

Abstract
The EPXH2 gene encodes for the soluble epoxide hydrolase (sEH), a homodimeric enzyme with each monomer containing two domains with distinct activities. The C-terminal domain, containing the epoxide hydrolase activity (Cterm-EH), is involved in the metabolism of arachidonic acid epoxides, endogenous chemical mediators that play important roles in blood pressure regulation, cell growth, and inflammation. We recently demonstrated that the N-terminal domain contains a Mg2+-dependent lipid phosphate phosphatase activity (Nterm-phos). However, the biological role of this activity is unknown. The inability of known phosphatase inhibitors to inhibit the Nterm-phos constitutes a significant barrier to the elucidation of its function. We describe herein sulfate, sulfonate, and phosphonate lipids as novel potent inhibitors of Nterm-phos. These compounds are allosteric competitive inhibitors with KI in the hundred nanomolar range. These inhibitors may provide a valuable tool to investigate the biological role of the Nterm-phos. We found that polyisoprenyl phosphates are substrates of Nterm-phos, suggesting a possible role in sterol synthesis or inflammation. Furthermore, some of these compounds inhibit the C-terminal sEH activity through a noncompetitive inhibition mechanism involving a new binding site on the C-terminal domain. This novel site may play a role in the natural in vivo regulation of epoxide hydrolysis by sEH.
The EPXH2 gene encodes the soluble epoxide hydrolase (sEH),1 a ubiquitous enzyme in vertebrates that transforms epoxides to their corresponding vicinal diols (1, 2). While highly expressed in the liver, sEH is also expressed in other tissues including vascular endothelium, leukocytes, some smooth muscle and the proximal tubule (3-5). This localization reflects the importance of sEH in the metabolism of epoxy fatty acids, such as epoxyeicosatrienoic acids (EETs) generated by cytochrome P450 epoxygenases (6), with critical roles in the regulation of cardiovascular, renal, and inflammatory biology (7-10). The hydrolysis of epoxy fatty acids modulates their intracellular fate (11, 12) and biological activity (8, 10, 13, 14). Pharmacological inhibition of sEH has resulted in blood pressure reduction in the spontaneously hypertensive rat (SHR) and in the angiotensin II-induced hypertensive rat model (15, 16). In this latter model, sEH inhibition also protects the kidney from hypertension-induced damage (17). Additionally, the deletion of this gene reduces blood pressure in male mice to female levels (18), further supporting the role of sEH in blood pressure regulation. However, it should be realized that the sEH-null phenotype is a double knockout.
At the molecular level, sEH is a homodimer with a monomeric unit of 62.5 kDa (2). Analysis of the primary structure suggests that the sEH gene (EPXH2) was produced by the fusion of two primordial dehalogenase genes; the C-terminal sEH domain has high homology to haloalkane dehalogenase, while the N-terminal domain is similar to haloacid dehalogenase (19). Interestingly, both domains possess catalytic activity. The C-terminus is the site of the epoxide hydrolysis activity (Cterm-EH) which is responsible for the described biology associated with sEH (2, 20, 21), while a magnesium-dependent hydrolysis of phosphate esters (Nterm-phos) was recently associated with the N-terminal domain of sEH (22, 23). Recent X-ray crystal structures of the mouse and human sEH confirmed the gene fusion hypothesis and showed that sEH exhibits a domain-swapped architecture (20, 21), suggesting a structural role for the N-terminal domain. The C-terminal domain of one subunit interacts with both the C- and N-terminal domain of the other monomer, while the N-terminal domain of one subunit interacts only with the C-terminal domain of both monomers. Aside from the physical interaction between the two C-terminal domains, no cooperative allosteric effects have been reported for the Cterm-EH activity (2). Kinetic analysis revealed a positive cooperative Hill coefficient of ∼2 for the hydrolysis of the monophosphate of dihydroxystearic acid, suggesting an allosteric interaction between the two monomers (23).
While the role and function of the C-terminal EH activity are well-known (3), nothing is known about the biological function of the Nterm-phos activity. It has been postulated that this activity could participate in xenobiotic metabolism (22) or in the regulation of the physiological functions associated with sEH (23). Obtaining potent inhibitors of the Nterm-phos activity will provide tools to better understand the biological role(s) of this enzyme. Common commercial phosphatase inhibitors do not influence Nterm-phos activity (23), and to our knowledge there are no known small molecule inhibitors for this activity. While sulfates are not substrates for the Nterm-phos activity (22), such compounds were recently shown to inhibit two tyrosine phosphatases (24, 25). This paper describes the evaluation of lipid sulfates as inhibitors for the Nterm-phos catalytic site and their effects on the Cterm-EH activity. A series of commercially available phophonates were evaluated in parallel. In addition, an improved assay for the Nterm-phos is reported.
MATERIALS AND METHODS
Reagents. Fatty acids were purchased from NuChek Prep (Elysian, MN). HPLC-grade chloroform (CHCl3), triethylamine (TEA), and glacial acetic acid were purchased from Fisher Scientific (Pittsburgh, PA). OmniSolv acetonitrile (ACN) and methanol (MeOH) were purchased from EM Science (Gibbstown, NJ). Compounds 1-5 were synthesized through the in situ generation of an activated sulfoimidate which was used to sulforylate hydroxy fatty acids following a method similar to the one used previously to synthesize lipid phosphates (23, 27). As an example, synthesis of compound 1 is described below. In addition, reaction yield and high-resolution mass spectrometry data for compounds 1-5 are given in Table 1. 1H NMRs were performed using a Mercury 300 NMR (Varian; Walnut Creek, CA). High-resolution mass spectra were acquired on a time-of-flight mass spectrometer (Micromass LCT, Manchester, U.K.) using negative mode electrospray ionization (ESI) and leucine-enkephalin as a lock mass compound. Chemical purity was estimated at >95% for each compound on the basis of 1H NMR spectra and ESI-LC/MS analyses. Negative mode electrospray ionization showed a single peak, while positive mode confirmed TEA as the only ESI-LC/MS detectable secondary component. Compound 6 was synthesized previously in the laboratory (23). Compounds 7-37 were purchased from either Sigma (St. Louis, MO) or Aldrich Chemical Co. (Milwaukee, WI), except for compound 9 which was provided by Promega (Madison, WI), compound 10 which was provided by City Chemicals (West-Haven, CT), and compound 33 which was purchased from Polycarbon Industries, Inc. (Devens, MA).
Table 1:
Hydroxy Lipid Sulfate Characterizationa
| Name | Structure | No. | Yield (%) | Mass ([M-H]-) |
|---|---|---|---|---|
| 10-Suflonooxy-octadecanoic acid | 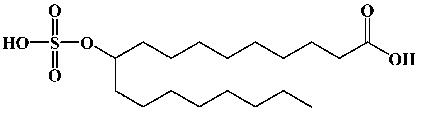 |
1 | 25 | 379.2165 (379.2233) |
| 9/10-Sulfonooxy-hydroxy-octadecanoic acid | 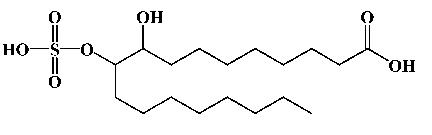 |
2 | 16 | 395. 2104 (395.2182) |
| 9-Octadecanylsulfate | 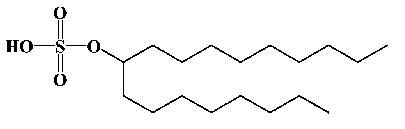 |
3 | 2 | 349.2384 (349.2491) |
| 12-Sulfonoxy--9-octadecenoic acid | 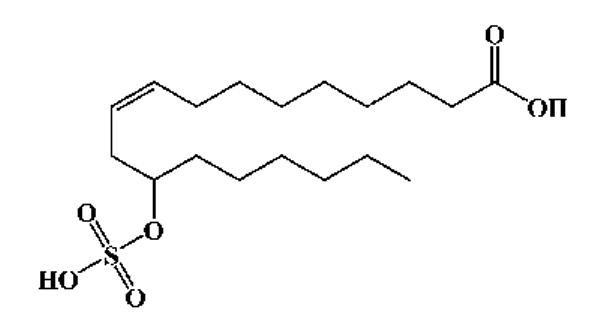 |
4 | 7 | 377.2051 (377.2076) |
| 12-Sulfonoxy--9-octadecenoic acid | 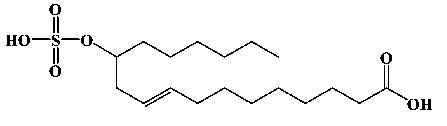 |
5 | 1 | 377.1929 (377.2076) |
Analyte purity was > 95%. While a single struture is shown, diol sulfate 2 is a 1:1 mixtuer of the monosulfate of each possible alcohol. Measured anionic masses are shown with theoretical masses in parentheses.
10-Sulfonooxyoctadecanoic Acid (1). In a small reaction vial, 100 mg of 10-hydroxyoctanoic acid was dissolved in 0.8 mL of acetonitrile and enriched with 150 μL of triethylamine, followed by 60 μL of trichloroacetonitrile and 40 μL of 100% sulfuric acid. The mixture was stirred at 50 °C for 2 h. The acetonitrile was then evaporated, and the resulting residue was dissolved in 10 mL of 1:4 methanol/water (v/v). The mixture was purified using a 1 g C18 solid-phase extraction cartridge (SPE; Varian, Walnut Creek, CA) equilibrated with water. The sulfurylated product was eluted from the column with 2:3 methanol/water (v/v). Fractions were screened for purity by ESI-LC/MS, and solvent was removed under vacuum to yield 32 mg (25% yield) of a yellow-brown waxy solid. Analysis revealed that the target compound was obtained as a triethylamine (TEA) salt. 1H NMR (CDCl3/CD3OD, 1:1): δ 4.35 (m, 1H, C10), 3.18 (dd, J) 7.5 Hz, 6H, CH2s of TEA), 2.29 (t, J) 7.2 Hz, 2H, C2), 1.63 (m, 11H, CH3s on TEA and C3), 1.30 (m, 26H, C4-C9 and C11-C17), and 0.88 (t, J) 6.9 Hz, 3H, C18). High-resolution MS: m/z 379.2165 (theoretical 379.2233).
Tandem Quadrupole Mass Spectrometry. The quantification of geraniol, farnesol, and geranylgeraniol, the products from dephosphorylation of compounds 34-37, was performed using HPLC with ESI and tandem mass spectrometric detection (MS/MS). The Shimadzu ASP10 HPLC system (Shimadzu Scientific Instruments, Columbia, MD) was set at a flow rate of 0.2 mL/min, and a 2.1× 30 mm XTerra MS C18 3.5 μm column (Waters, Milford, MA) was held at 20 °C. The samples were kept at 10 °C in the autosampler. The injection volume was 2 μL. A solvent system consisting of water with 0.1% formic acid (solvent A) and acetonitrile with 0.1% formic acid (solvent B) was used and set at a flow rate of 0.25 mL/min. The analytes were separated using a gradient program starting with a solvent composition of 40% solvent B ramped using a linear gradient for 7 min to 100% solvent B and held for 0.5 min. Compound 37 was analyzed by direct injections of a 3 μL sample into the mass spectrometer at a 0.25 mL/min flow rate of 10% solvent A and 90% solvent B. Pyrophosphate was analyzed by direct injection of a 5 μL sample into the mass spectrometer at a 0.05 mL/min flow rate of 50% solvent A and 50% solvent B.
Analytes were detected by electrospray ionization—tandem quadrupole mass spectrometry in the multiple reaction monitoring mode (MRM) using a Quattro Premier tandem quadrupole mass spectrometer (Micromass, Manchester, U.K.). Nitrogen gas flow rates were fixed with a cone gas flow of 25 L/h and a desolvation gas flow of 700 L/h. Electrospray ionization of geraniol, farnesol, and geranylgeraniol was performed in positive mode with a capillary voltage fixed at 3.20 kV and a cone voltage fixed at 25 V using a source temperature of 125 °C and a desolvation temperature of 350 °C. Capillary voltage and cone voltage were optimized in an infusion experiment. Intensities of analyte molecular ions [M + H]+ were low at 10 μM concentration of infused standards. However, intense [M + H - 18]+ ions were produced in the source due to water loss. Therefore, these ions were selected as precursor ions to set the MRM acquisition mode. Monitored transitions were m/z 137 > 95 for geraniol, m/z 205 > 121 for farnesol, and m/z 273 > 149 for geranylgeraniol at a collision voltage of 15 V for all analytes. Argon was used as collision gas (2.2 × 10-3 torr). Electrospray ionization of compound 37 and pyrophosphate was performed in negative ionization mode at the same instrument conditions described above using the MRM transition m/z 301 > 97 and m/z 177 > 79, respectively.
Concentrations of geraniol, farnesol, geranylgeraniol, and compound 37 were quantified using external standard calibration. Calibration curves for geraniol, farnesol, and geranylgeraniol contained six points from 0.03 to 10.0 μM and were linear (r2 > 0.99). The calibration curve for compound 37 contained seven points from 0.15 to 15 μM and had a good linear fit (r2 = 0.97). Chromatogram integration and analyte quantification were performed with the QuantLynx module of the MassLynx 4.0 software (Micromass, Manchester, U.K.). The limit of detection for pyrophosphate was found by injecting serial dilutions of the standard and was estimated to be 0.01 μM at 5 μL sample volume.
Enzyme Preparations. Recombinant human sEH (HsEH) was produced in a baculovirus expression system (28) and purified by affinity chromatography (29). The preparations were at least 97% pure as judged by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and scanning densitometry. No detectable esterase or glutathione transferase activities were observed. Recombinant mouse-eared cress sEH was produced in a baculovirus expression system and purified as described (30). The nucleotide sequence of the C-terminal region (Met237-Met554) of human sEH was amplified by PCR using 5′-CCGGAATTCATGAGCCATGGGTACGTGA-3′ as forward primer and 5′-ACGCGTCGACCTACATCTTTGAGACCACCG-3′ as reverse primer. The resulting band was gel purified and restricted with EcoRI and SalI, which were introduced by the primers, and the restricted fragment was subcloned into the multiple cloning site of the pFastBac1 vector (Invitrogen). After verification of the obtained nucleotide sequence, the recombinant pFastBac1 plasmid was introduced in competent DH10bac cells leading to the formation of a recombinant plasmid containing the DNA insert. The isolated recombinant plasmid was used to produce recombinant baculovirus in Sf21 cells following procedures recommended by the manufacturer. The truncated HsEH was produced in high-5 Trichloplusia ni cell cultures following published procedures (28). Seventy-two hours post infection, the cells were collected by centrifugation (2000g × 20 min). The cell pellet was then suspended in a sodium phosphate buffer (76 mM, pH 7.4) containing 1 mM phenylmethanesulfonyl fluoride, EDTA, and dithiothreitol. The suspension was then homogenized by Polytron at 9000 rpm for 1 min and centrifuged (10000g × 20 min). The resulting supernatant was frozen at -80 °C until used for experiments. The human placental alkaline phosphatase (APHP) was obtained from Sigma. Protein concentrations were quantified with the Pierce BCA assay (Pierce, Rockford, IL) using fraction V bovine serum albumin (BSA) as the calibrating standard.
Enzymatic Assays. Nterm-phos activity was measured in Bis-Tris HCl buffer (25 mM, pH 7.0) containing 0.1 mg/mL fraction V BSA and 1 mM MgCl2 (buffer A) at 30 °C. For compounds 8 and 9, the appearance of the fluorescent products was followed kinetically for 5-10 min on Spectromax M2 (Molecular Devices, Sunnyvale, CA) at the emission and excitation wavelengths recommended by the manufacturers. APHP activity was measured as described using 7 as substrate (23). Compounds 34-37 were incubated separately with the enzyme for a given time, and then the reaction was stopped by the addition of 100 μL of methanol. The reaction products for compounds 34-37 were extracted with 500 μL of ethyl acetate. The quantification of the alcohol products was performed by LC/MS analysis of 2 μL of the organic phase. The Cterm-EH activity was measured as described previously in buffer A, using either racemic 4-nitrophenyl-trans-2,3-epoxy-3-phenylpropyl carbonate (31) or racemic [3H]-trans-1,3-diphenylpropene oxide as substrate (32). This latter substrate was also used to measure the activity of the cress sEH and the truncated human sEH.
Inhibition Experiments. IC50s for the Nterm-phos activity were determined using Attophos (9) as substrate. Human sEH (400 nM) was incubated with inhibitors for 5 min in buffer A at 30 °C prior to substrate addition ([S]) 50 μM). IC50s for the Cterm-EH activity were determined using racemic 4-nitrophenyl-trans-2,3-epoxy-3-phenylpropyl carbonate as substrate as described (31, 33). Human sEH (100 nM) was incubated with inhibitors for 5 min in buffer A at 30 °C prior to substrate addition ([S]) 50 μM). By definition, IC50 is the concentration of inhibitor that reduces enzyme activity by 50%. The IC50 was determined by regression of at least five datum points with a minimum of two points in the linear region of the curve on either side of the IC50 value.
Kinetic Assay Conditions. For the Nterm-phos activity, dissociation constants were determined using Attophos as substrate. Compound 1 at concentrations between 0 and 25 μM was incubated in triplicate for 5 min at 30 °C with 200 μL of purified human sEH at 40 nM in buffer A. Substrate (3.1 ≤ [S]final e 100 μM) was then added. Velocity was measured as described above. The results were fitted to eq 1 corresponding to a competitive allosteric inhibition of two
| (1) |
catalytic sites (34), allowing the simultaneous determination of KI, R, and KS. Resolution of the nonlinear equation was performed using Sigma Plot (SPSS Science, Chicago, IL). For the Cterm-EH activity, dissociation constants were determined using racemic [3H]-trans-1,3-diphenylpropeneoxide as substrate. Compound 1 at concentrations between 0 and 50 μM was incubated in triplicate for 5 min at 30 °C with 100 μL of purified human sEH at 1 nM in buffer A. Substrate (2.5 ≤ [S]final ≤ 30 μM) was then added. Velocity was measured as described (32). For each inhibitor concentration, the plots of the velocity as a function of the substrate concentration allowed the determination of apparent kinetic constants (KMapp and VMapp) (34). Resolution of the nonlinear Michaelis equation was performed using Sigma Plot (SPSS Science, Chicago, IL). The plot of 1/VMapp as a function of the inhibitor concentration allows the determination of KI when 1/VMapp) 0. Results are the mean (standard deviation of three separate determinations of KI.
RESULTS
Nterm-Phos Assay Optimization. Phosphate esters of dihydroxy fatty acids such as compound 6 are good substrates for Nterm-phos; however, they are difficult to synthesize (reaction yield ∼1%), and detection of the hydrolysis products requires chromatographic separation and mass spectral detection (23). On the other hand, the readily available p-nitrophenyl phosphate (7) is a relatively poor substrate for the targeted activity with a low VM to Km ratio (Table 2). Therefore, to obtain a more facile assay to test for Nterm-phos activity, we tested two fluorescent phosphatase substrates. The 4-methylumbeliferol phosphate (8) is a poor substrate for the human sEH, as it was for the rat sEH (22). On the other hand, Attophos (9) is a good substrate for the Nterm-phos, with a Km value 5-fold lower than that for compound 6, the best substrate previously reported (23). While 9 is hydrolyzed 50-fold slower than 6, the high sensitivity of the fluorescent reporter allows the use of 5-fold less enzyme (40 nM instead of 180 nM). Furthermore, we were able to execute the fluorescent assay in a 96-well format, permitting the rapid screening of chemicals for Nterm-phos inhibition.
Table 2:
Catalytic Activity of Human sEH for Several Phosphate Substrates
| name | no. | Km(μM) | Vm(nmol·min-1·mg-1) | Hill coeff | VM/Km(nmol·min-1mg-1·μM-1) |
|---|---|---|---|---|---|
| threo-9/10-phosphonooxyoctadecanoic acid | 6 | 20.9a | 338a | 1.9a | 16.1 |
| p-nitrophenyl phosphate | 7 | 1600a | 57.6a | 1.0a | 0.04 |
| 4-methylumbeliferyl phosphate | 8 | 210 ± 30 | 7.9 ± 1.7 | 1.1 ± 0.1 | 0.04 ± 0.01 |
| Attophos | 9 | 3.6 ± 0.8 | 7.0 ± 0.1 | 1.6 ± 0.1 | 1.9 ± 0.2 |
Data from ref 23.
Nterm-Phos Inhibition. Phosphoesters of hydroxy fatty acids, such as 6, are good substrates for the Nterm-phos activity (23). Moreover, sulfates acting as inhibitors of phosphatases have also been reported (24-26). Therefore, we hypothesize that replacing the phosphate moiety by a sulfate would yield potent inhibitors for Nterm-phos activity. Following a procedure similar to that used to make phosphoesters (23), we synthesized five sulfate derivatives of hydroxy fatty acids (Table 1). Using Attophos as a reporting substrate, we measured the effects of these compounds as well as a series of commercial sulfates and sulfonates on the Nterm-phos activity. As hypothesized, lipid sulfates are effective inhibitors of Nterm-phos (data shown in Table 3). Interestingly, the structure activity obtained with the sulfate inhibitors differs from what was observed with the corresponding phosphate substrates (23). Compared to compound 1, the presence of the hydroxyl group R to the sulfate in compound 2 does not increase the potency while a corresponding R-hydroxy improved the substrate affinity of lipid phosphates (23). The removal of the acid function in compound 3 did not affect the inhibition potency. Furthermore, the sulfate of trans-ricinelaidate, 5, gives a 10-fold higher inhibition than the cis isomer ricinoleate, 4, and the phosphate of the cis isomer is hydrolyzed 6-fold faster than the trans isomer (23). In comparison to compound 1, removal of the sulfate group from the middle of the alkyl chain and placing it on the carbon next to the acid function, as in compound 10, resulted in a 2-fold loss of inhibition potency. A terminal sulfate function with a shorter alkyl chain, 11, resulted in a potent inhibitor, demonstrating the importance of the presence of a hydrophobic group to inhibitor potency and that the acid function is not necessary. Compared to 11, the replacement of the sulfate group by a sulfonate, as in compound 12, results in an inhibitor with similar potency, suggesting that sulfonates and sulfates are both potent inhibitors of Nterm-phos. Compared to compound 11, the replacement of the alkyl chain by an aromatic group, as in compound 13, resulted in a total loss of potency. Interestingly, compound 13 was found not to be a substrate for the rat sEH, and the corresponding phosphate, compound 7, is a poor substrate (22, 23). Good inhibition was obtained for compound 14, which has a sulfate group on position 3 of the A ring and a sulfonate on the alkyl tail of the sterol structure. The fact that taurocholic acid, compound 15, has only the sulfonate function suggests that the observed inhibition by compound 14 is due to the presence of the sulfate group on the A ring. Compared to compound 11, the replacement of the alkyl chain by hydrophilic groups, such as compounds 16-19, resulted in a total loss of inhibition potency.
Table 3:
EffectofSulfatesonHumansEHNterm-PhosActivity
| name | no. | IC50 (μM)a |
|---|---|---|
| 10-sulfonooxyoctadecanoic acid | 1 | 5.9 ± 1.6 |
| 9/10-hydroxysulfonooxyoctadecanoic acid | 2 | 17.5 ± 1.6( |
| 9-octadecanyl sulfate | 3 | 4.7 ± 1.2( |
| 12-sulfonoxy-cis-9-octadecenoic acid | 4 | >100 |
| 12-sulfonoxy-trans-9-octadecenoic acid | 5 | 9.7 ± 1.3( |
| R-sulfostearic acid | 10 | 9.6 ± 0.6( |
| sodium dodecyl sulfate | 11 | 5.2 ± 0.6( |
| sodium dodecyl sulfonate | 12 | 3.7 ± 0.5 |
| 4-nitrophenyl sulfate | 13 | >100 |
| taurolithocholic acid 3-sulfate | 14 | 5 ± 4 |
| taurocholic acid | 15 | >100 |
| estrone 3-sulfate | 16 | >100 |
| D-galactose 6-sulfate | 17 | >100 |
| L-ascorbic acid 2-sulfate dipotassium salt | 18 | >100 |
| N-acetyl-D-galactosamine 4-sulfate | 19 | >100 |
Results are the average ± SD of three separate experiments.
Since phosphonates are also used to inhibit phosphatases (35, 36), we investigated the inhibition potency of commercially available phosphonates on Nterm-phos activity (Table 4). Significant inhibition was obtained for three of the phosphonates tested, compounds 27, 32, and 33. The first one is very hydrophobic. Interestingly, the second one, compound 32, is a mimic of farnesyl pyrophosphate (36). Compound 33 is structurally similar to 11 and 12 and has a higher IC50 than the sulfur-containing compounds (11 and 12), suggesting that sulfonates and sulfates are better inhibitors of Nterm-phos than phosphonates.
Table 4:
Effect of Phosphonates on Human sEH Nterm-Phos Activity
| name | no. | IC50 (μM)a |
|---|---|---|
| tetraisopropyl methylenediphosphonate | 20 | >100 |
| diethyl vinylphosphonate | 21 | >100 |
| diethyl benzoylphosphonate | 22 | >100 |
| diethyl cyclopropyl methylphosphonate | 23 | >100 |
| diethyl trans-cinnamyl phosphonate | 24 | >100 |
| diethyl 4-methylbenzyl phosphonate | 25 | >100 |
| diethyl allyl phosphonate | 26 | >100 |
| dioctyl phenyl phosphonate | 27 | 13 ± 1 |
| dibenzyl phosphate | 28 | >100 |
| dimethyl 2-oxoheptyl phosphonate | 29 | >100 |
| diethyl 2,2,2-trifluoro-1-hydroxyethyl | 30 | >100 |
| phosphonate | ||
| diethyl ethylthiomethyl phosphonate | 31 | >100 |
| R-hydroxyfarnesylphosphonic acid | 32 | 73 ± 5 |
| dodecyl phosphonic acid | 33 | 40 ± 4 |
Results are the average ± SD of three separate experiments.
To verify that the observed inhibition is not an artifact, we tested the effect of a 10-fold increase in the BSA concentration in the buffer on the inhibition potency. No change in IC50 values was observed, suggesting that these inhibitors do not act by forming nonspecific aggregates with the enzyme (37). Because some of the compounds tested, such as compound 11, are used as detergents, one could suggest that the observed inhibition is due to a surfactant effect. However, the range of IC50s observed (3-100 μM) is far lower than the critical micelle concentrations of these compounds, which is generally in the low to mid-millimolar range (25), suggesting that the observed effect is not due to a detergent effect. To test the specificity of the inhibitors for Nterm-phos, we tested the inhibition of alkaline phosphatase from human placenta on compounds 1-5 and 10-33. No significant inhibition was obtained for any compounds at 100 μM (results not shown).
Mechanism of Inhibition. To understand the mode of action of these new inhibitors, we first tested if compounds 1 and 2 were substrates for the Nterm-phos. We incubated the enzyme (400 nM) with the compounds (100 μM) for an hour and analyzed the mixture by LC-MS to detect any alcohol or diol formed (23). We were not able to detect the formation of any alcohol or diol. We then determined IC50s for compounds 1-5, 11, 12, and 14 for several incubation times (0, 5, 15, and 30 min) with the enzyme before addition of the substrate; no changes in IC50s were observed. These results support the fact that sulfates are not substrates for Nterm-phos, which was previously demonstrated for compound 13 (22).
Next, we determined the kinetic constant for compound 1. A variety of kinetic models were evaluated using Sigma Plot. The simple and mixed-type inhibition models fit poorly to our data (r2 < 0.4). For each inhibitor concentration, we obtained sigmoidal velocity curves which suggest an allosteric inhibition model, and we obtained similar VM results which suggest a competitive inhibition model, and in fact the data were best fitted with an allosteric competitive inhibition model. This mechanism of inhibition for an enzyme with two equivalent active sites is described by the equilibrium (34):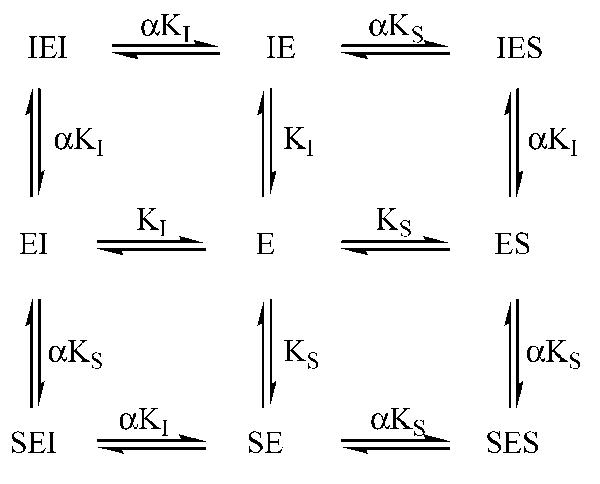 where the inhibitor, I, could bind at the same sites that the substrate, S, can bind the enzyme E. The binding of both the substrate and inhibitor changes the dissociation constant of the remaining vacant site for I or S by a factor R. The velocity for this type of inhibition is given by eq 1 (34). To determine KS, KI, and R, the velocity (V) results obtained for various concentration of I and S were fitted to eq 1 using the value of VM obtained in the absence of inhibitor. A typical result is shown in Figure 1A. We obtained an average (n) 3) KS of 9.8 (0.7 μM, an average KI of 0.7 (0.3 μM, and a factor R of 0.4 (0.1 and r2 above 0.91. Analysis of the curve fitting revealed that the residual variation was mainly observed for [I]) 5 μM; at this concentration we obtained twice as much inhibition than predicted, and calculated values are significantly different (p < 0.01) from obtained values. Removing the data for this concentration of inhibitor resulted in a significantly better fit of the model (r2 above 0.96), while obtaining similar kinetic parameter values [KS) 9.5 (0.8 μM, KI) 0.6 (0.2 μM, and R) 0.5 (0.1 (n) 3)]. Other allosteric inhibition models did not fit as well (r2 < 0.8). For the competitive allosteric inhibition model described herein, the inhibitor can also act as an activator (38). At a low substrate concentration (1 μM), we observed (Figure 1B) that low concentrations of compound 1 (<0.5 μM) increased the activity of the enzyme while an inhibitory effect was observed for higher concentrations of 1. These results suggest a homotropic cooperativity in the binding of 1 and that the inhibitors described herein bind at the Nterm-phos active site in a manner similar to substrate binding. Interestingly, the KS determined for Attophos is around 3-fold higher than its observed KM (Table 2), suggesting that the rate of Ntermphos dephosphorylation is the limiting step for the hydrolysis of this substrate (39).
where the inhibitor, I, could bind at the same sites that the substrate, S, can bind the enzyme E. The binding of both the substrate and inhibitor changes the dissociation constant of the remaining vacant site for I or S by a factor R. The velocity for this type of inhibition is given by eq 1 (34). To determine KS, KI, and R, the velocity (V) results obtained for various concentration of I and S were fitted to eq 1 using the value of VM obtained in the absence of inhibitor. A typical result is shown in Figure 1A. We obtained an average (n) 3) KS of 9.8 (0.7 μM, an average KI of 0.7 (0.3 μM, and a factor R of 0.4 (0.1 and r2 above 0.91. Analysis of the curve fitting revealed that the residual variation was mainly observed for [I]) 5 μM; at this concentration we obtained twice as much inhibition than predicted, and calculated values are significantly different (p < 0.01) from obtained values. Removing the data for this concentration of inhibitor resulted in a significantly better fit of the model (r2 above 0.96), while obtaining similar kinetic parameter values [KS) 9.5 (0.8 μM, KI) 0.6 (0.2 μM, and R) 0.5 (0.1 (n) 3)]. Other allosteric inhibition models did not fit as well (r2 < 0.8). For the competitive allosteric inhibition model described herein, the inhibitor can also act as an activator (38). At a low substrate concentration (1 μM), we observed (Figure 1B) that low concentrations of compound 1 (<0.5 μM) increased the activity of the enzyme while an inhibitory effect was observed for higher concentrations of 1. These results suggest a homotropic cooperativity in the binding of 1 and that the inhibitors described herein bind at the Nterm-phos active site in a manner similar to substrate binding. Interestingly, the KS determined for Attophos is around 3-fold higher than its observed KM (Table 2), suggesting that the rate of Ntermphos dephosphorylation is the limiting step for the hydrolysis of this substrate (39).
FIGURE 1:
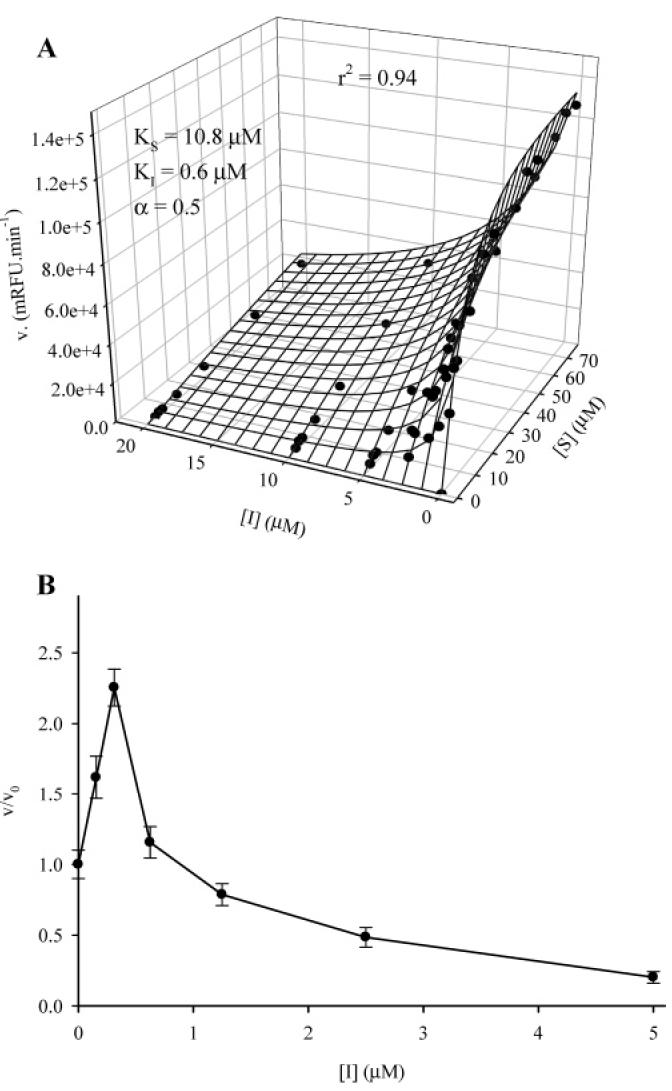
(A) Determination of the dissociation constant of 1 with human sEH using Attophos as substrate. The circles represent the collected data points. The mesh represents the curve resulting from the fitting of the data to eq 1. (B) Effect of 1 on human sEH Ntermphos activity at a low concentration (1 μM) of Attophos.
Nterm-Phos Endogenous Substrate. In a previous study, we reported that Nterm-phos prefers lipid phosphates as substrates (23). On the basis of the general inhibitor structure described herein, one could hypothesize that Nterm-phos endogenous substrates are terminal phospholipids such as polyisoprenyl phosphates which are important cellular signaling molecules (40). This is supported by the fact that compound 32, a farnesyl pyrophosphate mimic (36), is an inhibitor of Nterm-phos. To test this hypothesis, we assayed three isoprenyl pyrophosphates (compounds 34-36) that are important intermediates in the synthesis of sterols (41). Interestingly, these compounds are substrates for the sEH phosphatase (Table 5). For compound 34, we obtained a linear curve, indicating that the Km for this substrate is above the highest concentration tested (5 μM), and the relatively high VM/Km ratio suggests a high maximal velocity. Increasing the size of the terpene tail, as in compounds 35 and 36, resulted in a lower Km and a slower VM. Compounds 34-36 are hydrolyzed by an order of magnitude slower than the previously reported substrate (compound 6 in Table 2).
Table 5:
Kinetic Parameters of Human Nterm-Phos for Polyisoprenyl Phosphatesa
| name | no. | Km(μM) | VM(nmol.min-1.mg-1) | Hill coeff | VM/Km(nmol.min-1.mg-1.μM-1) |
|---|---|---|---|---|---|
| geranyl pyrophosphate | 34 | 5.3 ± 0.3 | |||
| farnesyl pyrophosphate | 35 | 10.1 ± 1.1 | 12.5 ± 1.0 | 1.0 ± 0.1 | 1.2 ± 0.2 |
| geranylgeranyl pyrophosphate | 36 | 3.4 ± 1.6 | 4.7 ± 1.1 | 1.0 ± 0.2 | 1.4 ± 0.8 |
| farnesyl monophosphate | 37 | 5.7 ± 0.4 | 303 ± 19 | 1.0 ± 0.1 | 53 ± 5 |
Results are the average ± SD of three separate experiments.
Because we detected only the alcohol form (see Materials and Methods), the kinetic parameters obtained could result from the direct removal either of pyrophosphate or of two phosphates successively with the formation of monophosphate as an intermediate. Concentrating on farnesyl pyrophosphate 35, over a 60 min incubation time (up to 80% hydrolysis of 35 by human sEH), we were not able to detect farnsyl monophosphate 37 by HPLC with ESI and tandem mass spectrometric detection. The estimated limit of detection for compound 37 was 0.1 μM (signal-to-noise ratio equals 3). We were able to detect 0.02 μM pyrophosphate in incubations of sEH with FPP; however, its concentration does not change with time of incubation, and it is present in buffer control, which suggests that the detected pyrophosphate is an impurity in the FPP standard. The kinetic parameters of 37 (Table 5) show that this compound is an excellent substrate for Nterm-phos. It is hydrolyzed approximately 50-fold faster than the corresponding pyrophosphate derivative 34. These results strongly suggest that Nterm-phos is a monophosphatase that hydrolyzes isoprenyl pyrophosphates to the corresponding alcohols and two phosphate molecules. It performs this reaction in two successive steps of phosphate removal with the second being much faster than the first one. Therefore, the kinetic parameters determined for the isoprenyl pyrophosphates (Table 5, compounds 34-36) most likely represent the removal of the first phosphate molecule, which is the rate-limiting step. Interestingly, compound 37 is hydrolyzed as fast as the previously reported substrate (compound 6 in Table 2).
Cterm-EH Inhibition. We then tested the effect of the Nterm-phos inhibitors on the Cterm-EH activity. As shown in Table 6, significant inhibition was obtained only for a small number of sulfates (1-3, 5, and 11). A slight inhibition was observed for the phosphonate 27. Interestingly, the pattern of inhibition of Cterm-EH is different than the one observed for Nterm-phos. For example, compound 12 is a far better inhibitor of Nterm-phos than compound 11, but it is not an inhibitor of Cterm-EH while compound 11 is. As observed above, increasing the concentration of BSA in the buffer by an order of magnitude does not alter the potency of the inhibitors, suggesting that these inhibitors do not act by forming nonspecific aggregates with the enzyme (37). Thus, sulfates represent a new class of inhibitors for Cterm-EH activity; previous potent inhibitors described include ureas, amides, and carbamates (42).
Table 6:
Effect of Sulfates, Pyrophosphates, and Phosphonates on Human sEH Cterm-EH Activity
| no. | IC50 (μM)a | no. | IC50 (μM)a | no. | IC50 (μM)a |
|---|---|---|---|---|---|
| 1 | 28 ± 2 | 15 | >100 | 25 | >100 |
| 2 | 90 ± 5 | 16 | >100 26 | 26 | >100 |
| 3 | 21 ± 5 | 17 | >100 | 27 | 92 ± 3 |
| 4 | >100 | 18 | >100 | 28 | >100 |
| 5 | 16 ± 3 | 19 | >100 | 29 | >100 |
| 10 | >100 | 20 | >100 | 30 | >100 |
| 11 | 50 ± 5 | 21 | >100 | 31 | >100 |
| 12 | >100 | 22 | >100 | 32 | >100 |
| 13 | >100 | 23 | >100 | 33 | >100 |
| 14 | 90 ± 5 | 24 | >100 |
Results are the average ± SD of three separate experiments.
To understand the mode of action of these compounds, we determined kinetic constants for compound 1. The best fit was obtained for a noncompetitive inhibition mechanism (Figure 2). This result suggests that the inhibitor does not bind at the active site, or least not exclusively at the active site, as previously described inhibitors do (42), thus showing that these new inhibitors of Cterm-EH act at a different site on the enzyme. We found for compound 1 at the Cterm-EH a KI of 31 (2 μM (n) 3), which is roughly 100-fold the KI obtained for this compound at the Nterm-phos (see above). This result suggests that the inhibition of Cterm-EH by compound 1 does not come from its binding to the pocket of Nterm-phos. To confirm this hypothesis, we tested the effect of compounds 1-5 on the EH activity of the full-length and N-terminally truncated (with only the C-terminus present) human sEH and the cress sEH, which does not contain a mammalian-like N-terminal domain (19). Similar patterns of inhibition (Table 7) were obtained for both full-length and truncated human sEH, while no inhibition was observed for the plant sEH. The small differences observed between the full-length and truncated human sEH are probably linked to the use of a truncated enzyme from crude extract versus purified full-length human sEH. These results suggest that sulfates inhibit Cterm-EH by binding to a site on the C-terminal domain that is distinct from the EH active site, and this site is not present on the plant sEH.
FIGURE 2:
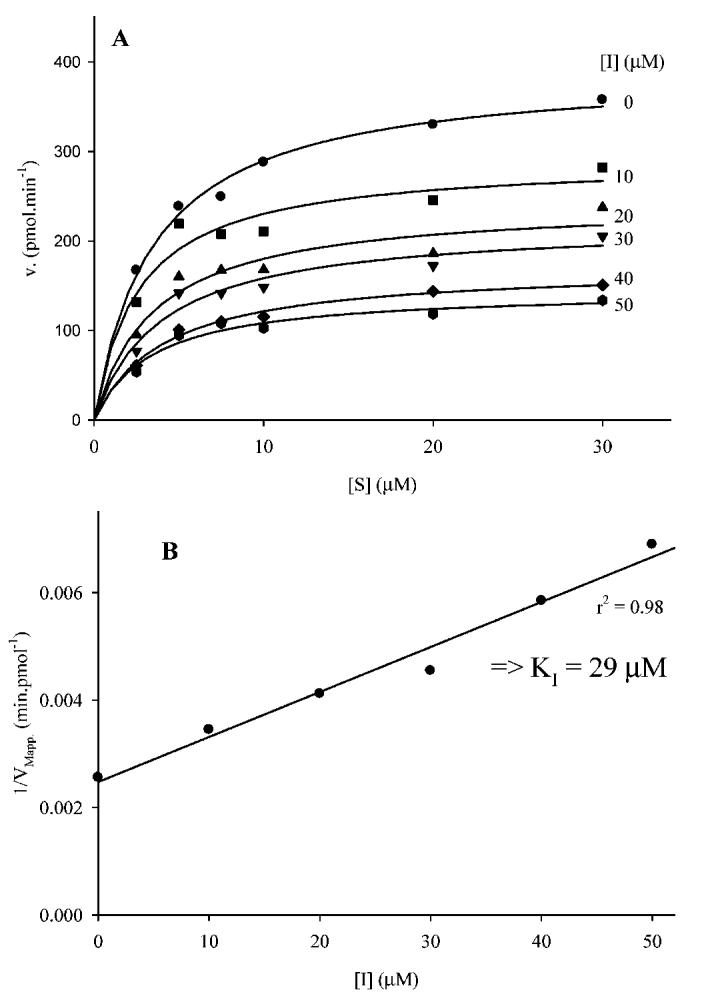
Determination of the dissociation constant of 1 with human sEH Cterm-EH activity using tDPPO as substrate. (A) For each inhibitor concentration (0-50 μM), the velocity is plotted as a function of the substrate concentration (0-30 μM) allowing the determination of an apparent maximal velocity (VMapp). (B) The plotting of 1/VMapp as a function of the concentration of inhibitor permits the determination of KI.
Table 7:
Effect of 100 μM Lipid Sulfates on EH Activity of Full-Length and Truncated Human sEH and Cress sEH
| inhibition at 100 μM (%)a |
|||
|---|---|---|---|
| human sEH |
|||
| no. | full length | truncated | cress sEH |
| 1 | 86 ± 2 | 89 ± 3 | <2 |
| 2 | 54 ± 3 | 58 ± 2 | <2 |
| 3 | 72 ± 2 | 67 ± 3 | <2 |
| 4 | 26 ± 3 | 16 ± 2 | <2 |
| 5 | 68 ± 2 | 44 ± 4 | <2 |
Results are the average ± SD of three separate experiments.
DISCUSSION
The recent discovery of the Nterm-phos activity (22, 23) has revealed a gap in our knowledge about the functional role of this enzyme. To fill this gap, new tools are needed. We report herein the use of Attophos, compound 9, as a new surrogate substrate for this activity; not only does it have a Ks in the low micromolar range, it also displays a positive cooperative binding similar to compound 1 (23). Compared to the assay using the latter substrate, use of Attophos gives an assay that is more sensitive and easier to perform. Furthermore, we were able to execute the fluorescent assay in a 96-well format, permitting us to quickly screen chemicals for Nterm-phos inhibition.
Using this new assay format, we investigated the effect of several pharmacophores on the inhibition of the sEH phosphatase activity. The results clearly show that sulfates, sulfonates, and phosphonates represent a new class of potent inhibitors of the Nterm-phos activity of sEH. Moreover, the inhibition is enhanced by the presence of a hydrophobic linear or cyclic tail; the presence of a carboxylic function or a double bond reduced the inhibition potency only slightly, except for the presence of a cis double bond. While surprising, this latter result was confirmed by testing compound 4 from several synthetic batches and from commercial sources (City Chemicals). The inhibition caused by these compounds does not decrease over time. One of the more potent inhibitors tested, compound 1, has a high nanomolar KI that is roughly 20-fold the enzyme concentration tested and 10-fold lower than the KS of the substrate, indicating that this compound binds relatively tightly to the enzyme. One could envision that optimization of the structure will yield stoichiometric inhibitors of Nterm-phos activity. The exact mechanism by which the sulfates, sulfonates, and phosphonates inhibit the Nterm-phos is not known. The kinetic inhibition was best described by a competitive model for which the inhibitor has a positive allosteric effect, like that observed for the substrate. This strongly suggests that the inhibitors mimic the binding of the substrate to the active site (Figure 3). The inhibitors most likely establish hydrogen bonds between their hydrophilic heads and residues within the active site. Furthermore, the hydrophobic tail of the inhibitors most likely bind through van der Waals interactions to an ∼14 Å long hydrophobic tunnel with one end at the Nterm-phos active site and the other end near the interface of the N- and C-terminal domains (21). It is not known which part of the inhibitor or substrate binding is responsible for the observed homotrophic cooperativity. Clearly, future structure determination and site-directed mutagenesis experiments are required to probe the allosteric regulation of Nterm-phos.
FIGURE 3:
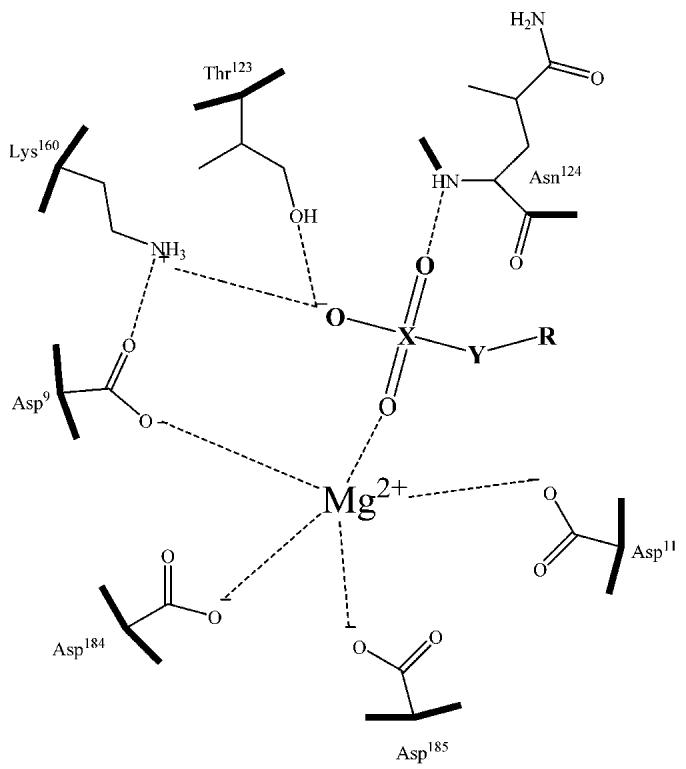
Hypothetical mechanism of Nterm-phos inhibition by sulfates, sulfonates, and phosphonates. The residue numbers are for the human sEH. Substituents: X) P or S, Y) O or CH2, and R) alkyl group.
Due to the allosteric effects observed for Nterm-phos and the fact that the two N-terminal domains of each homodimer do not form contacts with one another (20, 21), we believe that binding at the N-terminal active site could influence the Cterm-EH activity. While we found that some of the N-terminal inhibitors did affect the C-terminal activity, the results obtained clearly show that this effect is not through binding at the N-terminal domain. The data suggest the presence of a new binding site on the C-terminal domain that is distinct from the Cterm-EH catalytic site. The future discovery of inhibitors that bind exclusively to this latter site will be a valuable tool to probe the role of this site in the in vivo regulation of epoxide hydrolysis by sEH, which is an important process for blood pressure and inflammation regulation (3).
The mammalian soluble epoxide hydrolase is a unique enzyme in that it has the uncommon characteristic of having two enzymatic activities. While the role of the Cterm-EH activity in inflammation and hypertension, via epoxy fatty acid hydrolysis, is well documented (3), the role of the Nterm-phos remains to be elucidated. In a previous study, we reported that Nterm-phos prefers lipid phosphates as substrates (23). On the basis of the general inhibitor structure described herein, we found that polyisoprenyl phosphates are also good substrates for Nterm-phos. Polyisoprenyl phosphates are important cellular signaling molecules, thus suggesting a possible role for Nterm-phos in sterol synthesis or inflammation (40, 41). Alternatively, since a sterol sulfate, compound 14, inhibits the enzyme, sterol phosphates may also be substrates for Nterm-phos. Ultimately, the inhibitors developed and described herein provide valuable tools to investigate the biological role of the Nterm-phos.
Footnotes
This work was supported in part by NIEHS Grant R37 ES02710, NIEHS Superfund Basic Research Program Grant P42 ES04699, NIEHS Center for Environmental Health Sciences Grant P30 ES05707, and NIH/NINDS Grant R03 NS050841. K.L.T. was supported by Berlex Biosciences.
- sEH
- soluble epoxide hydrolase
- Cterm-EH
- C-terminal epoxide hydrolase
- Nterm-phos
- N-terminal phosphatase
REFERENCES
- 1.Gill SS, Hammock BD, Yamamoto I, Casida JE. Preliminary chromatographic studies on the metabolites and photodecomposition products of the juvenoid 1-(4′-ethylphenoxy-6,7-epoxy)-3,7-dimethyl-2-octene. In: Menn JJ, Beroza M, editors. Insect Juvenile Hormones: Chemistry and Action. Academic Press; New York: 1972. pp. 177–189. [Google Scholar]
- 2.Morisseau C, Hammock BD. Epoxide hydrolases: mechanisms, inhibitor designs and biological roles, Annu. Rev. Pharmacol. Toxicol. 2005;45:311–333. doi: 10.1146/annurev.pharmtox.45.120403.095920. [DOI] [PubMed] [Google Scholar]
- 3.Newman JW, Morisseau C, Hammock BD. Epoxide hydrolases: their roles and interactions with lipid metabolism. Prog. Lipid Res. 2005;44:1–51. doi: 10.1016/j.plipres.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 4.Draper AJ, Hammock BD. Soluble epoxide hydrolase in rat inflammatory cells is indistinguishable from sEH in rat liver. Toxicol. Sci. 1999;50:30–35. doi: 10.1093/toxsci/50.1.30. [DOI] [PubMed] [Google Scholar]
- 5.Yu Z, Davis BB, Morisseau C, Hammock BD, Olson JL, Kroetz DL, Weiss RH. Vascular localization of soluble epoxide hydrolase in human kidney. Am. J. Physiol. Renal Physiol. 2004;286:F720–F726. doi: 10.1152/ajprenal.00165.2003. [DOI] [PubMed] [Google Scholar]
- 6.Chacos N, Capdevila J, Falck JR, Martin-Wixtrom C, Gill SS, Hammock BD, Estabrook RA. The reaction of arachidonic acid epoxides (epoxyeicosatrienoic acids) with a cytosolic epoxide hydrolase. Arch. Biochem. Biophys. 1983;233:639–648. doi: 10.1016/0003-9861(83)90628-8. [DOI] [PubMed] [Google Scholar]
- 7.Capdevila JH, Falck JR. arachidonic acid monooxygenases: from cell signaling to blood pressure regulation. Biochem. Biophys. Res. Commun. 2001;285:571–576. doi: 10.1006/bbrc.2001.5167. [DOI] [PubMed] [Google Scholar]
- 8.Spector AA, Fang X, Snyder GD, Weintraub NL. Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog. Lipid Res. 2004;43:55–90. doi: 10.1016/s0163-7827(03)00049-3. [DOI] [PubMed] [Google Scholar]
- 9.Sun J, Sui X, Bradbury JA, Zeldin DC, Conte MS, Liao JK. Inhibition of vascular smooth muscle cell migration by cytochrome p450 epoxygenase-derived eicosanoids. Circ. Res. 2002;90:1020–1027. doi: 10.1161/01.res.0000017727.35930.33. [DOI] [PubMed] [Google Scholar]
- 10.Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999;285:1276–1279. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weintraub NL, Fang X, Kaduce TL, VanRollins M, Chatterjee P, Spector AA. Epoxide hydrolases regulate epoxyeicosatrienoic acid incorporation into coronary endothelial phospholipids. Am. J. Physiol. 1999;277:H2098–H2108. doi: 10.1152/ajpheart.1999.277.5.H2098. [DOI] [PubMed] [Google Scholar]
- 12.Greene JF, Williamson KC, Newman JW, Morisseau C, Hammock BD. Metabolism of monoepoxides of methyl linoleate: bioactivation and detoxification. Arch. Biochem. Biophys. 2000;376:420–432. doi: 10.1006/abbi.2000.1753. [DOI] [PubMed] [Google Scholar]
- 13.Greene JF, Newman JW, Williamson KC, Hammock BD. Toxicity of epoxy fatty acids and related compounds to cells expressing human soluble epoxide hydrolase. Chem. Res. Toxicol. 2000;13:217–226. doi: 10.1021/tx990162c. [DOI] [PubMed] [Google Scholar]
- 14.Chen J-K, Capdevila J, Harris RC. Heparin-binding EGF-like growth factor mediates the biological effects of P450 arachidonate epoxygenase metabolites in epithelial cells. Proc. Natl. Acad. Sci. U.S.A. 2002;99:6029–6034. doi: 10.1073/pnas.092671899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, Parker C, Graham L, Engler MM, Hammock BD, Zeldin DC, Kroetz DL. Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ. Res. 2000;87:992–998. doi: 10.1161/01.res.87.11.992. [DOI] [PubMed] [Google Scholar]
- 16.Imig JD, Zhao X, Capdevila JH, Morisseau C, Hammock BD. Soluble epoxide hydrolase inhibition lowers arterial blood pressure in angiotensin II hypertension. Hypertension. 2002;39:690–694. doi: 10.1161/hy0202.103788. [DOI] [PubMed] [Google Scholar]
- 17.Zhao X, Yamamoto T, Newman JW, Kim IH, Watanabe T, Hammock BD, Stewart J, Pollock JS, Pollock DM, Imig JDJ. Soluble epoxide hydrolase inhibition protects the kidney from hypertension-induced damage. Am. Soc. Nephrol. 2004;15:1244–1253. [PubMed] [Google Scholar]
- 18.Sinal CJ, Miyata M, Tohkin M, Nagata K, Bend JR, Gonzalez FJ. Targeted disruption of soluble epoxide hydrolase reveals a role in blood pressure regulation. J. Biol. Chem. 2000;275:40504–40510. doi: 10.1074/jbc.M008106200. [DOI] [PubMed] [Google Scholar]
- 19.Beetham JK, Grant D, Arand M, Garbarino J, Kiyosue T, Pinot F, Oesch F, Belknap WR, Shinozaki K, Hammock BD. Gene evolution of epoxide hydrolases and recommended nomenclature. DNA Cell Biol. 1995;14:61–71. doi: 10.1089/dna.1995.14.61. [DOI] [PubMed] [Google Scholar]
- 20.Argiriadi MA, Morisseau C, Hammock BD, Christianson DW. Detoxification of environmental mutagens and carcinogens: structure, mechanism, and evolution of liver epoxide hydrolase. Proc. Natl. Acad. Sci. U.S.A. 1999;96:10637–10642. doi: 10.1073/pnas.96.19.10637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gomez GA, Morisseau C, Hammock BD, Christianson DW. Structure of human soluble epoxide hydrolase reveals mechanistic interferences on bifunctional catalysis in epoxide and phosphate ester hydrolysis. Biochemistry. 2004;43:4716–4723. doi: 10.1021/bi036189j. [DOI] [PubMed] [Google Scholar]
- 22.Cronin A, Mowbray S, Dürk H, Homburg S, Fleming I, Fisslthaler B, Oesch F, Arand M. The N-terminal domain of mammalian soluble epoxide hydrolase is a phosphatase. Proc. Natl. Acad. Sci. U.S.A. 2003;100:1552–1557. doi: 10.1073/pnas.0437829100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Newman JW, Morisseau C, Harris TR, Hammock BD. The soluble epoxide hydrolase encoded by EPXH2 is a bifunctional enzyme with novel lipid phosphate phosphatase activity. Proc. Natl. Acad. Sci. U.S.A. 2003;100:1558–1563. doi: 10.1073/pnas.0437724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun J-P, Wu L, Fedorov AA, Almo SC, Zhang Z-Y. Crystal structure of the Yersinia protein-tyrosine phosphatase YopH complexed with a specific small molecule inhibitor. J. Biol. Chem. 2003;278:33392–33399. doi: 10.1074/jbc.M304693200. [DOI] [PubMed] [Google Scholar]
- 25.Granjeiro JM, Miranda MA, da Glória ST, Maia M, Ferreira V, Taga EM, Aoyama H, Volpe PLO. Effect of homologous series of n-alkyl sulfates and n-alkyl trimethylammonium bromides on low molecular pass protein tyrosine phosphatase activity. Mol. Cell. Biochem. 2004;265:133–140. doi: 10.1023/b:mcbi.0000044390.18530.39. [DOI] [PubMed] [Google Scholar]
- 26.Scott HM, Coughtrie MWH, Burchell A. Steroid sulphates inhibit the rat hepatic microsomal glucose-6-phosphatase system. Biochem. Pharmacol. 1991;41:1529–1532. doi: 10.1016/0006-2952(91)90572-m. [DOI] [PubMed] [Google Scholar]
- 27.Ullman B, Perlman RL. Chemically phosphorylated protamine: A new substrate for the study of phosphoprotein phosphatase activity. Biochem. Biophys. Res. Commun. 1975;63:424–430. doi: 10.1016/0006-291x(75)90705-6. [DOI] [PubMed] [Google Scholar]
- 28.Beetham JK, Tian T, Hammock BD. cDNA cloning and expression of a soluble epoxide hydrolase from human liver. Arch. Biochem. Biophys. 1993;305:197–201. doi: 10.1006/abbi.1993.1411. [DOI] [PubMed] [Google Scholar]
- 29.Wixtrom RN, Silva MH, Hammock BD. Affinity purification of cytosolic epoxide hydrolase using derivatized epoxy-activated Sepharose gels. Anal. Biochem. 1988;169:71–80. doi: 10.1016/0003-2697(88)90256-4. [DOI] [PubMed] [Google Scholar]
- 30.Morisseau C, Beetham JK, Pinot F, Debernard S, Newman JW, Hammock BD. Cress and potato soluble epoxide hydrolases: Purification, biochemical characterization, and comparison to mammalian enzymes. Arch. Biochem. Biophys. 2000;378:321–332. doi: 10.1006/abbi.2000.1810. [DOI] [PubMed] [Google Scholar]
- 31.Dietze EC, Kuwano E, Hammock BD. Spectrophotometric substrates for cytosolic epoxide hydrolase. Anal. Biochem. 1994;216:176–187. doi: 10.1006/abio.1994.1023. [DOI] [PubMed] [Google Scholar]
- 32.Borhan B, Mebrahtu T, Nazarian S, Kurth MJ, Hammock BD. Improved radiolabeled substrates for soluble epoxide hydrolase. Anal. Biochem. 1995;231:188–200. doi: 10.1006/abio.1995.1520. [DOI] [PubMed] [Google Scholar]
- 33.Morisseau C, Du G, Newman JW, Hammock BD. Mechanism of mammalian soluble epoxide hydrolase inhibition by chalcone oxide derivatives. Arch. Biochem. Biophys. 1998;356:214–228. doi: 10.1006/abbi.1998.0756. [DOI] [PubMed] [Google Scholar]
- 34.Segel IH. Enzyme kinetics: behavior and analysis of rapid equilibrium and steady-state enzyme systems. Wiley; New York: 1993. [Google Scholar]
- 35.Zabell AP, Corden S, Helquist P, Stauffacher CV, Wiest O. Inhibition studies with rationally designed inhibitors of the human low molecular weight protein tyrosine phosphatase. Bioorg. Med. Chem. 2004;12:1867–1880. doi: 10.1016/j.bmc.2004.01.042. [DOI] [PubMed] [Google Scholar]
- 36.Cheng F, Oldfield E. Inhibition of isoprene biosynthesis pathway enzymes by phosphonates, bisphosphonates, and diphosphates. J. Med. Chem. 2004;47:5149–5158. doi: 10.1021/jm040036s. [DOI] [PubMed] [Google Scholar]
- 37.McGovern SL, Caselli E, Grigorieff N, Shoichet BK. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J. Med. Chem. 2002;45:1712–1722. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- 38.Tricot C, Villeret V, Sainz G, Dideberg O, Stalon V. Allosteric regulation in Pseudomonas aeruginosa catabolic ornithine carbamoyltransferase revisited: association of concerted homotropic cooperative interactions and local heteroptropic effects. J. Mol. Biol. 1998;283:695–704. doi: 10.1006/jmbi.1998.2133. [DOI] [PubMed] [Google Scholar]
- 39.Fersht A. Enzyme Structure and Mechanism. W. H. Freeman and Co.; New York: 1985. 2nd ed. [Google Scholar]
- 40.Levy BD, Serhan CN. Polyisoprenyl phosphate signaling: topography in human neutrophils. Biochem. Biophys. Res. Commun. 2000;275:739–745. doi: 10.1006/bbrc.2000.3371. [DOI] [PubMed] [Google Scholar]
- 41.Holstein SA, Hohl RJ. Isoprenoids: remarkable diversity of form and function. Lipids. 2004;39:293–309. doi: 10.1007/s11745-004-1233-3. [DOI] [PubMed] [Google Scholar]
- 42.Morisseau C, Goodrow MH, Dowdy D, Zheng J, Greene JF, Sanborn JR, Hammock BD. Potent urea and carbamate inhibitors of soluble epoxide hydrolases. Proc. Natl. Acad. Sci. U.S.A. 1999;96:8849–8854. doi: 10.1073/pnas.96.16.8849. [DOI] [PMC free article] [PubMed] [Google Scholar]