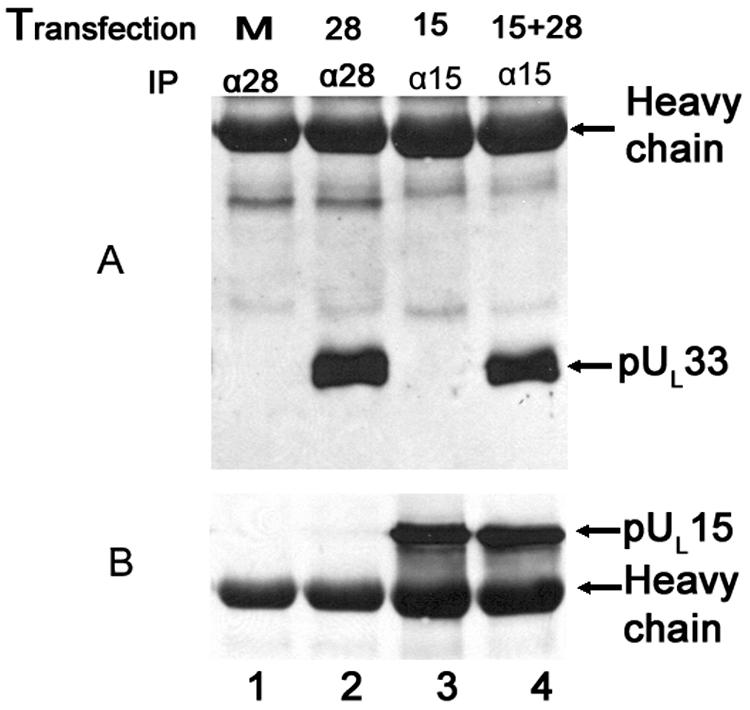
Immunoblots of CV33 cell lysates immunoprecipitated with pUL15N- and pUL28-specific antibodies. CV33 cells were mock transfected (M, lane 1) or transfected with plasmids expressing UL28 (28, lane 2), UL15 (15, lane 3), or UL15 and UL28 (15+28, lane 4). Twenty-four hours later, lysates were prepared and immunoprecipitations (IP) were performed with antibodies directed against pUL28 (α28, lanes 1 and 2) or the N terminus of pUL15 (α15, lanes 3 and 4). Immunoblots of the immunoprecipitated material were probed with antibody directed against pUL33 (A). The same blots were stripped as described in Materials and Methods and reprobed with antibody against the C terminus of pUL15 (B).
pUL28 can protect pUL33 from efficient degradation by the proteosome.
Preliminary evidence indicated that pUL33 in CV33 cells was difficult to detect by immunoblotting unless the cells were infected with wild-type HSV-1 (not shown). To determine whether the decreased amounts of pUL33 in uninfected CV33 cells were a consequence of its proteasomal degradation or poor overall expression, CV33 cells were untreated or treated with the proteosome inhibitor MG132 or lactacystin or with a similar amount of dimethyl sulfoxide (DMSO), which was used as a carrier in the MG132 treatment. As shown in Fig. 6A, treatment with various amounts of MG132 significantly increased the amount of detectable pUL33 in CV33 cells over levels obtained after mock treatment or treatment with DMSO. This suggested that pUL33 within CV33 cells was efficiently degraded by the proteosome. To confirm the results, we repeated this experiment with the proteosome inhibitor lactacystin and, as controls, chloroquine (an inhibitor of the lysosomal degradation pathway) and calpain inhibitor 2 (which blocks calpain proteases). As shown in Fig. 6B, both MG132 and lactacystin substantially increased the amount of detectable pUL33 above levels obtained in cells that were not treated, whereas chloroquine, DMSO, or calpain inhibitor 2 did not dramatically increase the levels of pUL33. We therefore conclude that pUL33 is efficiently degraded by the proteosome in uninfected CV33 cells.
FIG. 6.

Immunoblots of CV33 cells treated with proteosome inhibitors. CV33 cells (CV1 cells engineered to express pUL33) were treated for 6 h with the indicated compounds, at which time cell lysates were prepared and separated by SDS-polyacrylamide gel electrophoresis. After transfer to a nitrocellulose membrane, immunoblotting was performed using antibodies against pUL33 (upper panels) or actin as a loading control (lower panels).
Because the evidence presented above indicated that pUL28 and pUL33 interacted directly, we hypothesized that the interaction of pUL28 with pUL33 might increase the stability of the latter. To test this possibility, CV33 cells were transfected with expression plasmids containing UL28, UL15, or both, the cells were lysed 24 h later, and the presence of pUL33 and actin (as a loading control) in the lysates was determined by immunoblotting.
As shown in Fig. 7, the presence of pUL28 correlated with a greatly increased level of pUL33-specific immunoreactivity in the CV33 cells, whereas expression of pUL15 did not increase the amount of detectable pUL33 above that obtained upon mock transfection (compare Fig. 7A, lanes 1 to 3). Coexpression of pUL15 and pUL28 also increased levels of pUL33 immunoreactivity (Fig. 7A, lane 4). These data indicate that UL28 causes increased amounts of pUL33 to accumulate in the CV33 cell line, whereas UL15 did not induce such effects. Taken together with the knowledge that pUL33 is normally degraded by the proteosome in CV33 cells, these data suggest that the increased stability of pUL33 conferred by expression of pUL28 is a consequence of the interaction of these proteins.
FIG. 7.

Immunoblots of pUL33 in the presence and absence of UL28 and UL15. CV33 cells were mock transfected (Mock) or transfected with plasmids expressing the indicated open reading frames (28, UL28; 15, UL15; 15+28, UL15 and UL28). Twenty-four hours later, cell lysates were prepared and separated by SDS-polyacrylamide gel electrophoresis. After transfer to a nitrocellulose membrane, immunoblotting was performed using the polyclonal antibody against pUL33 (A), the C terminus of pUL15 (B), or pUL28 (C) or actin as a loading control (D). (E) Vero cells were infected with the indicated virus (Δ15, UL15 null virus; Δ28, UL28 null virus) or mock infected (Mock) and were incubated in the presence or absence of 10 mm lactacystin or the DMSO carrier from 12 to 18 h p.i. Immunoblots of lysates harvested at 18 h p.i. were probed with the antibody against pUL33. A cellular protein recognized by the pUL33 antibody served as a loading control.
To determine whether pUL28 or pUL15 could increase the stability of pUL33 in infected cells, Vero cells were mock infected or were infected with wild-type virus or deletion viruses lacking UL28 or UL15. At 12 h postinfection (p.i.), the infected cells were left untreated or were treated with 10 μM lactacystin or the equivalent concentration of the DMSO carrier, and lysates of the cells were prepared at 18 h p.i. Immunoblots of the lysates were then probed with pUL33 antibody. As shown in Fig. 7E, less pUL33 was detected in cells infected with the UL28 deletion mutant (Fig. 7E, lanes 2 and 3) than in cells infected with either the wild-type virus (Fig. 7E, lanes 8 and 9) or the UL15 deletion virus (Fig. 7E, lanes 5 and 6). Moreover, lactacystin treatment increased the accumulation of pUL33 in cells infected with the UL28 deletion mutant (Fig. 7E, lane 4) but did not affect accumulation of pUL33 in cells infected with wild-type HSV-1(F) (Fig. 7E, lane 9) or the UL15 deletion virus (Fig. 7E, lane 7). These data indicate that pUL28 is necessary for protection of pUL33 from proteasomal degradation in infected cells, whereas the presence of pUL15 does not affect pUL33 accumulation.
pUL33 augments coimmunoprecipitation of pUL15 and pUL28 in a pUL28-expressing cell line.
To determine if pUL33 could enhance the pUL15/pUL28 interaction in the absence of other HSV proteins, a UL28-expressing cell line (CV28) was mock transfected or transfected with expression plasmids containing UL33, UL15, or both. Twenty-four hours after transfection, cell lysates were prepared and reacted with antibody against pUL33, pUL15, or pUL28. The presence of pUL15 and pUL28 in the immunoprecipitated material was determined by immunoblotting.
The constitutively expressed UL28 protein was immunoprecipitated from lysates of mock-transfected CV28 cells by the pUL28-specific antibody (Fig. 8A, lane 2) and was coimmunoprecipitated with the pUL33-specific antibody from lysates of CV28 cells expressing pUL33 (Fig. 8A, lane 3). The UL28 protein of CV28 cells was not immunoprecipitated with the pUL33-specific antibody in the absence of pUL33 expression (Fig. 8A, lane 1). Surprisingly, expression of UL15 in CV28 cells followed by reaction with the pUL15N antibody immunoprecipitated pUL15 (Fig. 8B, lane 4) but not pUL28 (Fig. 8A, lane 4) from CV28 cells. These data suggest that in this cell line pUL33 augments the interaction of pUL15 and pUL28 in the absence of other viral proteins.
FIG. 8.

Immunoblots of proteins immunoprecipitated from a UL28-expressing cell line (CV28). CV28 cells were mock transfected (Mock, lanes 1 and 2) or transfected with plasmids expressing UL33 (33, lane 3), UL15 (15, lane 4), or UL15 and UL33 (15+33, lane 5). Twenty-four hours later, lysates of the cells were prepared and subjected to immunoprecipitation (IP) with antibodies directed against pUL33 (α33, lanes 1 and 3), pUL28 (α28, lane 2), or the N terminus of pUL15 (α15N, lanes 4 and 5). An immunoblot of electrophorectically separated material was probed with antibody to pUL28 (A). The same immunoblot was stripped as described in Materials and Methods and reprobed with antibodies against the C terminus of pUL15 (B).
To further investigate this conclusion, a reciprocal experiment was performed. Specifically, lysates of CV28 cells that were mock transfected or transfected with expression plasmids bearing UL33, UL15, or both were subjected to immunoprecipitation with pUL28-specific antibody, and the presence of pUL15 in the immunoprecipitated material was monitored by immunoblotting. As shown in Fig. 9A, lanes 7 and 8, pUL15 was coimmunoprecipitated with pUL28 antibody in CV28 cells whether or not pUL33 was expressed in the lysate; however, the presence of pUL33 significantly augmented the coimmunoprecipitation of pUL28 and pUL15. To quantify the extent of this augmentation, the amounts of pUL15 in the lysates of CV28 cells transfected with pUL15 or pUL15 and pUL33 together were determined using a Molecular Dynamics PhosphorImager. The level of pUL15 immunoreactivity in the immunoprecipitates (Fig. 9A, lanes 7 and 8) was then compared to that obtained from the lysates (Fig. 9A, lanes 3 and 4), and a ratio of the two values was calculated (Fig. 9B). These calculations revealed that expression of pUL33 enhanced coimmunoprecipitation of pUL15 and pUL28 approximately 9.5-fold.
FIG. 9.

Immunoblots of lysates and immunoprecipitates of CV28 cells expressing putative terminase proteins probed with pUL15-specific antibody. (A) CV28 cells (CV1 cells expressing pUL28) were mock transfected (Mock, lanes 1 and 5) or were transfected with UL33 (33, lanes 2 and 6), UL15 (15, lanes 3 and 7), or UL15 and UL33 (15+33, lanes 4 and 8). Lysates were prepared 24 h posttransfection. Fifteen microliters of the lysates (lanes 1 to 4) or material immunoprecipitated with the pUL28-specific antibody (lanes 5 to 8) was electrophoretically separated, and immunoblots of the separated proteins were probed with antibody directed against the C terminus of pUL15. pUL15-specific immunoreactivity in the blot was quantified with a Molecular Dynamics PhosphorImager before exposure to radiographic film. (B, left histogram) The amount of pUL15 immunoreactivity in lane 7 was divided by the amount in lane 3. (B, right histogram) The amount of pUL15 immunoreactivity in lane 8 was divided by the amount in lane 4. These ratios differed by approximately 9.5-fold.
DISCUSSION
The current results confirm previous results showing that pUL33 coimmmunoprecipitates with pUL15 and pUL28 in lysates of infected cells (6). Using pUL15, pUL28, and pUL33 overexpressed in insect cells, previous experiments demonstrated interactions between UL15 and UL33, pUL28 and pUL15, and pUL33 and pUL28 (1, 6). In contrast to some of the previous results, the current results with HSV-infected cells demonstrate that pUL15 interacts with pUL33 only upon coexpression of pUL28. It was also of interest to note that this coexpression greatly stabilized pUL33. This observation is consistent with the notion that the pUL33/pUL28 interaction precludes aberrant folding of pUL33, thereby protecting it from targeted degradation by the proteosome. We cannot exclude the possibility that pUL15 and pUL33 interact directly but view it likely that such an interaction is considerably weaker than that of pUL28 with pUL33. Weaker interactions may be revealed more readily when proteins are highly expressed, as might have occurred in the previous study when the very strong baculovirus polyhedron promoter was used to drive gene expression (6). Because the current study investigated the putative terminase proteins under conditions of their native environment, stoichiometry, and levels of expression, we give more credence to the current results.
Because all of the interactions in both studies were identified in the context of a cell lysate, a further caveat is that other proteins may affect the interactions. Because few viral proteins other than pUL15 and pUL33 coimmunoprecipitate with pUL28 (6), it seems most likely that any augmentation of the interaction would be mediated by cellular proteins or perhaps transiently by viral proteins yet to be identified.
It was of interest to find that pUL33 augmented the interaction of pUL15 and pUL28 both in infected cells and when transiently expressed in uninfected mammalian cells. This represents the first identification of an activity of pUL33 that might be relevant to its role in the HSV DNA cleavage/packaging reaction. The relatively small pUL33 has no obvious DNA or ATP binding motifs that might be expected of a terminase subunit (14), and this is consistent conceptually with its primary role as an adapter to augment interaction between the DNA binding (likely pUL28) and ATPase (likely pUL15) subunits of the terminase. Because pUL33 interacts primarily with pUL28, it seems most likely that pUL33 enhances the pUL15/pUL28 interaction by optimizing pUL28's capacity to bind pUL15, perhaps by optimizing pUL28 folding. On the other hand, given the multifunctionality of most HSV proteins it also seems likely that pUL33 will exhibit other interesting activities upon more extensive analyses.
That the pUL15/pUL28/pUL33 complex can form independently of the capsid or portal under physiological conditions in infected cells is supported by the observation here (not shown) and elsewhere that pUL6 does not coimmunoprecipitate with pUL15, pUL28, or pUL33 from infected-cell lysates (6). Although antibodies can interfere with coimmunoprecipitations, it seems unlikely that three different antibodies to three different proteins would all fail to pull down the portal if the terminase/portal protein complex was soluble and intact. On the other hand, the lysates employed in these studies would not be expected to contain abundant amounts of nuclear proteins. If the terminase requires the portal vertex within an intranuclear procapsid for binding, as seems likely, the current studies would not be expected to detect a portal/terminase interaction because the lysates should not contain intact procapsids. That pUL6 and putative terminase components pUL15 and pUL28 can interact is supported by studies using transient-expression systems (22). Clearly, further studies of the distributions of the relevant protein complexes in HSV-infected cells are necessary to determine the sites of assembly and interaction of the putative terminase and portal encoded by pUL6.
Acknowledgments
We thank Jarek Okulicz-Kozaryn for technical assistance and Andrew Davison and Fred Homa for mutant viruses used in these studies.
This work was supported by Public Health Service grant GM50740 from the National Institutes of Health.
REFERENCES
- 1.Abbotts, A. P., V. G. Preston, M. Hughes, A. H. Patel, and N. D. Stow. 2000. Interaction of the herpes simplex virus type 1 packaging protein UL15 with full-length and deleted forms of the UL28 protein. J. Gen. Virol. 81:2999-3009. [DOI] [PubMed] [Google Scholar]
- 2.Adelman, K., B. Salmon, and J. D. Baines. 2001. Herpes simplex DNA packaging sequences adopt novel structures that are specifically recognized by a component of the cleavage and packaging machinery. Proc. Natl. Acad. Sci. USA 98:3086-3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Al-Kobaisi, M. F., F. J. Rixon, I. McDougall, and V. G. Preston. 1991. The herpes simplex virus UL33 gene product is required for the assembly of full capsids. Virology 180:380-388. [DOI] [PubMed] [Google Scholar]
- 4.Baines, J. D., C. Cunningham, D. Nalwanga, and A. J. Davison. 1997. The UL15 gene of herpes simplex virus type 1 contains within its second exon a novel open reading frame that is translated in frame with the UL15 gene product. J. Virol. 71:2666-2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beard, P. M., and J. D. Baines. 2004. The DNA cleavage and packaging protein encoded by the UL33 gene of herpes simplex virus 1 associates with capsids. Virology 324:475-482. [DOI] [PubMed] [Google Scholar]
- 6.Beard, P. M., N. S. Taus, and J. D. Baines. 2002. DNA cleavage and packaging proteins encoded by genes UL28, UL15, and UL33 of herpes simplex virus type 1 form a complex in infected cells. J. Virol. 76:4785-4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Black, L. W. 1989. DNA packaging in dsDNA bacteriophages. Annu. Rev. Microbiol. 43:267-292. [DOI] [PubMed] [Google Scholar]
- 8.Cunningham, C., and A. J. Davison. 1993. A cosmid-based system for constructing mutants of herpes simplex virus type 1. Virology 197:116-124. [DOI] [PubMed] [Google Scholar]
- 9.Davison, A. J. 1992. Channel catfish virus: a new type of herpesvirus. Virology 186:9-14. [DOI] [PubMed] [Google Scholar]
- 10.Dittmer, A., J. C. Drach, L. B. Townsend, A. Fischer, and E. Bogner. 2005. Interaction of the putative human cytomegalovirus portal protein pUL104 with the large terminase subunit pUL56 and its inhibition by benzimidazole-d-ribonucleosides. J. Virol. 79:14660-14667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koslowski, K. M., P. R. Shaver, J. T. I. Casey, T. Wilson, G. Yamanaka, A. K. Sheaffer, D. J. Tenny, and N. E. Pedersen. 1999. Physical and functional interactions between the herpes simplex virus UL15 and UL28 DNA cleavage and packaging proteins. J. Virol. 73:1704-1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koslowski, K. M., P. R. Shaver, X.-Y. Wang, D. J. Tenny, and N. Pedersen. 1997. The pseudorabies virus UL28 protein enters the nucleus after coexpression with the herpes simplex virus UL15 protein. J. Virol. 71:9118-9123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krosky, P. M., M. R. Underwood, S. R. Turk, K. W. Feng, R. K. Jain, R. G. Ptak, A. C. Westerman, K. K. Biron, L. B. Townsend, and J. C. Drach. 1998. Resistance of human cytomegalovirus to benzimidazole ribonucleosides maps to two open reading frames: UL89 and UL56. J. Virol. 72:4721-4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McGeoch, D. J., M. A. Dalrymple, A. J. Davison, A. Dolan, M. C. Frame, D. McNab, L. J. Perry, J. E. Scott, and P. Taylor. 1988. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J. Gen. Virol. 69:1531-1574. [DOI] [PubMed] [Google Scholar]
- 15.Mitchell, M. S., S. Matsuzaki, S. Imai, and V. B. Rao. 2002. Sequence analysis of bacteriophage T4 DNA packaging/terminase genes 16 and 17 reveals a common ATPase center in the large subunit of viral terminases. Nucleic Acids Res. 30:4009-4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Newcomb, W. W., R. M. Juhas, D. R. Thomsen, F. L. Homa, A. D. Burch, S. K. Weller, and J. C. Brown. 2001. The UL6 gene product forms the portal for entry of DNA into the herpes simplex virus capsid. J. Virol. 75:10923-10932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reynolds, A. E., Y. Fan, and J. D. Baines. 2000. Characterization of the UL33 gene product of herpes simplex virus 1. Virology 266:310-318. [DOI] [PubMed] [Google Scholar]
- 18.Salmon, B., and J. D. Baines. 1998. Herpes simplex virus DNA cleavage and packaging: association of multiple forms of UL15-encoded proteins with B capsids requires at least the UL6, UL17, and UL28 genes. J. Virol. 72:3045-3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salmon, B., D. Nalwanga, Y. Fan, and J. D. Baines. 1999. Proteolytic cleavage of the amino terminus of the UL15 gene product of herpes simplex virus type 1 is coupled with maturation of viral DNA into unit-length genomes. J. Virol. 73:8338-8348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tengelsen, L. A., N. E. Pedersen, P. R. Shaver, M. W. Wathen, and F. L. Homa. 1993. Herpes simplex virus type 1 DNA cleavage and capsidation require the product of the UL28 gene: isolation and characterization of two UL28 deletion mutants. J. Virol. 67:3470-3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trus, B. L., N. Cheng, W. W. Newcomb, F. L. Homa, J. C. Brown, and A. C. Steven. 2004. Structure and polymorphism of the UL6 portal protein of herpes simplex virus type 1. J. Virol. 78:12668-12671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.White, C. A., N. D. Stow, A. H. Patel, M. Hughes, and V. G. Preston. 2003. Herpes simplex virus type 1 portal protein UL6 interacts with the putative terminase subunits UL15 and UL28. J. Virol. 77:6351-6358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu, D., and S. K. Weller. 1998. Genetic analysis of the UL 15 gene locus for the putative terminase of herpes simplex virus type 1. Virology 243:32-44. [DOI] [PubMed] [Google Scholar]
- 24.Yu, D., and S. K. Weller. 1998. Herpes simplex virus type 1 cleavage and packaging proteins UL15 and UL28 are associated with B but not C capsids during packaging. J. Virol. 72:7428-7439. [DOI] [PMC free article] [PubMed] [Google Scholar]