

Abstract
We use Langevin dynamics simulations to study the process by which a coarse-grained DNA chain is packaged within an icosahedral container. We focus our inquiry on three areas of interest in viral packing: the evolving structure of the packaged DNA condensate; the packing velocity; and the internal buildup of energy and resultant forces. Each of these areas has been studied experimentally, and we find that we can qualitatively reproduce experimental results. However, our findings also suggest that the phage genome packing process is fundamentally different than that suggested by the inverse spool model. We suggest that packing in general does not proceed in the deterministic fashion of the inverse-spool model, but rather is stochastic in character. As the chain configuration becomes compressed within the capsid, the structure, energy, and packing velocity all become dependent upon polymer dynamics. That many observed features of the packing process are rooted in condensed-phase polymer dynamics suggests that statistical mechanics, rather than mechanics, should serve as the proper theoretical basis for genome packing. Finally we suggest that, as a result of an internal protein unique to bacteriophage T7, the T7 genome may be significantly more ordered than is true for bacteriophage in general.
INTRODUCTION
Ubiquitous and intertwined with life throughout human history, viruses are of central importance in biology. In the past century, giant strides have been made in the epidemiology, pathology, and biochemistry of viruses, yet because of their subcellular size, our knowledge of viruses has necessarily been indirect (1). With the advent of recent technological innovations, particularly single molecule and atomic force experiments, we are for the first time capable of studying viral properties directly, down to the smallest length scale of the biological spectrum, the individual macromolecule. In the quest to uncover the physical nature of viruses, the stage has been reached where polymer physics and molecular biology merge.
The specific aspect of viruses on which we focus in this article, the packaging of the viral genome within its rigid protective coat, or capsid, is a remarkable physical process. Linear dimensions of capsids are typically tens of nanometers, whereas the length of the genome to be packaged is generally three to four orders of magnitude longer (2). The persistence length—the length scale along the chain contour for which directionality remains correlated—is ∼50 nm in physiological conditions, the same order of magnitude as the linear dimensions of a typical viral capsid. The bending required of the genome as it is wound tightly within the capsid thus leads to a buildup of energy that is large in comparison with kBT. Additionally, the presence of phosphate linkages leads to high linear charge density along the DNA backbone and gives rise to large repulsive energy inside the densely packed capsid. This most simple physical picture of the process of viral genome packing reveals a fundamental conflict of scales, wherein a long molecule must be compressed within a length scale on which it resists bending and to a density at which it must also overcome strong repulsive forces. The packing process has long been a subject of interest because of the tremendous pressures thought to arise inside the capsid.
We are interested in addressing two fundamental topics regarding the packing of dsDNA into capsids. The first topic is the configuration of the collapsed chain within the capsid, in particular how the structure evolves in the course of the packing process. In free solution, dsDNA is known to collapse in the presence of multivalent salts into highly compacted doughnut-shaped toroids (3,4), an example of a process termed “DNA condensation” (5). The positively charged multivalent salt ions mediate an apparent attraction between the negatively charged strands of DNA. The final toroidal structure results from a compromise between attractive interactions and bending energy. The viral packing process involves a similar structural transformation of DNA from a free solution configuration to a highly compressed packaged structure. It is worth exploring how closely the compaction in virus heads parallels the formation of free solution toroids.
For nearly 30 years, wide angle x-ray diffraction data have been available for various bacteriophage (T7, T2, λ) demonstrating that the presence of packaged DNA gives rise to a strong ring at 1/24 Å (6,7). Images of cross sections of free-solution toroids obtained from cryo-transmission electron microscopy (TEM) reveal hexagonal close packing of the DNA strands, indicative of a highly compact structure within the body of the toroid (3). Most current theoretical studies of the viral packing problem cite TEM images of packed virus heads. One cryo-TEM study in particular presented striking images of order obtained as a composite of individual T7 viruses in cross section (8). The virus images reveal, depending upon the angle of viewing, close-packed or banded structures that have been widely interpreted as arising from some toroidal or highly ordered structure. Other TEM studies have proposed a more modest ordering within viruses, with a local nematic-type order restricted to small regions of the capsid (9–15).
Regardless of how much order can be inferred from TEM studies, packaged ds-DNA structures are not likely to correspond to free solution toroid structures. Despite the many similarities between viral packing and polyelectrolyte condensation, the effect of confinement, unique to the packing process, is likely to prevent the formation of the same structures seen in free-solution toroids within the capsid. The outer diameter of the observed free solution toroidal structures are generally too large to fit inside virus capsids. In addition, a free solution toroidal structure, with its fairly large inner unfilled core (the diameter is set by the relatively large persistence length of dsDNA), could not fill the capsid interior to known packing densities.
The structure now most commonly attributed to packaged dsDNA, and more reasonably in accord with the notion of confinement than a free-solution toroid, is the so-called “inverse spool” structure (16,17). In fact, the inverse spool idea not only indicates a final packaged structure, but also suggests the process by which the DNA is wound. Inverse spooling is essentially a prescription for minimizing the bending energy of the chain, where the chain is treated as a mechanical string with bending resistance. The chain winds about and fills the outer bounds of confinement in an orderly fashion, with the most recently packed strands stacked upon previous strands. Eventually a complete new layer is formed that then serves as the outer bounds of confinement. As the process repeats, the chain subsequently winds progressively inwards toward some central axis. This process is very similar to the inverse of the process by which a length of thread is wrapped about a spool, in this case toward rather than away from a central axis.
However appealing the notion, experimental evidence has not conclusively demonstrated radial ordering within bacteriophage in general. One of the earlier theories of viral packing posited a much less-ordered structure akin to a ball of yarn (18) and many other structures have been proposed over the years. How and to what extent the DNA is ordered is not known.
The second question we wish to address involves the interplay of various forces, e.g., bending, electrostatic, and entropic, that arise during translocation (19,20) and compaction. Trying to deduce the interplay of these forces presents an even larger challenge than trying to deduce the packaged structure itself and is well beyond current experimental capabilities. The study of the interplay of forces has thus far been addressed only by analytical theory (16,17,21–24). Unfortunately, theories to date have required that some final structure be assumed. Additionally, forces in polyelectrolyte solutions are also notoriously difficult to treat with analytical theory, because they arise from both short and long-range potentials. Although we can currently infer that forces involved in packaging are very large, we cannot detail which forces are important and how the forces dictate the structure of the condensed viral genome. Progress in the understanding of these two questions would be of great help in understanding this crucial step of the viral replication cycle.
Two recent publications on genome packing in bacteriophage have had great influence, particularly in advancing a more physical approach to the subject. The publications were the result of remarkable experiments, each providing clues to the physical nature of individual virus particles. The first to appear was the cryo-TEM study of bacteriophage T7 of Cerritelli et al. (8). More recently, Smith et al. published their now famous single molecule optical tweezer study of the φ29 bacteriophage (25). Taken together, the articles give us information about the packaged structure and internal force buildup of two different viruses, yet the findings do not necessarily represent complementary findings. Although the work of Smith et al. (25) addresses the buildup of force within the capsid, it gives us no indication of corresponding packaged structure. Conversely, whereas Cerritelli et al. (8) literally give us a picture of the packaged structure, nothing can be said from this picture of internal force.
Further, we do not know that T7 and φ29 are representative of bacteriophage in general. The images of pervasive radial order in T7 have not been seen in any other bacteriophage that we are aware of. That order has yet to be detected in other phage may suggest that the radial order in T7 is not characteristic of bacteriophage in general, but rather is the result of a structural feature unique to T7. Additionally, there is some question as to the degree of ordering implied by the cryo-TEM images of Cerritelli et al. (8), as the striking order followed only from a great deal of averaging of individual images.
We feel that current available data do not point conclusively to any general packaged configuration or process whereby it is formed. In light of this uncertainty, the usefulness of Langevin dynamics simulation becomes evident. Motivated by these considerations, we have preformed Langevin dynamics simulations, where we can simultaneously follow the evolution of packaged DNA structure and the resulting internal energies, without having to assume a final structure or bias the packing process arbitrarily.
SIMULATION METHOD
Simulation of viral DNA
Because genomes are giant macromolecules composed of an enormous number of individual atoms, all-atom molecular dynamics simulations of entire viral genomes are currently not possible. To model ds-DNA, the type of viral genome with which we are concerned, we follow the traditional course-graining approach in which the chemically detailed double helix is replaced by a wormlike statistical chain of identical beads. Detail of the DNA structure on a length scale smaller than the bead diameter is lost. However, our goal is to describe global, rather than local, properties of the DNA chain. The simulated polymer is parameterized by the bead diameter d, an effective “bond” length, lo, the number of beads N, and the effective charge density α. The bead diameter, d, is a measure of the range of excluded volume interaction, which is itself determined by how many physical basepairs are subsumed in each bead. We wish to represent the roughly cylindrical or tubular geometry of the DNA double helix by a linear assembly of spherical beads, which we do in the following way: we fix d to be 2.5 nm, the hydrated diameter of ds-DNA in physiological conditions and we set lo equal to half of d. Consecutive spherical beads along the chain are thus overlapped to form a more approximately cylindrical structure. To avoid unphysical repulsion, the excluded volume interaction does not apply to consecutive beads along the chain. The resulting space-filling chain has a diameter that matches ds-DNA. Eight-hundred such beads are required to simulate each micrometer of phage genome. Throughout most of this article, we set α = 0. However, we will consider the case of a self-attractive chain, in which we are simulating indirectly the electrostatic bridging of DNA mediated by polyvalent cations. We will also present a brief section using Debye-Huckel interactions to model electrostatics, taking into consideration a range of values of α. For now, ignoring electrostatics greatly simplifies our calculations. We find that our results are qualitatively insensitive to the specific functional forms of the repulsive interactions.
Simulation of viral capsid
Confinement is imposed on each polymer bead in the simulation by the application of an icosahedrally symmetric force field. Twenty planes are defined, corresponding to the 20 faces of an icosahedron. The interior of the capsid includes the spatial region between these 20 planes, where beads are not subject to any confinement forces. Beads that traverse the confining planes in the outward direction are subjected to an inward-pointing restoring force normal to the confining plane. The magnitude of the restoring force is a function of the distance of encroachment of polymer beads into the confining potential and is given identically by the Lennard-Jones potential (to be given explicitly in the following section) for two polymer beads encountering each other. The capsid is parameterized by a single scalar size parameter (the distance from capsid center to the center of any face of the icosa) and the Lennard-Jones parameters borrowed from the bead-bead interactions.
Our representation of the capsid as a static force field is appropriate for the focus of this study: the structure of the confined DNA itself. We are interested in the capsid only indirectly, i.e., for the role it plays in determining the packaged DNA structure through confinement. Mechanical properties of the capsid, such as elasticity and ultimate strength, are beyond the scope of this article.
Force and energy calculations
DNA beads in our simulation are subject to interactions intended to capture the relevant physical forces involved in the packing process. There are two classes of bead potentials in our model: DNA-DNA and DNA-capsid.
First, we describe interactions between beads of our simulated DNA.
Excluded volume interaction
The excluded volume interaction is modeled by a Lennard-Jones potential, ULJ:
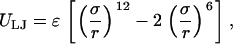 |
(1) |
where r is the center-of-mass distance between two beads, σ is the hard-core diameter, and ɛ is the strength of the potential. The energy minimum is at r = σ. For r < σ, a repulsive force arises, whereas for r > σ, an attractive force is found. For the case of a self-repulsive chain, the Lennard-Jones potential is truncated at r = σ.
Bond stretching interaction
The energy required to extend the bond between two consecutive beads along the chain is given by:
 |
(2) |
where kstretch is the stretch modulus and lo is the natural bond length.
Bending interaction
The work required to introduce a bend of angle θ along the backbone of the polymer is given by the three-body interaction:
 |
(3) |
where θ is the bond angle formed by three consecutive beads, θo is the effective equilibrium bond angle, and kbend is the bending modulus. The bending resistance of the genome is of central importance in this work. Therefore, from here on, we refer to kbend simply as k.
Now we describe the DNA-capsid interactions.
Repulsive interaction between the capsid wall and DNA
This interaction is modeled by the purely repulsive portion of the potential form used for DNA-DNA excluded volume interactions discussed previously, ULJ(r;σwall,ɛwall). The distance r refers to the distance from the center-of-mass of an encroaching polymer bead to the nearest restraining wall of the capsid. Parameters are chosen so that the repulsion felt by a polymer bead as it encroaches upon a capsid wall matches that for a polymer bead as it encroaches upon another polymer bead.
Dynamics
The molecular motor that drives the packing is simulated by introducing a force to the portion of the DNA that occupies the position of the opening at the top of the capsid. As packing proceeds, successive portions along the chain contour occupy this position and are thus subject to the packing force. The simulation algorithm entails solving the Langevin equation for each bead of the polymer chain:
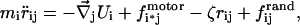 |
(4) |
where i is the bead index and j is the component index. Thus, rij is the jth component of the position vector of the ith bead. Each bead is assigned a mass, mi. The first term on the right-hand side of Eq. 4 represents the sum, for each bead, of the configuration-dependent forces. These forces are derived from the intra-DNA and DNA-capsid interactions described in the previous section. The second term describes the effect of the molecular motor as a downward force on the polymer bead i* located at capsid mouth. The effect of solvent, acting primarily through frequent collision of the relatively tiny and very fast-moving solvent molecules with much larger polymer segments, is described by the third and fourth terms. The third term is the velocity-dependent viscous drag force, where ζ is the coefficient of viscous drag. The fourth term is the random force acting on each bead, which is assumed to obey a Gaussian distribution centered at zero. The square of the standard deviation of the distribution is given by the fluctuation-dissipation theorem, relating the magnitude of the deterministic frictional force to the random thermal bath force in thermal equilibrium,
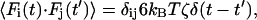 |
(5) |
where F(t) is the random force at time t, kB is the Boltzmann constant, and ζ is the coefficient of viscous drag. We have chosen as characteristic units the thermal energy available at room temperature, kBT298 = 4.1 pN nm, the Bjerrum length in water at room temperature, lB = 0.7 nm, and ζ = 7.5 × 10−13 Ns/m, for a sphere of diameter 2.5 nm in water at room temperature. The characteristic timescale Δt in our simulation is the minimum interval over which thermal bath forces are uncorrelated, which is determined by the fluctuation-dissipation theorem as 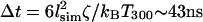 . Each Langevin dynamics step corresponds to a time δt = 0.0003Δt = 12.9 ps.
. Each Langevin dynamics step corresponds to a time δt = 0.0003Δt = 12.9 ps.
The Langevin dynamics simulation proceeds through the solution of the equations of motion of the N particles in the system, which are integrated using the standard velocity verlet algorithm (26)
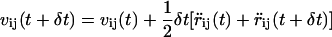 |
(6) |
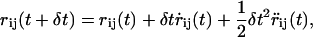 |
(7) |
where rij is as previously defined and vij is the jth component of the velocity of the ith bead.
Varying the number of particles in the simulation
The simulation does not proceed with a constant number of beads, N. At any given time step in the simulation, N comprises only those beads located within or at the opening of the capsid. Two beads are defined to be in the opening: i*, the bead occupying the location at which the motor force is applied, and the bead directly above i*. The external (i.e., unpackaged) length of the chain is simply excluded from consideration. The inherent assumption—that the external portion of the chain does not play a significant role in determining the course of packing—is based on both our initial simulation work that included external chain beads and the experimental work of Smith et al. (25), where the external portion of DNA is attached to a 2.2-μm polystryrene sphere and straightened by tension. Therefore, for the external portion of the chain, we expect that the bending energy is minimal and that the viscosity is dominated by the giant polystyrene sphere and is thus relatively invariant to degree of packing. In our preliminary simulation work, we found that an effective tension, acting through the downward force on the chain at the capsid opening and the opposing viscous drag, maintained the initially vertically aligned configuration of the external beads throughout the course of packing. Thus inclusion of the external beads in our initial simulation work, while demanding in terms of computing time, offered in return very little insight into the packing process. In our current simulations, the initial chain is simply a small (3,4) number of beads aligned vertically at and below the capsid opening. Each time the motor force pushes a bead into the capsid, a new chain-end bead is created at the upper end of the chain, to fill the resulting vacancy created at the capsid opening. N is thus increased throughout the packing process. At any given time, the terminal bead is given a large frictional coefficient, ζterminal = 75ζ, to maintain a large and constant external viscous resistance to packing, a simple approximation of the external chain and PS sphere. This procedure allows us to expend computing resources solely on our stated focus of interest, the evolution of packaged structure and its relationship to the internal buildup of forces.
Determination of persistence length
The limited flexibility of ds-DNA plays a crucial role in the current theories of viral packing. A simple statistical mechanical calculation of persistence length can be obtained by assuming each bond angle along the chain to be independent and calculating the ensemble average bond angle as
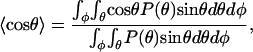 |
(8) |
where P(θ) is the distribution function. The persistence length is then given as (27),
 |
(9) |
where lo is the natural bond length and θ is complementary to the bond angle φ formed by consecutive bonds. From Eqs. 8 and 9, we have calculated predicted values of persistence length for various values of k, as shown in Fig. 1 a.
FIGURE 1.

Persistence length determination. (a) Predicted persistence lengths, based upon Eqs. 8 and 9, for a number of different values of k. (b) The projection of end-to-end distance onto the initial bond vector is shown in black for k = 750. The solid horizontal line is the average value of the projection of end-to-end distance onto the initial bond vector, which defines the persistence length. (c) The average of cosθ is used at each time step to determine the persistence length, given as 1/(1 + 〈cosθ〉). The average over all time steps is shown as a solid horizontal line.
To make a fair comparison with theory, we must verify that our basic simulated chain (kend = 750) has a persistence length of ∼50 nm. We have performed simulations of our chain in free solution to calculate the persistence length. The calculation involves periodically projecting the end-to-end distance of the chain along the initial bond vector of the chain. The time average of the projected vector (see Fig. 1 b) is the chain persistence length (28). In addition, we monitor the instantaneous average bond angle throughout the simulation and using Eq. 9 to calculate a persistence length (see Fig. 1 c).
RESULTS AND DISCUSSION
In the previous section, we have outlined a number of simplifying assumptions that have allowed us to define our simulations through the selection of a small number of parameters. In all simulations, a constant motor force of 55 pN was applied, approximately the value of the average “stall” force of the φ29 portal motor. As the chain becomes compressed within the capsid, our molecular motor packs against the internal resistive force with the same force as does the φ29 molecular motor. The simulated capsid is 0.7 times the height of the φ29 capsid, corresponding to ∼1/3 the volume; δt = 13 ps for all simulations. For bending constants we have used a series of values that are integral multiples of the value, k = 750, corresponding roughly to a free solution persistence length of 60 nm.
Packing velocity
Our simulation program outputs the loaded length, or more precisely, the number of beads packaged, as a function of time. The experimentally measurable quantity is the tether length, or the length of viral DNA external to the capsid. For the sake of comparison with experiments, we define for the simulations a tether length: tether length = (number of beads packed upon completion) − (number of beads currently packed). The packing velocity can be determined either by taking the slope of the number of beads packed versus time or, equivalently, the negative of the slope of the tether length versus time. We plot the tether length versus simulation time in Fig. 2 and the packing velocity versus loaded length in Fig. 3. In Fig. 4, we plot the loaded length versus simulation time. Chains parameterized with four different bending constants, k = 0, 750, 1500, 2250, have been used. The constants are integer multiples (from 0–3) of the value k = 750 corresponding roughly to the persistence length of ds-DNA in physiological conditions. Qualitatively, the curves of simulated tether length (Fig. 2) and packing velocities (Fig. 3) are strikingly similar to those obtained experimentally.
FIGURE 2.

Simulated tether length versus time. The tether length for individual runs is plotted as a function of simulation time. The bending stiffness parameters for the runs (k = 0; k = 750; k = 1500; and k = 2250) are indicated in the figure.
FIGURE 3.

Average packing velocity versus loaded length. Averages of packing velocity versus loaded length for a number of identically parameterized runs is shown in black. In each case, the value of the stiffness parameter, k, is indicated in the upper right-hand portion of the figure.
FIGURE 4.

Loaded length versus simulation time. Results of loaded length versus simulation time for a number of individual runs are shown for each k used in our simulations. In the cases where the chain has finite bending stiffness; i.e., for k > 0, characteristics of stochastic packing can be seen.
However, by observing packing velocities of individual runs (Fig. 5), we can see that the process of averaging has masked the presence of distinct pauses, which are common in our packing simulations. In addition, the plot of number of beads packaged versus time (Fig. 4) does not fit very closely with current theories of viral packing. The packing curve (Fig. 4) reveals two qualitatively different modes of packing. For at least the first half of the packing process, the packing curve is highly regular, both in the constancy of packing velocity and in the indistinguishable and uniform behavior from run to run. During the final stage of packing, the packing becomes a highly irregular function of time. Comparing different runs at a given time, we find that one run may be stalled while another is undergoing rapid packing. Similarly, given a single run, the packing can be paused, immediately followed by a burst of packing. Simulations have been run for very long times and we find that in most cases additional, albeit miniscule, increases in packing can be observed by simply waiting long enough. This suggests that packing in the later stages depends upon the internal chain configuration undergoing some slow relaxation process, allowing additional space for additional beads. Here we have our first hint of one of our major themes in this article, that polymer dynamics play a crucial role in the process of chain confinement.
FIGURE 5.

Packing velocity versus simulation time for individual runs; k = 750 in both cases. The presence of pauses, as indicated by circles, is clearly visible in individual runs.
We also note that the magnitudes of packing rates in our simulations are not directly comparable with experiment, as high packing velocities are required in simulations to complete the simulation in a reasonable time. The initial plateau region (Fig. 3) of the velocity curves is similar for all stiffnesses, indicating that the initial rate of packing is not strongly dependent upon chain stiffness. The packing rate is level until some critical degree of packing, at which point there is a decrease in the slope seen as packing nears completion. In the case of k = 0, where the chain does not have bending stiffness, the initial velocity of 0.4 simulation units is faster than for cases of finite bending stiffness. However, there does not appear to be significant difference in initial velocity for the chains with k = 750, 1500, or 2250. There is a noticeably more abrupt decrease toward zero packing velocity found in the cases of nonzero bending stiffness.
Fig. 2 shows that the tether length decreases very rapidly in the initial stage of packing and then much more slowly in the final stage of packing. (In the work of Smith et al. (25), the tether length is much longer, as it includes both the portion of the chain remaining to be packed, as well as an additional spacer length.) The tether length curves are in qualitative agreement with experimental results, with one obvious difference: our simulated curves have more prominent plateau regions where packing is temporarily halted. Although pauses are also visible in the experimental results, they are not as prominent as those in our simulations. Recall that our simulations proceed with a constant packing force and thus the pauses cannot be related to lack of evenness of the motor. The pauses are then most likely temporary blockages due to crowding of the chain within the capsid, with the duration of the pauses being related to the relaxation dynamics of the confined polymer. Relaxation processes of polymers are quite slow on the timescales accessible in simulation of polymers. Thus, the relatively exaggerated duration of pauses in simulation results can likely be explained by the small timescale of our simulations. High packing velocity gives a steeper slope in the tether length curve, whereas on the other hand the chain relaxation process is a function only of chain stiffness, not packing velocity. The high packing velocity thus may explain the exaggerated pause duration as compared to experimental results.
The pauses in our simulations and the pauses seen experimentally may in fact both be the result of chain relaxation phenomena. This idea is supported by further examination of the pauses. The duration of the pauses and the loaded length at which they arise do not appear to fit any regular pattern, except that no pauses are seen until at least 25% of the genome has been packaged. This behavior is true both in our simulations (Fig. 4) and in experimental results. The fact that pauses are not seen in the early stage of packing supports the idea that the pauses are due to relaxation in the crowded capsid environment. A possible rivaling theory, that experimentally observed pauses are related to specific DNA sequences, is disputed by the experimental results themselves, which indicate no correlation between pauses in the later stages of packing. Smith et al. (25) attribute pauses entirely to choppiness of the molecular motor. However, the absence of pauses in the early stage of packing points more plausibly toward internal chain relaxation, which become slower at higher packing density.
Finally, note in Fig. 4 that the final degree of packing, even for identically parameterized simulations, varies from run to run. Thus, the final degree of packing in our simulations is apparently not predetermined, as it would be in the case of highly ordered packing. This is particularly true for the chains with finite stiffness. Interestingly, in the experiments of Smith et al. (25) also, the overall change in tether length varies from run to run. Their interpretation is that the results do not indicate variable degree of final packing, but rather are artifactual, arising from DNA-bead attachment position. On the other hand, we believe that the long polymer is essentially forced into metastable conformations in a short period of time. As the polymer is packed at high velocity, the packaged portion of the chain is likely not attaining a stable equilibrium structure. Instead it constantly searches for lower energy configurations (to be addressed in more detail later) by undergoing various relaxation processes. The end of the pauses seen in our simulations is likely related to relaxation of chain configuration, which once achieved frees up additional volume for packing.
The explanation of stochastic packing results in terms of polymer relaxation implies that the velocity at which the chain is being fed into the capsid causes the resulting packaged polymer structure to diverge from a corresponding hypothetical equilibrium structure, i.e., the structure that would be obtained from an infinitely slow packing rate. The relaxation processes result from coordinated displacements of chain segments relative to each other and represent incremental steps back toward the “equilibrium” configuration. The evolution of nonequilibrium chain structure and the punctuated steps back toward equilibrium are what give the packing its stochastic character. The most notable implication of the stochastic packing is that there cannot be a one-to-one correspondence between loaded length and packaged structure. Nondeterministic packaging is a notion quite at odds with current theoretical models of force buildup and the inverse-spool model on which they are based.
Because the chain is very long and its motion severely constrained by confinement, relaxation phenomena are quite slow, more particularly for the larger-scale rearrangements. Even with the coarse-graining approach outlined in the previous section, the time span accessible in our simulation is smaller by orders of magnitude than the experimental timescale (approximate milliseconds to minutes, respectively). It is an unavoidable consequence that the velocity of packing in simulations is higher than it is in experiment. The packing velocities measured by Smith et al. (25) were of the order of 10−2 μm/s. Our initial packing velocities were slightly <5000 μm/s, similar to the packing rate used in previous simulation work (16). Thus, it is likely the case for simulations that, should the polymer configuration fall out of its equilibrium in the course of packing, it may never be able to recover fully in the course of simulations of reasonable time duration. In fact, the same may be true in nature as well. Smith et al. measured a characteristic packaging time of the order of a minute. However, in their experiments, the φ29 motor is burdened with an unnaturally large load: spliced to the end of the wild-type φ29 genome is additional piece of DNA approximately equal in length to the entire φ29 genome. The resulting strand of DNA is then anchored to a 2.2 μm polystyrene sphere. Such additional drag surely reduced the packing velocity greatly as compared to wild-type packing, assuming wild-type packing occurs on times of the order of seconds, significantly shorter than the time for large-scale polymer relaxations. The packing velocity in nature likely falls somewhere in between experimental and simulation packing velocities. Our results suggest that the pauses and variations in final loaded length, both in simulations and in the experiments of Smith et al. (25), may be attributable to nonequilibrium evolution of packaged structure. This nonequilibrium nature of packaging may be an important, and widely unrecognized, consideration.
Chain configuration and order
It is widely believed, based on the bending rigidity of ds-DNA, that a well-ordered ds-DNA structure is formed during confinement within bacteriophage capsids. The ordered structure is thought to result from the minimization of chain bending energy. The idea is supported, partly, by the cryo-TEM images that show radially periodic density in phage T7(8). Indeed, when bending energy is allowed to act in the absence of any solvent, it has been shown that energy minimization leads to well-ordered structures (23). However, at finite temperatures, chaotic thermal motion of solvent molecules constantly agitates the chain toward a state of higher disorder; there is constant competition between disorder-inducing thermal fluctuations and the order-inducing bending potential. The degree of order in the case of viral packing should depend upon the degree to which thermal disordering is overmatched by bending energy, of which the persistence length is a quantitative measure.
It is often assumed, because of the similarity of the persistence length and the capsid dimensions, that the bending forces acting on the packaged DNA dominate thermal forces. However, even on the length scale of the persistence length, a molecule exhibits a good deal of thermal fluctuation. This principle is illustrated in Fig. 6, where we show snapshots from a simulation of a chain undergoing Brownian motion in free solution. The chain was originally perfectly straight, i.e., in its minimum energy configuration, and aligned along the z axis, to which the viewing axis is perpendicular. The first two beads were held fixed, as is evident in the images. The chain, composed of a nonoverlapping string of beads, has been parameterized such that its persistence length is 20 beads. Only the first 20 beads of the chain are shown, so that we are observing chain configuration of a chain segment whose contour length is equal to the persistence length. As can be seen, the chain retains significant flexibility even below the length scale of its persistence length, in obvious contrast with its energy minimized structure, i.e., unbent. Thus, confinement of a chain to a container of linear dimensions of the persistence length does not effectively render the chain inflexible as is commonly presumed.
FIGURE 6.

Snapshots of DNA. The images show the gyrations of a chain with stiffness constant k = 750. The contour length of the chain shown is equal to its persistence length. The figure demonstrates the flexibility a polymer exhibits, even when considering only a short segment of contour length, equal to the persistence length.
In light of this insight, we will now proceed by addressing two basic assumptions of ordered packing. First, should we expect order when we apply the mechanical treatment of DNA at finite temperature and second, do we really see such order in cryo-TEM images? Our treatment of DNA, as a mechanical body weakly coupled to a thermal bath, allows us to address the first question, that is, whether an ordered structure can be expected from simple bending and confinement considerations only. In Fig. 7, a and b, periodic snapshots from two simulations, with stiffness parameters, k = 750 (Lp = 60 nm) are shown. For each simulation, three snapshots of DNA structure are shown, at the beginning, middle, and late stages of packing. The capsid height in both cases, 35 nm, is less than the persistence length of either of the chains. Interactions between different segments of the chain are (a) purely repulsive (σ = 1.0, ɛ = 1.0) and (b) attractive (σ = 1.5, ɛ = 1.0). Clearly, there is no discernable symmetry in the purely repulsive case (a). In Fig. 7 b, we show a structure formed with chain-chain attraction (σ = 1.5, ɛ = 1.0). In the case of the self-attractive chain, we find a structure like the folded toroid proposed by Hud (29), where chain strands are locally aligned in a nematic-type fashion. However, there is no radially symmetric order aligned with the packing axis.
FIGURE 7.

Evolution of packaged structure throughout the course of packing. Simulation snapshots demonstrate the progression of structures taken by the packaged genome. Each row corresponds to one of the packing conditions considered in this study. Hollow capsids (no Serwer core) are shown in panels a and b. Capsids with Serwer cores present are shown in panels c and d. (a and c) Interactions between DNA strands are purely repulsive; (b and d) DNA strands are self-attracting. In all cases, the bending stiffness is k = 750, corresponding to the bending stiffness of dsDNA in physiological conditions. The effect of the Serwer core on the order of the packaged structure is evident from these pictures.
In a related set of experiments, we considered the situation where the persistence length of the chain exceeds the height of the capsid by more than an order of magnitude. In this case, as was also seen by Kindt et al., the chain packed in an inverse-spool fashion initially, wrapping in a helical layer around its outer layer of confinement. However, the inverse-spool process was not maintained after this initial layer was formed, with the inner core filling in a disordered fashion. The inability to predict an ordered structure with the mechanical DNA model of our simulations is a significant result because it indicates that the mechanical approach to modeling DNA is contradicted by, rather than supported by, the experimentally observed order (8). It is possible that the mechanical model for DNA chain rigidity in bacteriophage is too simplistic. For example the possibility of kinking or melting of short stretches of DNA is not accounted for in our simple mechanical model of DNA. However, these effects would likely lead to less, not more, spool-like order within the capsid.
Another possibility is that the case for interpreting the cryo-TEM images of Cerretelli et al. (8) as representing spool-like order within phage has been overstated. A considerable number of other studies, including cryo-TEM, on a variety of bacteriophage have revealed considerably less order, showing for example that where striations exist, they are limited to patches, or domains, of the order of 20 nm (9–15). Further, these individual domains are not oriented with respect to each other. This more modest ordering agrees more with that found in many of our simulations; see Fig. 7, a and b.
In the case of T7, a considerable amount of disorder could be masked in the procedure by which Cerritelli et al. (8) generated images of radial symmetry. Many individual images were averaged into a single composite, which was then subsequently radially averaged. We were interested in determining if our disordered but tightly packed structures give rise to similar bands of radial order when subjected to a similar averaging process. X-ray diffraction studies have demonstrated that, at the high packing density found in a fully packaged phage, chains are on average locally close-packed. With respect to any long axis of an icosahedron, the average radial distance to a confining wall is of fairly constant radius, except near the poles. That is, along the majority of the axis, the chain experiences approximately cylindrical confinement. For a densely packaged structure, we expect close-packed layers to be pressed against confining walls. Our question was whether, when viewed down the packing axis, circularly symmetric bands of fluctuating particle density might be discernable radiating inwards from the radius of the wall. Diffraction between wall-induced close-packed layers has been speculated to be the cause of the low wavelength ripple seen superposed on the 1/2.4 nm ring in x-ray diffraction data.
To obtain a quantitative measure of circularly symmetric ordering, the bead density of a given configuration from our simulation was projected onto the plane perpendicular to the packing axis. Projected densities obtained in this way have been presented as contour plots, or further averaged azimuthally to obtain a radial distribution function. By recreating contour plots from radial distribution functions and taking the negative of the resulting images, we obtain images that can be compared to the composite TEM images of Cerretelli et al. (8).
We have found that the radial distribution functions obtained for our disordered packaged structures do not consistently show radially periodic density fluctuations (Fig. 8 a). We do, however, consistently see at least a single faint band at the perimeter of the capsid. This is similar to what has been seen in reconstructions of the T4 virus (14). Apparently the circular order induced by the wall decays very rapidly and is therefore unlikely to explain the extent of order indicated by the T7 images. In the case of the self-attractive chain, there is clearly more local nematic type packing, although with no alignment to the packing axis (see Fig. 7 b). This local order occasionally, but not consistently, leads to striking radial order.
FIGURE 8.

Probability density of packaged structures. Projected bead density on the xy plane (perpendicular to packing axis) is shown in the first column. Projected bead density is averaged over polar angle and presented as radial probability density in columns 2 and 3. (a–d) These correspond to the same packing conditions as in Fig. 7.
We thus suspect that there is some genuine radial symmetry to the packaged T7 genome with respect to the packing axis, perhaps more than for other ds-DNA bacteriophage studied. T7 may be unique in possessing an internal structural protein connected to the capsid at the portal vertex and protruding inwards toward the center of the icosahedron. This hollow cylindrical protein within the T7 capsid was first identified by Serwer in 1976 (30). We have performed simulations to determine whether this internal structure might somehow function as an essential order-inducing feature of the T7 capsid. The “Serwer core” was modeled as a hollow cylindrical tube through which the beads of the chain were extruded into the capsid. We found that, even when a purely repulsive chain was considered, the packaging order was noticeably increased compared to the core-less capsid. When intrastrand attractive interactions were allowed to act, the internal structural core became yet more effective at inducing order.
In Fig. 8, we show density contour plots of projected bead density for a number of different simulations we have performed. Increased radial order can clearly be seen in cases where packing was performed into capsids possessing the internal cylindrical core (the Serwer core). The Serwer core seems to function mainly by altering the position at which the chain emerges inside the capsid. The chain is extruded from the bottom of the cylindrical core, located near the center of the icosahedron. Because of limited flexibility, the most recently extruded portion of the chain is directed toward the lower vertex of the icosahedron. The potential of the confining walls in this lower region of the capsid is symmetric about the packing axis, causing the most recently extruded portion of chain to form a small-radius circular loop perpendicular to the packing axis. Subsequent loops are forced underneath previously formed loops, which consequently rise upwards along the packing axis. The loops are driven up also in search of a wider cross-sectional radius into which they can expand, available in the region away from the poles of the icosahedron. As this process continues, a uniaxial assembly of stacked hoops forms, with most recent added hoops located at the bottom. This assemblage stabilizes as it is skewered onto the projecting core, which then serves as a structural axis. The structures formed from this procedure are shown in Fig. 7, c and d. The shading gradient along the contour of the chain helps to reveal the history of packing, with recently packaged beads lighter, earlier packaged beads darker. The shading gradient verifies that the Serwer core forces more recently packaged strands of DNA to the bottom of the capsid, as described above.
Finally, we have defined a toroidal order parameter to quantify the degree to which the packaged DNA forms a toroid-like assemblage of stacked hoops aligned with the packing axis. To do this we considered individual arcs of 15 consecutive beads along the chain. A unit normal vector for each arc was defined by normalizing the cross-product of the two vectors connecting the middle bead to each of the end beads. For each arc along the chain contour (neighboring arcs overlap), we calculated the unit arc normal and projected its length along the packing axis. The order parameter was determined by dividing the sum of the magnitude of projected arc normals by the number of arcs. The order parameter approaches unity for a perfect stack of hoops, whereas a group of randomly oriented arc normals gives a value of 0.5. In Fig. 9, we give the order parameter versus loaded length for each of the major classes of simulations in this study—hollow capsid/repulsive chain, hollow capsid/attractive chain, Serwer core/repulsive chain, Serwer core/attractive chain. In the case of the hollow capsid filled by a purely repulsive chain, the order parameter approaches 0.5 during packing, indicating that the loops of packaged genome are not aligned on average with any particular axis, as is evident also in Fig. 7 a. Packing carried out with a Serwer core present can be seen to give rise to increased toroidal order. The case of self-attractive/Serwer core gave the highest overall toroidal ordering, with order parameter values in excess of 0.9. For the hollow capsid/attractive chain simulations, in the early stages of packing, structures formed appeared toroid-like. However, as the packing proceeded, the toroid-like structures did not maintain alignment with the packing axis and ultimately often lost their toroid-like appearance. This behavior is visible in Fig. 9, where the corresponding value of the order parameter is initially as large as the values corresponding to packaging with a Serwer core.
FIGURE 9.

Toroidal order parameter. The ordering of packaged structures into hoops aligned with the packing z axis is given by the toroidal order parameter. The order parameter is followed throughout the course of packing for the major packing conditions considered in this work. (▪, Serwer core/self-attractive chain; •, Serwer core/repulsive chain; ▴, no Serwer core/ self-attractive chain; and ♦, no Serwer core/repulsive chain.
Energy, force, and pressure
The energy increase associated with packaging of DNA results from bending of the semiflexible DNA chain and repulsive interactions that arise during compression, both between the chain and the wall and between different segments of the chain. Knowing these energies as a function of loaded length allows us to calculate a quantitative measure of force resisting further packing. This force is obtained by taking the slope of the internal energy versus number of beads packaged averaged over a number of identically parameterized runs. Note that this force ignores entropic contributions. In averaging energy and force for a group of runs, the range of loaded length considered is determined by the run with the shortest span of total loaded length. In Fig. 10, the resistive force calculated from average energy difference is plotted versus loaded length of viral genome. The total force versus length packed, as well as the decomposition of the energies and forces into bending and repulsive components, are given for chains with four different bending stiffnesses, k = 0, 750, 1500, and 2250. There are a number of interesting observations to be made of the calculated resistive force. Early in the packaging process, the force is dominated by resistance to bending, which remains fairly constant. The force arising from repulsion is negligible until the later stages of packing, where it increases drastically. These findings are in agreement with earlier simulation studies. The bending and repulsive forces appear to be equal at the termination of packing for k = 750, but not for the less flexible chains.
FIGURE 10.

Force versus loaded length. Repulsive, bending, and total energy are shown as a function of loaded length. Chains of three stiffness parameters were used: (top) k = 750; (middle) k = 1500; (bottom) k = 2250. For each k considered, the force versus loaded length curves represent averages over a number of identically parameterized runs.
The idea of nonequilibrium structure may be essential in explaining the next observation, that in no case does the resistive force increase to a value equal to the packing force. The simplest explanation for this is that, as the chain is packaged and the conditions inside the capsid become crowded, the chain becomes kinetically frozen in a metastable nonequilibrium state. (The neglect of the entropic contribution to the resistive force may also be a factor. However, it has been argued that the entropic contribution is negligible as compared to the bending and repulsive components in genome packing. We have no reason to believe that this situation is any exception.) Recall that we cannot distinguish for certain between paused and completed packing, i.e., whether packing has truly stopped, or has merely temporarily stalled. By waiting long enough, we find that the chain can eventually relax, so that the packing undergoes a subsequent step-like increase. Because the simulations may have been terminated before packing was absolutely completed, it may be tempting to conclude that we have ignored some final increment of crucial resistive force. However, evidence arising from our work suggests that the final packaged structure is inherently nonequilibrium or “glassy”. Termination of packing is apparently not caused by a static force balance, but rather by some combination of resistive force and restricted mobility of the packaged chain. Packing does not occur the same way every time and the dynamics of chain relaxation largely determine chain packing, with energy considerations relegated to secondary importance. In this light, we are not surprised to find that the final resistive force has not reached the same magnitude as the packing force. We propose that there is no one final packaged structure, predetermined by orderly packing and energy minimization. Again, we can point to experimental results to support our ideas. Smith et al. (25) found that the φ29 motor can package more DNA than is required by the full φ29 genome. Considering the intricate crafting of natural systems, this cushion should not be assumed to be unimportant. This seemingly “excess” packing capacity might not be excess at all, but rather necessary for packing structures that pack to different degrees.
The stochastic nature of packing is further revealed by examining the evolution of bending energy for individual runs. In Fig. 11, we follow bending energy as a function of loaded length for chains with bending modulus k = 750. Three identically parameterized runs are shown, each given a unique symbol (squares, circles, triangles) and differing only in the value of random seed used.
FIGURE 11.

Bending energy versus packaged length. The bending energy of packaged structures is given as a function of loaded length. In all cases k = 750. Three identically parameterized runs are shown, with each run given a unique symbol. Each data point represents the average bending energy of the enormous number of configurations associated with an incremental loading of several tens of nanometers. For a given loaded length, average bending energy varies from run to run. The loaded length and associated bending energy at which the packaging stalls for all six runs we have performed is plotted in the inset figure. The final configurations do not correspond to any particular loaded length or internal bending energy; the termination of packing is evidently unrelated to the bending energy at which it occurs.
Each data point represents the average bending energy of all chain configurations associated with an incremental loading of 25 nm. Thus, a difference in average bending energy between different runs indicates a difference in average structure, not attributable to thermal fluctuation. Bending energy is clearly not a single valued function of loaded length.
For each run, the loaded length at which the packaging stalls varies. The final configurations do not correspond to any particular loaded length or internal bending energy. The final loaded length appears to be unrelated to bending energy, supporting the nonequilibrium notion of packaged structures.
We have also calculated the resistive force directly, by measuring the average force exerted upwards on the bead in the neck. The calculation involves coordinates extracted periodically throughout the course of a packing simulation. Each of these coordinates become the starting configurations for a new set of simulations. The motor force was turned off and the bead corresponding to the neck position was held in place. During each simulation, the beads below in the capsid undergo Brownian motion and exert a force on the bead in the neck, which is measured and averaged over a large number of time steps. We have found that the force attains a constant, pseudoequilibrium value within reasonable amounts of processing time (see Fig. 12).
FIGURE 12.

Direct force measurements of the bead in the neck of the capsid for a single run. (See text for procedure.) Each point represents the pseudoequilibrium force due to interactions with packaged beads. The noisiness of the force with incremental change in loaded length is not due to uncertainty in the measurement, but rather is a genuine demonstration of nonequilibrium behavior, particularly the local conformational fluctuations near the neck, of the packaged structures.
We have presented numerous pieces of evidence that packing is not driven exclusively by an equilibrium buildup of resistive force. It would be surprising, then, if our direct-force results indicated a smooth increase in internal resistive force, culminating in a final value countering the motor force. Indeed, we do not see such well-behaved direct force measurements. In Fig. 12, we give a plot of direct resistive force versus loaded length for a single packing run, with k = 750. At lower loaded lengths, we find a very small and well-behaved monotonic increase in resistive force. As packing proceeds we find that the direct measurement of resistive force yields erratic values. As new portions of chain are forced through the neck into the capsid, previously packaged strands of the chain near the neck region must locally rearrange to accommodate the newly packaged portion; the chain becomes crowded within the capsid and the steric resistance to packing becomes determined by random local motions, which are likely the cause of both the irregular packing and erratic force measurement.
Note that the direct resistive forces display enormous positive fluctuations, up to twice the magnitude of the motor force, before the final configuration. The fluctuations in force with very small change in loaded length fits very well with the idea that direct force measurement values are dictated only by local configurations in the neck region. As proof of this idea, we have calculated—simultaneously with the direct force measurements—the average pressure over the entire interior capsid walls. Our interpretation of the direct force measurement as a local/nonenergetic effect and the energy difference force determination as a global energy effect is strongly supported by measurements of the internal pressure buildup within the capsid. The pressure is determined as the force per unit area exerted by the polymer beads on the capsid walls, averaged over the entire inner surface of the capsid. We expect the pressure to reflect the global energy state within the capsid, i.e., we expect that the pressure behavior will be a relatively well-behaved monotonically increasing function of loaded length. In Fig. 12 b, we plot on the same axis both the hold force measurements and the corresponding pressure measurements. Although direct force values fluctuate wildly with loaded length, the internal pressure inside the capsid is fairly constant and well-behaved.
We find that when the final configuration of any packing run is used as a basis for a hold-force simulation, the resistive force measured is comparable to the motor force. The results of our direct force measurement thus seem to contradict the findings of Kindt et al. (16), both in that our force values become erratic and that they do in fact attain the magnitude of the motor force at the completion of packing. However, we believe the apparent discrepancies can be reconciled by the simple consideration that the packing simulations of Kindt et al. (16) were performed with constant velocity conditions. For a constant velocity simulation, the irregular portion of the packing shown in Fig. 4 is inaccessible. Recall that in the irregular phase, pauses occur, for which we must wait for relaxation processes to occur. A constant velocity simulation instead stops at the first pause. Kindt et al. (16) never measure irregular or large forces essentially because their simulations proceeded to lower loaded lengths.
Despite the fact that our direct force measurement values attain the magnitude of the motor force at completion of packing, we do not believe that this either: 1), contradicts our previous energy difference results showing that the internal force does not attain the magnitude of the packing force; or 2), supports the idea that the packing process is determined primarily by energetic considerations. We believe that the packing stalls due to local steric blockage in the neck region, a kinetically driven phenomena. The configuration is locally frozen and so with a motor force pushing against it, of course a reaction force equal and opposite to the motor force will arise. The direct resistive force that we measure for our final configurations is then simply a reaction force arising from the application of the packing force to a locally glass-like solid. Our energy difference force determination, on the other hand, was based on an accounting of the energy of the entire packaged structure.
In Fig. 13, we see that there is no final or equilibrium pressure, as might be erroneously concluded from the direct force measurements. If the direct force measurements were truly showing equilibrium in the final packaged states, the pressure would not display this behavior.
FIGURE 13.

Pressure versus loaded length. The average measured pressures versus loaded lengths are shown for six identically parameterized runs. In all cases k = 750 and there is no Serwer core present. For a given loaded length, the pressure does not display the noisiness from run to run as seen for the direct measurements of resistive force (Fig. 12). The measured pressures, in tens of atmospheres, are in agreement with previous work. The terminal values of pressure and loaded length for each individual run is shown in the inset. As was the case with bending energy, the termination of packing does not correlate with the buildup of internal pressure to any specific terminal value.
Finally, we show in Fig. 14 the bending energy versus loaded length for the different classes of simulations in this study. It is clear that the presence of the Serwer core during packing leads ultimately to lower bending energy structures. This provides definitive proof that packaged structures formed in the absence of the Serwer core are not dictated by bending energy minimization. It is interesting to note further that the structures formed in the presence or absence of the Serwer core are not distinguishable in the early stages of packing by their bending energies. Thus, the Serwer core acts to guide the chain to a more ordered structure initially, which is essential in the subsequent formation of lower bending energy final structures.
FIGURE 14.

Average bending energy versus loaded length—effect of Serwer. No Serwer core/self-repulsive chain (•), Serwer core/self-repulsive chain (▴); and Serwer core capsid/attractive chain (▪) are shown. In all cases, k = 750. The presence of the Serwer core guides the chain to lower energy configurations, particularly at higher degrees of packing. Two points are interesting: 1), bending energy in absence of Serwer core is clearly not minimized; and 2), the structures during early stages of packing are not distinguishable in terms of bending energy, although we know that the structures formed in the presence of the Serwer core are more ordered. Without an energy difference to select for the more ordered initial structures, a more disordered initial structure arises. At higher degrees of packing, the more ordered structures are clearly of lower bending energy. However, the initially unordered structures are unable to rearrange in the crowded capsid interior to form lower energy structures and so we find that the final configurations in the absence of the Serwer core remain stuck in a state of relatively high energy.
Role of electrostatics
In this section, we present a brief calculation of electrically charged DNA beads packaged into a smaller capsid and observe that the findings presented in this article are qualitatively insensitive to electrostatics. The experiments of Smith et al. (25) were done at 50 mM NaCl and 5 mM MgCl2. The Debye screening length, determined predominantly by NaCl under these conditions, is ∼1.5 nm. The bond length of our coarse-grained dsDNA is 1.25 nm (bead diameter = 2.5 nm; consecutive beads overlap to produce more “tubular” structure). There are ∼8 phosphate groups per 1.25 nm along the contour of dsDNA. Thus, we assume that there are 8 phosphate groups per bead in our simulations. However, it is not physically realistic to give a full charge per phosphate group, as we know that counterion condensation significantly reduces the effective linear charge density of dsDNA. From previous work in our group (32,33), we know that only a small fraction, 0 < α < 0.2, of phosphate groups remain unneutralized by counterion (or salt) condensation. We have performed a series of three packing simulations (α = 0.0, 0.10, 0.20; namely 0.0, 0.8, and 1.6 charges per bead, respectively) on a capsid with the center-to-vertex distance being 7.5 nm. We use Debye-Huckel interactions with a screening length of 1.5 nm in these simulations. The effect of electrostatics on the time evolution of loaded length is given in Fig. 15. The presence of charge manifests itself only in reducing the final degree of packing (no attractive bridging effects in the presence of Mg2+ are considered) as the repulsive energy reduces the final allowed packing density. However, the reduction in the eventual packing density is only modest for the experimentally relevant values of α. More importantly, the generic features of the packing pathway in our simulations (such as the presence of irregularities in the packing rate and packaged order) are independent of electrostatics. These generic features match well with the experimental features and we conclude that their existence is independent of electrostatics but crucially depends on the bending energy, excluded volume, and confinement, under the experimental conditions. The relatively minor role played by electrostatics on the packing process is consistent with the results presented in Fig. 10, where the sign and strength of the Lennard-Jones potential play minor roles. Additionally, we found in Figs. 7 and 8 that consideration of a self-attracting chain (simulating the bridging effect of polyvalent cations) led to no qualitative effect on the degree of toroidal ordering of the packaged structure. Therefore, we conclude that, regardless of the presence or absence of charge, packing proceeds in an irregular manner, leading to disordered packaged structures and pauses due to configurational rearrangement in the packing process. Given the enormous computational expense of considering detailed electrostatics, we have instead directed our computational resources in this article toward modeling more accurately the physical dimensions and packing rates in phage genome packaging. Charge may play an interesting and important role in other aspects of viral packaging, which we will explore in future work, but for the findings we present in this article, the bending energy, excluded volume, confinement alone lead to the chain configurations becoming nonequilibrium.
FIGURE 15.

Influence of electrostatics on loaded length versus simulation time. A smaller scale system was used to allow for inclusion of electrostatics: the vertex-to-vertex size of the capsid was 15 nm, the bending stiffness constant, k, was 350, and the packing force was 38 pN. The values used for α, the fraction of bare charge, are given in the legend. Loaded length is given per bead (bond length = 1.25 nm). The units of simulation time for this reduced scale system do not match those for other simulations presented in this article. Pauses, the fingerprint of irregular packing, are clearly present in each case. The only feature distinguishing the different runs is the final degree of packing, which for the physically relevant values of α shown, was reduced, likely a consequence of the introduction of additional repulsive energy within the capsid.
CONCLUSION
We have simulated the packing process of a ds-DNA bacteriophage. The geometry of the confining vessel, the intrinsic bending resistance of the simulated chain, the diameter of the chain, and the applied motor force were all selected based upon physical relevance. In the analysis of simulated packing rate data we have noted a number of stochastic phenomena. The stochastic features do not seem to match with the idea of spool-like packing, yet are in general accord with experimental results of Smith et al. (25). In addition, we have found that packaged structure does not evolve as predicted by spool-like packing. Specifically, stacked hoops do not form and align with the packing axis. The presence of the internal Serwer core does guide the packaging process toward more spool-like ordered final structures. The Serwer core appears to be crucial in the initial stage of packing, selecting for more ordered structures that are indistinguishable in terms of bending energy from disordered arrangement of loops. Simulated TEM images of self-attractive chains packaged in the presence of a Serwer core display the experimentally observed radial ordering. However, the simulated TEM images have proven unreliable in confirming spool-like packing, as radially symmetric structures were also observed in packaged structures showing no discernable orientation with the packing axis. Our future work will explore the implications of packaged structure on the release of the viral genome and the role of electrostatics, specifically how various charge distributions on the interior capsid wall affect the packaging process.
Upon completion of our work, a simulation study closely related to this work has been published (31).
Acknowledgments
We acknowledge National Institutes of Health grant No. 1R01HG002776-01, National Science Foundation grant No. DMR-0209256, and the Materials Research Science and Engineering Center at the University of Massachusetts at Amherst.
References
- 1.Alberts, B., D. Bray, J. Lewis, M. Raff, K. Roberts, and J. D. Watson. 2002. Molecular Biology of the Cell. Garland Science, New York.
- 2.Knipe, D. M. Howley, P.M. 2001. Fundamental Virology. Lippincott Williams and Wilkins, Philadelphia, PA.
- 3.Hud, N. V., and K. H. Downing. 2001. Cryoelectron microscopy of lambda phage DNA condensates in vitreous ice: the fine structure of DNA toroids. Proc. Natl. Acad. Sci. USA. 98:14925–14930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hud, N. V., K. H. Downing, and R. Balhorn. 1995. A constant radius of curvature model for the organization of DNA in toroidal condensates. Proc. Natl. Acad. Sci. USA. 92:3581–3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bloomfield, V. A. 1996. DNA condensation. Curr. Opin. Struct. Biol. 6:334–341. [DOI] [PubMed] [Google Scholar]
- 6.Earnshaw, W., S. Casjens, and S. C. Harrison. 1976. Assembly of head of bacteriophage P22: x-ray-diffraction from heads, proheads and related structures. J. Mol. Biol. 104:387–410. [DOI] [PubMed] [Google Scholar]
- 7.Earnshaw, W. C., J. King, S. C. Harrison, and F. A. Eiserling. 1978. Structural organization of DNA packaged within heads of T4 wild-type, isometric and giant bacteriophages. Cell. 14:559–568. [DOI] [PubMed] [Google Scholar]
- 8.Cerritelli, M. E., N. Q. Cheng, A. H. Rosenberg, C. E. McPherson, F. P. Booy, and A. C. Steven. 1997. Encapsidated conformation of bacteriophage T7 DNA. Cell. 91:271–280. [DOI] [PubMed] [Google Scholar]
- 9.Booy, F. P., W. W. Newcomb, B. L. Trus, J. C. Brown, T. S. Baker, and A. C. Steven. 1991. Liquid-crystalline, phage-like packing of encapsidated DNA in herpes-simplex virus. Cell. 64:1007–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richards, K. E., R. C. Williams, and R. Calendar. 1973. Mode of DNA packing within bacteriophage heads. J. Mol. Biol. 78:255–259. [DOI] [PubMed] [Google Scholar]
- 11.Lepault, J., J. Dubochet, W. Baschong, and E. Kellenberger. 1987. Organization of double-stranded DNA in bacteriophages: a study by cryoelectron microscopy of vitrified samples. EMBO J. 6:1507–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serwer, P., S. J. Hayes, and R. H. Watson. 1992. Conformation of DNA packaged in bacteriophage-T7: analysis by use of ultraviolet light-induced DNA-capsid cross-linking. J. Mol. Biol. 223:999–1011. [DOI] [PubMed] [Google Scholar]
- 13.Aubrey, K. L., S. R. Casjens, and G. J. Thomas. 1992. Secondary structure and interactions of the packaged DsDNA genome of bacteriophage-P22 investigated by Raman difference spectroscopy. Biochemistry. 31:11835–11842. [DOI] [PubMed] [Google Scholar]
- 14.Olson, N. H., M. Gingery, F. A. Eiserling, and T. S. Baker. 2001. The structure of isometric capsids of bacteriophage T4. Virology. 279:385–391. [DOI] [PubMed] [Google Scholar]
- 15.Poy, S. 1994. Molecular Biology of Bacteriophage T4. J. D. Karam, editor. American Society for Microbiology, Washington, DC.
- 16.Kindt, J., S. Tzlil, A. Ben-Shaul, and W. M. Gelbart. 2001. DNA packaging and ejection forces in bacteriophage. Proc. Natl. Acad. Sci. USA. 98:13671–13674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Purohit, P. K., J. Kondev, and R. Phillips. 2003. Mechanics of DNA packaging in viruses. Proc. Natl. Acad. Sci. USA. 100:3173–3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Black, L. W. 1989. DNA packaging in DsDNA bacteriophages. Annu. Rev. Microbiol. 43:267–292. [DOI] [PubMed] [Google Scholar]
- 19.Muthukumar, M. 1999. Polymer translocation through a hole. J. Chem. Phys. 111:10371–10374. [Google Scholar]
- 20.Kong, C. Y., and M. Muthukumar. 2004. Polymer translocation through a nanopore. II. Excluded volume effect. J. Chem. Phys. 120:3460–3466. [DOI] [PubMed] [Google Scholar]
- 21.Odijk, T. 1998. Hexagonally packed DNA within bacteriophage T7 stabilized by curvature stress. Biophys. J. 75:1223–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tzlil, S., J. T. Kindt, W. M. Gelbart, and A. Ben-Shaul. 2003. Forces and pressures in DNA packaging and release from viral capsids. Biophys. J. 84:1616–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arsuaga, J., R. K. Z. Tan, M. Vazquez, D. W. Sumners, and S. C. Harvey. 2002. Investigation of viral DNA packaging using molecular mechanics models. Biophys. Chem. 101:475–484. [DOI] [PubMed] [Google Scholar]
- 24.Purohit, P. K., M. M. Inamdar, P. D. Grayson, T. M. Squires, J. Kondev, and R. Phillips. 2005. Forces during bacteriophage DNA packaging and ejection. Biophys. J. 88:851–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith, D. E., S. J. Tans, S. B. Smith, S. Grimes, D. L. Anderson, and C. Bustamante. 2001. The bacteriophage phi 29 portal motor can package DNA against a large internal force. Nature. 413:748–752. [DOI] [PubMed] [Google Scholar]
- 26.Allen, M. P., and D. J. Tildesley. 1987. Computer Simulation of Liquids. Clarendon Press, Oxford, UK.
- 27.Yamakawa, H. 1971. Modern Theory of Polymer Solutions. Harper & Row, New York.
- 28.Stevens, M. J., and K. Kremer. 1995. The nature of flexible linear polyelectrolytes in salt-free solution: a molecular-dynamics study. J. Chem. Phys. 103:1669–1690. [Google Scholar]
- 29.Hud, N. V. 1995. Double-stranded DNA organization in bacteriophage heads: an alternative toroid-based model. Biophys. J. 69:1355–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Serwer, P. 1976. Internal proteins of bacteriophage-T7. J. Mol. Biol. 107:271–291. [DOI] [PubMed] [Google Scholar]
- 31.Spakowitz, A. J., and Z. G. Wang. 2005. DNA packaging in bacteriophage: is twist important? Biophys. J. 88:3912–3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu, S., and M. Muthukumar. 2002. Langevin dynamics simulation of counterion distribution around isolated flexible polyelectrolyte chains. J. Chem. Phys. 116:9975–9982. [Google Scholar]
- 33.Liu, S., K. Ghosh, and M. Muthukumar. 2003. Polyelectrolyte solutions with added salt: a simulation study. J. Chem. Phys. 119:1813–1823. [Google Scholar]