

SUMMARY
The catabolite activator protein (CAP) bends DNA in the CAP-DNA complex, typically introducing a sharp DNA kink, with a roll angle of ∼40° and a twist angle of ∼20°, between positions 6 and 7 of the DNA half-site, 5′-A1A2A3T4G5T6G7A8T9C10T11-3′ (“primary kink”). In previous work, we showed that CAP recognizes the nucleotide immediately 5′ to the primary-kink site, T6, through an “indirect-readout” mechanism involving sequence effects on energetics of primary-kink formation. In this work, to understand further this example of indirect readout, we have determined crystal structures of CAP-DNA complexes containing each possible nucleotide at position 6. The structures show that CAP can introduce a DNA kink at the primary-kink site with any nucleotide at position 6. The DNA kink is sharp with the consensus pyrimidine-purine step T6G7 and the nonconsensus pyrimidine-purine step C6G7 (roll angles of ∼42°, twist angles of ∼16°), but is much less sharp with the nonconsensus purine-purine steps A6G7 and G6G7 (roll angles of ∼20°, twist angles of ∼17°). We infer that CAP discriminates between consensus and non-consensus pyrimidine-purine steps at positions 6-7 solely based on differences in the energetics of DNA deformation, but that CAP discriminates between the consensus pyrimidine-purine step and non-consensus purine-purine steps at positions 6-7 both based on differences in the energetics of DNA deformation and based on qualitative differences in DNA deformation. The structures further show that CAP can achieve a similar, ∼46° per DNA half-site, overall DNA bend through a sharp DNA kink, a less sharp DNA kink, or a smooth DNA bend. Analysis of these and other crystal structures of CAP-DNA complexes indicates that there is a large, ∼28° per DNA half-site, out-of plane, component of CAP-induced DNA bending in structures not constrained by end-to-end DNA lattice interactions and that lattice contacts involving CAP tend to involve residues in or near biologically functional surfaces.
Keywords: catabolite activator protein (CAP), cAMP receptor protein (CRP), protein-DNA interaction, protein-induced DNA bending, indirect readout
INTRODUCTION
Sequence-specific DNA-binding proteins primarily recognize DNA sites through “direct readout,” involving hydrogen bonds and van der Waals interactions with DNA base pairs.1-3 However, sequence-specific DNA-binding proteins also can recognize DNA sites through “indirect readout,” without direct contact with DNA base pairs.2-9 Indirect readout is less well characterized than direct readout. Mechanisms for indirect readout include, in principle, water-mediated hydrogen bonds, sequence effects on DNA conformation, and sequence effects on susceptibility to DNA deformation.
One clear example of indirect readout involves the Escherichia coli catabolite activator protein (CAP; also known as the cAMP receptor protein, CRP), which recognizes the consensus DNA half-site 5′-A1A2A3T4G5T6G7A8T9C10T11-3′.10-12 CAP makes no direct contacts to the T:A base pair at position 6 of the consensus DNA half-site13-15; nevertheless, CAP displays strong specificity for T:A at position 6, preferring T:A over A:T, C:G, and G:C by 1.5, 1.4, and 1.4 kcal/mol per half-site, respectively.12 In the CAP-DNA complex, CAP sharply bends DNA, typically introducing a DNA kink, with a roll angle of ∼40° and a twist angle of ∼20°, between positions 6 and 7 (“primary kink”).13-18 In the consensus half-site, positions 6 and 7 are part of a T:A/G:C base-pair step, a base-pair step that is associated with high roll angles19-23 and that has high susceptibility to roll deformation.24-27 It has been proposed that the specificity for T:A at position 6 is a consequence of the primary kink between positions 6 and 7 and of the effects of the T:A/G:C step on DNA deformation and/or DNA deformability.14 Two specific mechanisms for discrimination against nonconsensus base pairs at position 6 have been considered: (i) altered DNA deformation, wherein non-consensus base pairs at position 6 partly or fully preclude formation of the primary kink and thus partly or fully preclude formation of CAP-DNA interactions distal to the primary kink (e.g., interactions with DNA phosphates at positions −3 through 5) and (ii) altered energetic susceptibility to DNA deformation, wherein both consensus and nonconsensus base pairs at position 6 permit formation of the primary kink, but nonconsensus base pairs impose a higher energetic cost for formation of the primary kink.
In previous work, in a first effort to understand this example of indirect readout, we determined crystal structures of complexes of CAP with a DNA site containing the nonconsensus base pair C:G at position 6.28 We observed, in each of two crystal forms, that complexes containing the consensus base pair T:A or the nonconsensus base pair C:G exhibited essentially identical overall DNA bend angles and local geometries of DNA kinking. We inferred that discrimination between the consensus base pair T:A or the nonconsensus base pair C:G does not involve differences in the deformation of DNA, but, rather, solely differences in the energetics of DNA kinking.
In this work, to understand further this example of indirect readout, we have extended the analysis to complexes of CAP with DNA sites containing all possible base pairs at position 6. The results show that DNA kinking can occur with any base at position 6, but reveal qualitative differences in DNA kinking with pyrimidine-purine steps at positions 6-7 vs. with purine-purine steps at positions 6-7 and thus suggest differences in mechanisms of discrimination against nonconsensus pyrimidine-purine steps vs. discrimination against nonconsensus purine-purine steps.
RESULTS
Structure determination
CAP-DNA complexes were prepared using 38 bp DNA fragments containing the consensus DNA site for CAP and derivatives of the consensus DNA site with T:A → A:T, T:A → G:C, or T:A → C:G substitutions at position 6 of each DNA half-site ([6A;17T]DNA, [6G;17C]DNA, and [6C;17G]DNA) (Figure 1). The complexes yielded crystals belonging to space group P212121, in new crystal forms for CAP-DNA (crystal forms C and C′; Table 1). Diffraction data were collected using synchrotron radiation to 2.8 Å for the form-C structures and to 2.1 Å for the CAP-[6C;17G]DNA form-C′ structure (Table 2).
Figure 1.

DNA fragments used in crystallization of CAP-DNA complexes. (a) DNA fragments used in this work. (b) DNA fragments used in prior work by us and others.13-15,28,30,36 Filled ovals, two-fold symmetry axes; vertical bars, two-fold-symmetry-related single-phosphate gaps; boldface, two-fold-symmetry-related positions 6 and 17.
Table 1.
Summary of all CAP-DNA crystal structures
| Crystal Form | Space group | Average Unit Cell (Å) | Base-pairs | Protein | DNAa | Reference |
|---|---|---|---|---|---|---|
| A | C2221 | 30 | CAP | GC31 | [13] | |
| a=137 | CAP | GCE31 | [14] | |||
| b=153 | CAP | ICAP30 | [15] | |||
| c=76 | [E181F]CAP | ICAP30 | [15] | |||
| CAP | [6C;17G]ICAP30 | [28] | ||||
| B | P3121 | a=78 | 38-46 (26 modeled) | CAP | DNA46 | [30] |
| CAP | ICAP38 | [28] | ||||
| b=78 | CAP | [6C;17G]ICAP38 | [28] | |||
| c=142 | [E181D]CAP | [6C;17G]ICAP38 | [36] | |||
| C | P212121 | a=61 | 38 | CAP | ICAP38 | this work |
| b=76 | CAP | [6A;17T]ICAP38 | this work | |||
| c=180 | CAP | [6G;17C]ICAP38 | this work | |||
| C′ | P212121 | a=61 | 38 (35 modeled) | |||
| b=78 | CAP | [6C;17G]ICAP38 | this work | |||
| c=155 | ||||||
DNA sequences are defined in Figure 1
Table 2.
Crystallographic data and refinement statistics
| CAP-ICAP38 | CAP-[6A;17T]ICAP38 | CAP-[6G;17C]ICAP38 | CAP-[6C;17G]ICAP38 | |
|---|---|---|---|---|
| A. Data collection parameters | ||||
| Space Group | P212121 | P212121 | P212121 | P212121 |
| Unit Cell Parameters | ||||
| a (Å) | 61.55 | 61.29 | 61.35 | 60.79 |
| b (Å) | 75.55 | 75.35 | 75.57 | 77.74 |
| c (Å) | 180.90 | 178.08 | 179.98 | 154.81 |
| Resolution (Å) | 2.8 | 2.8 | 2.8 | 2.1 |
| Observed reflections | 680,778 | 446,431 | 363,974 | 1,344,496 |
| Unique reflections | 21,651 | 21,638 | 21,876 | 43,254 |
| Completeness (%) | 99.6 | 82.9 | 98.6 | 97.3 |
| Rmergea (%) | 9.1 | 9.1 | 9.0 | 10.9 |
| I/σ(I) | 23.4 | 13.4 | 17.0 | 28.7 |
| B. Refinement | ||||
| Atoms included in refinement | 4746 | 4746 | 4746 | 5055 |
| Solvent molecules | 42 | 31 | 62 | 266 |
| Reflections (working + test) | 20395 | 16375 | 21159 | 42032 |
| Test set size (%) | 10 | 10 | 10 | 10 |
| Deviations from ideal geometry | ||||
| Bond lengths (Å) | 0.0078 | 0.0071 | 0.0077 | 0.0060 |
| Bond angles (Å) | 1.33 | 1.31 | 1.28 | 1.14 |
| Torsion angles (°) | 21.8 | 22.0 | 22.0 | 21.5 |
| Improper torsion angles (°) | 1.04 | 0.97 | 0.99 | 0.89 |
| Average B-factor (Å2) | 75.2 | 74.3 | 73.8 | 45.9 |
| R-factorb (%) | 24.2 | 23.1 | 24.0 | 22.5 |
| R-freec (%) | 29.5 | 30.2 | 28.9 | 26.6 |
Rmerge = ∑h∑k |< Ik − Iki >| / ∑h∑iIk × 100
R-factor = ∑∥Fo| − |Fc∥ / ∑|Fo|, where Fo and Fc are the observed and calculated structure factor amplitudes, respectively.
R-free = ∑T∥Fo| − |Fc∥ / ∑T|Fo|, where T is the test set; random 10% of observations excluded from refinement.
Each structure contains two non-equivalent half-complexes, consisting of one CAP protomer and one DNA half-site, related by a non-crystallographic pseudo two-fold axis (denoted half-complex A and half-complex B) (Figure 2). The CAP-DNA, CAP-[6A;17T]DNA, and CAP-[6G;17C]DNA form-C structures contains one bound cAMP molecule per CAP subunit. The CAP-[6C;17G]DNA form-C′ structure contains two bound cAMP molecules per CAP subunit, with the second binding site 50% occupied. Five half-complexes display a sharp kink between positions 6 and 7: half-complex A of CAP-DNA, half-complex A of CAP-[6A;17T]DNA, half-complex A of CAP-[6G;17C]DNA, and half-complexes A and B of CAP-[6C;17G]DNA. Three half-complexes display a smooth bend between positions 6 and 7: half-complex B of CAP-DNA, half-complex B of CAP-[6A;17T]DNA, and half-complex B of CAP-[6G;17C]DNA.
Figure 2.
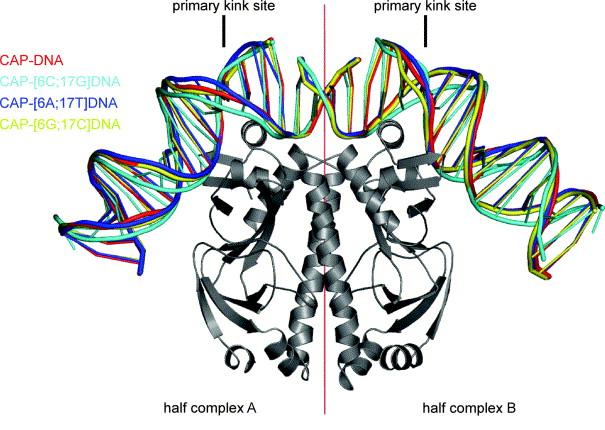
Superimposed structures of complexes of CAP with DNA sites containing all possible base pairs at position 6 of the primary-kink site obtained in this study. CAP is presented in a ribbon representation. DNA is colored as follows: cyan, CAP-DNA; blue, CAP-[6A;17T]DNA; yellow, CAP-[6G;17C]DNA; red, CAP-[6C;17G]DNA. The figure was generated using PyMol (http://pymol.sourceforge.net/) and nuccyl (http://www.mssm.edu/students/jovinl02/research/nuccyl.html).
Kinked half-complexes
Half-complexes A of CAP-DNA, CAP-[6A;17T]DNA, and CAP-[6G;17C]DNA, and half-complexes A and B of CAP-[6C;17G]DNA each exhibits a kink at the primary-kink site (Figures 3 and 4; Table 3). The kink is sharp--and nearly identical in geometry--with the consensus pyrimidine-purine step T6-G7 and the non-consensus pyrimidine-purine step C6-G7 (roll angles of ∼41° and ∼41°, twist angles of ∼16° and ∼18°). In contrast, the kink is less sharp for the purinepurine steps A6-G7 and G6-G7 (roll angles of ∼19° and ∼21°, twist angles of ∼22° and ∼12°). The kinked half-complexes exhibit overall DNA bend angles of ∼50° (Table 3). Despite the difference in local DNA geometry at positions 6-7, the overall DNA bend angles are similar with pyrimidine-purine steps at positions 6-7 and with purine-purine steps at position 6-7 (∼53° vs. ∼46°).
Figure 3.
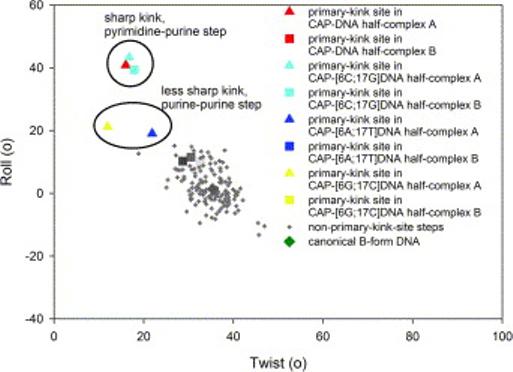
DNA helical parameters of complexes of CAP with DNA sites containing all possible base pairs at position 6 of the primary-kink site obtained in this study. The figure presents a plot of DNA roll angle versus DNA twist angle for each dinucleotide step of each half-complex herein (triangles, steps in half-complexes A; squares, steps in half-complexes B). DNA sequences are as in Figure 1. Colors are as in Figure 2.
Figure 4.
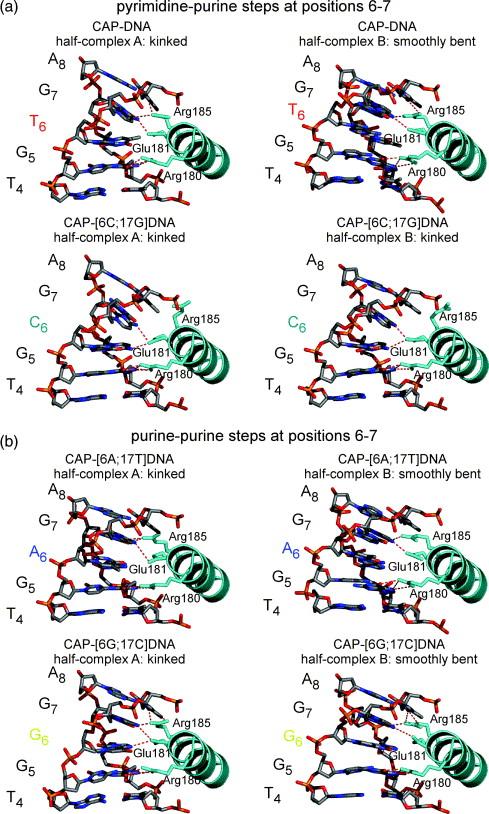
CAP-DNA interactions at the primary-kink site in complexes of CAP with DNA sites containing all possible base pairs at position 6 of the primary-kink site. Panels illustrate the second α-helix of the helix-turn-helix motif of CAP (“recognition helix”) and positions 4 to 8 of the DNA half-site. For reference, the primary kink is located between positions 6 and 7 of the DNA half-site. Hydrogen bonds between CAP side chains and DNA base pairs are indicated by red dashed lines (4.0 Å cutoff for assignment of hydrogen bonds). (a) Complexes with DNA sites containing pyrimidine-purine steps at positions 6-7. (b) Complexes with DNA sites containing purine-purine steps at positions 6-7.
Table 3.
Kinked CAP-DNA half-complexes
| Pyrimidine-purine step | |||||
|---|---|---|---|---|---|
| Complex | Half-complex | Base pair step | Roll | Twist | Overall bend |
| CAP-DNA | A | T6G7 | 41° | 16° | 48° |
| CAP-[6C;17G] DNA | A | C6G7 | 43° | 17° | 52° |
| CAP-[6C;17G] DNA | B | C6G7 | 39° | 18° | 58° |
| Purine-purine step | |||||
|---|---|---|---|---|---|
| Complex | Half-complex | Base pair step | Roll | Twist | Overall bend |
| CAP-[6A;17T] DNA | A | A6G7 | 19° | 22° | 46° |
| CAP-[6G;17C] DNA | A | G6G7 | 21° | 12° | 46° |
The protein in each kinked half-complex is structurally similar to that in each of other kinked half-complexes (RMSD ∼0.7 Å for Cα atoms) and to that in previous CAP-DNA structures (RMSD ∼1.0 Å for Cα atoms) (data not shown). The pattern of hydrogen bonding between the amino acids of the recognition helix of CAP and the DNA half-site (positions 4 to 8) in the kinked half-complexes appears to be independent of the class of step at positions 6-7 (Table 4; Figure 4). In half-complexes A of CAP-DNA (pyrimidine-purine step) and CAP[6A;17T]DNA (purine-purine step), Arg180, Glu181, and Arg185 of CAP make direct hydrogen-bonded interactions with G:C at position 5, G:C at position 7, and A:T at position 8 of the DNA half-site. In half-complexes A of CAP-[6G;17C]DNA and CAP-[6C;17G]DNA, an additional hydrogen bond is observed between Glu181 and the cytosine N4 atom at position 6 (see Chen et al.28). Protein-phosphate interactions are similar to previously reported CAP-DNA complexes14, with no observed sequence related differences.
Table 4.
Key amino-acid to base interactions distances (Å) at the primary kink sites of all CAP-DNA structures.
| Complex: half-complex | Crystal Form | Arg 180 | Glu 181 | Arg 185 | PDB ID Reference | ||
|---|---|---|---|---|---|---|---|
| 5G | 6C/6C′ | 7C′ | 7G | 8T′ | |||
| CAP-GC31: A | A | 2.9, 3.0, 2.7 | 3.3 | 3.7, 3.2 | 2.8 | 1CGP13 | |
| CAP-GC31: B | A | 3.3, 4.0, 3.8, 2.7 | 2.9 | 3.4 | 2.6 | 1CGP13 | |
| CAP-GCE31: A | A | 3.4, 2.5 | 2.7 | 2.8, 3.5 | 3.8, 3.7 | 1J5914 | |
| CAP-GCE31: B | A | 2.6, 3.4 | 3.5 | 3.9 | 3.5, 3.5 | 1J5914 | |
| CAP-ICAP30: A | A | 2.7, 3.1, 3.1 | 2.7 | 3.4, 2.9 | 2.9 | 1RUN15 | |
| CAP-ICAP30: B | A | 3.0, 2.5, 3.2 | 3.5, 3.0 | (4.7) | 3.4 | 1RUN15 | |
| [E181F]CAP-ICAP30: A | A | 3.6, 2.6, 2.8, 3.7 | 3.1* | 3.9, 3.3 | 3.6 | 1RUO15 | |
| [E181F]CAP-ICAP30: B | A | 2.6 | 3.2* | 3.2 | 3.7 | 1RUO15 | |
| CAP-[6C;17G]ICAP30: A | A | (7.3) | 4.0, 3.6 | 3.7 | 4.0, 3.4 | 1O3T28 | |
| CAP-[6C;17G]ICAP30: B | A | 3.6, 2.6, 2.4 | 2.8 (C) | 3.0, 3.4 | 3.5 | (5.1) | 1O3T28 |
| CAP-DNA46 | B | 3.5, 2.9 | 3.2 | 2.8 | 3.0, 2.6 | 2CGP30 | |
| CAP-ICAP38 | B | 3.0, 3.2, 2.9 | 3.3 | 3.2 | 2.7 | 1O3Q28 | |
| CAP-[6C;17G]ICAP38 | B | 2.8, 3.5, 2.8, 3.9 | 3.3 (C) | 2.8 | 2.9, 3.1 | 3.2 | 1O3R28 |
| [E181D]CAP-[6C;17G]ICAP38 | B | 3.8, 3.3 | 3.0+(C) | 3.5+ | 2.7, 3.7 | 3.0 | 1O3S36 |
| CAP-ICAP38: A | C | 3.6 3.2, 2.9 | 2.8 | 3.1 | 3.1 | 1ZRC | |
| CAP-ICAP38: B | C | 3.8, 2.9, 2.5 | 3.2 | 2.7, 3.5 | 3.5 | 1ZRC | |
| CAP-[6A;17T] ICAP38: A | C | 3.3, 2.6, 3.7 | 3.5 | 3.1, 3.3 | 3.5 | 1ZRD | |
| CAP-[6A;17T] ICAP38: B | C | 3.6, 2.8, 3.4 | 2.8 | 3.7, 3.7 | (4.2) | 1ZRD | |
| CAP-[6G;17C] ICAP38: A | C | 3.8, 3.0, 2.9 | 3.2 (C′) | 3.6 | 2.7 | 2.7, 3.9 | 1ZRE |
| CAP-[6G;17C]ICAP38: B | C | 3.4, 2.4, 2.8 | (4.6)(C′) | 3.9 | 2.6 | 3.6, 3.4 | 1ZRE |
| CAP-[6C;17G]ICAP38: A | C' | 3.9, 3.1, 2.8, 3.9 | 3.0 (C) | 3.2 | (6.5) | (5.6) | 1ZRF |
| CAP-[6C17;G] ICAP38: B | C' | 3.3, 2.8, 4.0 | 3.3 (C) | 2.8 | (8.8) | (7.6) | 1ZRF |
Key interaction distances at each primary kink site, with a 4.0 Å distance cutoff. Distances in parentheses are greater then 4.0 Å, but represent the nearest distance for that interaction. Unless noted, CAP is wild-type sequence and the DNA sequence at the primary kink site is 5′-T4G5T6G7A8-3′ / 5′-T′4C′5A′6C′7A′8-3′.
Aromatic hydrogen bond between Phe181 and Cyt 7′ 15.
Hydrogen bonding between Asp181 and Cyt 7′/Cyt 6 36.
Smoothly bent half-complexes
Half-complexes B of CAP-DNA, CAP-[6A;17T]DNA, and CAP-[6G;17C]DNA each exhibits a smooth bend at the primary-kink site (Figures 3 and 4; Table 5). The smoothly bent half-complexes have roll angles of ∼11° and twist angles of ∼31° at positions 6-7. These values are typical for unkinked B-DNA (Figure 3). There is no significant difference in roll angles or twist angles for smoothly bent half-complexes with a pyrimidine-purine step (T6-G7) vs. with purine-purine steps (A6-G7 or G6-G7). The smoothly bent half-complexes exhibit overall DNA bend angles of ∼42° (Table 5). The overall DNA bend angles are similar with pyrimidine-purine steps at positions 6-7 and with purine-purine steps at positions 6-7 (38° vs. ∼44°).
Table 5.
Smoothly bent CAP-DNA half-complexes
| Pyrimidine-purine step | |||||
|---|---|---|---|---|---|
| Complex | Half-complex | Base pair step | Roll | Twist | Overall bend |
| CAP-DNA | B | T6G7 | 12° | 31° | 38° |
| Purine-purine step | |||||
|---|---|---|---|---|---|
| Complex | Half-complex | Base pair step | Roll | Twist | Overall bend |
| CAP-[6A;17T] DNA | B | A6G7 | 10° | 29° | 43° |
| CAP-[6G;17C] DNA | B | G6G7 | 10° | 33° | 44° |
The protein in each smoothly bent half-complex is structurally similar to that in each other smoothly bent half-complex (RMSD ∼0.4 Å for Cα atoms) and to that in each kinked half-complex (RMSD ∼1.0 Å for Cα atoms) (data not shown). The pattern of hydrogen bonding between the amino acids of the recognition helix of CAP and the DNA half-site (positions 4 to 8) in the smoothly bent half-complexes appears to be independent of the class of step at positions 6-7 (Table 4; Figure 4). In all smoothly bent half-complexes, Arg180 and Glu181 of CAP make hydrogen bonds with G:C at position 5 and G:C at position 7. In smoothly bent half-complexes B of CAP-DNA (pyrimidine-purine step) and CAP-[6G;17C]DNA (purine-purine step), Arg185 of CAP makes an additional hydrogen bond A:T at position 8. Protein-phosphate interactions are similar to previously reported CAP-DNA complexes14, with no observed sequence related differences.
DISCUSSION
Indirect readout at position 6
Our structures demonstrate that CAP can introduce a DNA kink at the primary-kink site with any nucleotide at position 6. However, our structures also reveal qualitative differences in DNA kinking with pyrimidine-purine steps vs. purine-purine steps at positions 6-7, suggesting differences in the mechanisms of discrimination against nonconsensus pyrimidine-purine steps vs. discrimination against nonconsensus purine-purine steps. These structural differences are considered significant because all four structures were obtained from a common starting model, using identical refinement procedures.
We observe, in agreement with Chen et al.28, that CAP can introduce a sharp DNA kink--with the same extent of kinking, and the same geometry of kinking--in complexes with the consensus pyrimidine-purine step T6-G7 and the non-consensus pyrimidine-purine step C6-G7. We infer, in agreement with Chen et al.28, that indirect-readout-mediated discrimination against the non-consensus base C at position 6 does not involve differences in the geometry of kinking, but, rather, solely differences in the energetics of kinking.
In contrast, we observe that CAP can introduce only a much less sharp DNA kink--with less than one-half the roll angle (∼20° vs. ∼42°)--in complexes with the non-consensus purine-purine steps A6-G7 and G6-G7. We infer that indirect-readout-mediated discrimination against the non-consensus bases A and G at position 6 involves qualitative differences in the geometry of kinking as well as differences in the energetics of kinking.
CAP-induced DNA bending: kinking
The CAP-DNA and CAP-[6C;17G]DNA structures display sharp kinks at the primary-kink site, with roll angles of ∼42° and twist angles of ∼16°. These structures contain pyrimidine-purine steps (consensus T6-G7 and non-consensus C6-G7, respectively), which are known to be highly susceptible to deformation,19-27 at the primary-kink site. The CAP-[6A;17T]DNA and CAP-[6G;17C]DNA structures provide the first opportunity to observe purine-purine steps at the primary kink site. Purine-purine steps are known to be less highly deformed than pyrimidine-purine steps in protein-DNA structures19 and to exhibit significantly lower dynamic flexibility than pyrimidine-purine steps.29 Our observations are consistent with these patterns (Figure 3; Table 5). The kinked half-complexes with purine-purine steps at positions 6-7, A6-G7 and G6-G7, display roll angles that are only approximately one-half those for the kinked half complexes with pyrimidine-purine steps (∼20° vs. ∼41°).
CAP-induced DNA bending: smooth bending
The new CAP-DNA crystal form-C permits visualization of wild-type CAP bending the consensus DNA site with a kink at the primary kink site in one half-complex and with a smooth bend at the primary-kink site in the other, pseudo-two-fold-related, half-complex (Figures 2 and 3; Tables 3 and 5). The results establish that wild-type CAP can bend the consensus DNA site to the essentially the same overall DNA bend angle (∼47° per half-complex) through a kink or through a smooth bend (Tables 3 and 5; the observed differences in bend angle are not considered significant due to the resolution of each structure and crystal packing variations). In addition, there is no correlation between DNA-end lattice contacts and kinked or smooth bending (The wild-type CAP-DNA structure in ref. 30 [CAP-DNA46 (B) in Table 1] has no DNA-end lattice contacts, and also displays a smooth bend.). In our structures, as in the structure in ref. 30, smooth bending is accomplished by an ∼10° increase in roll angle at positions 6-7 and modest, ∼4°, increases in roll angle at the neighboring T4-G5, G5-N6, G7-A8, and A8-T9 steps. We infer that the kinked and smoothly bent DNA observed in CAP-DNA complexes likely correspond to two states of a dynamic complex.
CAP-induced DNA bending: out-of-plane component
The CAP-DNA structures herein, like CAP-DNA structures in the literature,13-15,28,30 exhibit an out-of-plane component of the DNA bend, such that the path of the DNA forms a non-zero dihedral angle. The dihedral angles range from −24° to −62°, with CAP-DNA (C) having the smallest angle and CAP-[6C;17G]DNA (C′) having the largest angle. These dihedral angles fall within the range observed in previous wild-type CAP-DNA structures (−24° to −62°).
A plot of dihedral angle versus overall bend angle for all wild-type CAP-DNA structures herein and in the literature reveals two distinct groups (Figure 5a). One group consists of crystal forms A and C and exhibits a relatively modest mean dihedral angle: −30° (±5°). The second group consists of forms B and C′ and exhibits a large mean dihedral angle: −56° (±8). The basis for the difference between the two groups appears to be a difference in DNA-DNA crystal lattice contacts. In crystal structures in the first group (forms A and C), which have a modest mean dihedral angle of −30°, DNA fragments in the crystal lattice pack end-to-end to form a pseudo-continuous DNA helix and thus are constrained by end-to-end DNA lattice interactions. In contrast, in crystal structures in the second group (forms B and C′), which have a large mean dihedral angle of −56°, DNA fragments in the crystal lattice do not pack end-to-end to form a pseudo-continuous DNA helix and thus are not to be constrained by end-to-end DNA lattice interactions. The crystal structures in the second group, which lack constraining end-to-end DNA crystal-lattice interactions, are likely to more accurately reflect DNA conformation in solution. Therefore, we suggest that, in solution, there is a large out-of-plane component to CAP-induced DNA bending, with a dihedral angle on the order of −56°.
Figure 5.
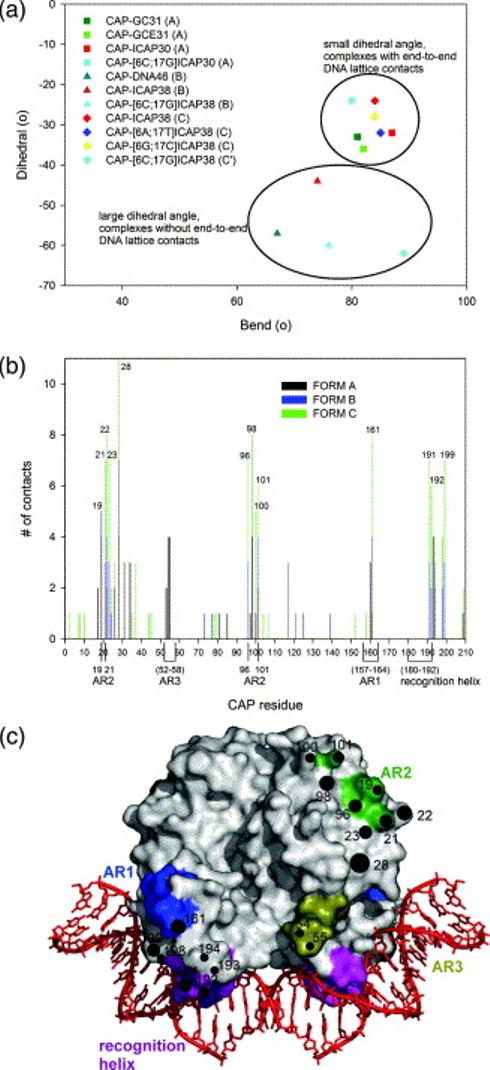
Analysis of crystal structures of wild-type CAP-DNA complexes herein and in the literature. (a) Plot of DNA dihedral angle versus DNA bend angle in crystal structures of wild-type CAP-DNA complexes (squares, crystal form A; triangles, crystal form B; diamonds, crystal form C; circles, crystal form C′). (b) Plot of CAP residue number versus frequency of involvement in crystal-lattice contacts in crystal structures of wild-type CAP-DNA complexes. (c) Surface representation of CAP showing residues involved in crystal-lattice contacts in at least 35% of crystal structures of wild-type CAP-DNA complexes (circles, with higher diameters indicating higher frequencies of involvement in crystal-lattice contacts), DNA recognition helices (magenta), and transcription activating regions 1 (AR1, blue), 2 (AR2, dark green), and 3 (AR3, olive green).
Lattice interactions
All four P212121 CAP-DNA complexes have similar protein-protein crystal packing contacts (Figure 5b). An analysis of crystal lattice contacts in all wild-type CAP-DNA herein and in the literature shows that similar residues are involved in lattice contacts in all crystal forms (Figure 5b,c). Interestingly, most conserved CAP-DNA crystal lattice contacts involve residues within or near surfaces of CAP that make protein-protein interactions with RNA polymerase during transcription activation (activating regions 1, 2, and 3; AR1, AR2, and AR3; reviewed in Lawson et al.31 and Busby & Ebright32). The physicochemical properties of the surface patches that mediate critical transient protein-protein interactions in this system--namely, high surface accessibility and high hydrophilicity--apparently make them favored determinants for crystal lattice contact. It is conceivable that the physicochemical properties of surface patches that mediate critical transient protein-protein interactions in other systems also may make them favored determinants for crystal lattice contacts--and thus that clues to identification of such surfaces may be obtained from analysis of crystal lattice contacts.
MATERIALS AND METHODS
CAP
CAP was purified using cAMP-affinity chromatography.33 Immediately before crystallization, the protein was further purified by gel-filtration chromatography on a 2.5 ml Bio-Gel P-6DG column (Bio-Rad, Inc.) in 10 mM Tris-HCl (pH 7.5), 200 mM NaCl, 0.1 mM EDTA, 2 mM dithiothreitol, 0.02% (w/v) NaN3, and concentrated to 0.1-0.2 mM by centrifugal filtration.14
DNA fragments
To produce each DNA duplex used in this work, two half-site oligodeoxyribonucleotides duplexes were annealed via overhanging 5′ ends (see Shultz et al.13 and Parkinson et al.14,15). The use of two half-site oligodeoxyribonucleotide duplexes minimizes hairpin formation and has been used in numerous CAP-DNA crystal structures.13-15,28,34-36 The following oligodeoxyribonucleotides were purchased from Integrated DNA Technologies, Inc. and purified by reversed-phase HPLC as described:14,15 for DNA fragment ICAP38, 5′-ATTTCGAAAAATGTGAT-3′ (17mer) and 5′-CTAGATCACATTTTTCGAAAT-3′ (21mer); for DNA fragment [6A;17T]ICAP38, 5′-ATTTCGAAAAATGAGAT-3′ (17mer) and 5′-CTAGATCTCATTTTTCGAAAT-3′ (21mer); for DNA fragment [6G;17C]ICAP38, 5′-ATTTCGAAAAATGGGAT-3′ (17mer) and 5′-CTAGATCCCATTTTTCGAAAT-3′ (21mer); for DNA fragment [6C;17G]ICAP38, 5′-ATTTCGAAAAATGCGAT-3′ (17mer) and 5′-CTAGATCGCATTTTTCGAAAT-3′ (21mer).
Crystallization
Crystals of CAP-DNA, CAP-[6A;17T]DNA, CAP-[6G;17C]DNA complexes were grown by vapor diffusion at 22 °C as 2 μl hanging drops in a solution containing 0.13 mM CAP-DNA complex, 2mM cAMP, 0.1 M MES pH 6.2, 0.2 M NaCl, 0.05 M Mg2Cl, 5% PEG 8000, and 14-15% Dioxane. Initial crystals of CAP-[6C;17G]DNA were obtained by microseeding with CAP-DNA or CAP-[6A;17T]DNA crystals. Subsequent crystals were seeded from CAP-[6C;17G]DNA crystals. The variation in the CAP-[6C;17G]DNA crystals (crystal form C′ vs. C) does not appear to be a consequence of the DNA sequence. Instead, other variables such as a new protein preparation, and new crystallization solutions likely contributed to the change.
Data collection and analysis
The CAP-[6A;17T]DNA, CAP-[6G;17C]DNA, and CAP-[6C;17G]DNA crystals were soaked in increasing concentrations of PEG 3350 and Glycerol (final concentration: 10% PEG 3350, 30% Glycerol) over a period of one hour before cooling them in liquid nitrogen. The CAP-DNA crystal was flash-cooled directly in the cryogenic stream subsequent to an identical cryoprotection procedure. Data were collected on the X25 beamline at the Brookhaven National Synchrotron Light Source with an ADSC Q315 CCD detector (λ=1.100 Å, temperature=108K). The data were indexed and scaled using the HKL2000 software package.37 The data collection and processing statistics for each crystal are summarized in Table 2.
The CAP-DNA structure was solved by molecular replacement using the AMORE program.38 Using the CAP-DNA structure28 in crystal form B (space group P3121) as a complete biological unit (CAP dimer, 26 base-pairs of DNA), a molecular replacement solution was found with a starting R value of 52.3%. Initial models for the CAP-[6A;17T]DNA and CAP-[6G;17C]DNA complexes were derived from the refined CAP-DNA structure in crystal form C (P212121). The CAP-[6C;17G]DNA (C′) structure was solved by molecular replacement using the AMORE program38 and confirmed with Phaser.39 The search model was the refined CAP-DNA structure in crystal form C (P212121), truncated to contain only 20 base-pairs of DNA. The starting R value was 47%.
Models were refined with CNS40 using overall anisotropic B-factor correction and bulk solvent correction including nucleic acid parameters41. Simulated annealing with torsion angle dynamics42 was employed. Composite omit maps displayed positive electron density for the DNA beyond the limits of the starting model. Manual model building was performed using XtalView43. Following energy minimization, waters were gradually added initially using the automated water picking of CNS, followed by manual inspection and addition of waters. Individual isotropic temperature factors were refined. The CAP-[6C;17G]DNA crystal displayed positive electron density for 5 ordered dioxane molecules (from the crystallization solution) and a partially occupied second cAMP binding site (50%, as determined by occupancy refinement). Analysis of stereochemistry by PROCHECK44 revealed that 87.4%, 89.9%, 89.9%, and 95.8% of the main chain torsion angles of CAP-DNA, CAP-[6A;17T]DNA, CAP-[6G;17C]DNA, and CAP-[6C;17G]DNA, respectively, fall within the most favored regions of the Ramachandran Plot. The remaining residues had torsion angles in the allowed regions of the Ramachandran Plot.
DNA helical parameters
DNA helical parameters were analyzed using 3DNA.45 Overall DNA bend angles were calculated by measuring the angles subtended by canonical B-form DNA helices superimposed on the same 3 bp segments on each complex (superimposition on base-pairs at positions –2 → 1 and 22 → 24). Dihedral angles were determined by calculating the angle between two planes, each consisting of one point at the center of the helical axis of each terminal base-pair and two points from the helical axis of the central 8 base-pairs.
Protein Data Bank accession codes
Atomic coordinates and structure factors have been deposited in the Protein Data Bank46 with accession codes 1ZRC (CAP-DNA), 1ZRD (CAP-[6A;17T]DNA), 1ZRE (CAP-[6G;17C]DNA), and 1ZRF (CAP-[6C;17G]DNA).
Acknowledgements
We thank Shengfeng Chen for his advice on CAP-DNA crystallization, Wilma K. Olson for helpful discussions about DNA deformation, and NSLS X25 beamline personnel, especially Michael Becker, for assistance during data collection. This work was supported by National Institutes of Health grants GM21589 to H.M.B and C.L.L. and GM41376 to R.H.E., a National Institutes of Health traineeship GM08319 to A.A.N., and a Howard Hughes Medical Institute investigatorship to R.H.E.
Abbreviations
- CAP
catabolite activator protein
- CRP
cAMP receptor protein
- RMSD
root mean square deviation
References
- 1.von Hippel PH, Berg OG. On the specificity of DNA-protein interactions. Proc Natl Acad Sci U S A. 1986;83:1608–12. doi: 10.1073/pnas.83.6.1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brennan RG, Matthews BW. Structural basis of DNA-protein recognition. Trends Biochem Sci. 1989;14:286–90. doi: 10.1016/0968-0004(89)90066-2. [DOI] [PubMed] [Google Scholar]
- 3.Pabo CO, Sauer RT. Transcription factors: structural families and principles of DNA recognition. Annu Rev Biochem. 1992;61:1053–95. doi: 10.1146/annurev.bi.61.070192.005201. [DOI] [PubMed] [Google Scholar]
- 4.Otwinowski Z, Schevitz RW, Zhang RG, Lawson CL, Joachimiak A, Marmorstein RQ, Luisi BF, Sigler PB. Crystal structure of trp repressor/operator complex at atomic resolution. Nature. 1988;335:321–9. doi: 10.1038/335321a0. [DOI] [PubMed] [Google Scholar]
- 5.Gartenberg MR, Crothers DM. DNA sequence determinants of CAP-induced bending and protein binding affinity. Nature. 1988;333:824–9. doi: 10.1038/333824a0. [DOI] [PubMed] [Google Scholar]
- 6.Lesser DR, Kurpiewski MR, Jen-Jacobson L. The energetic basis of specificity in the Eco RI endonuclease--DNA interaction. Science. 1990;250:776–86. doi: 10.1126/science.2237428. [DOI] [PubMed] [Google Scholar]
- 7.Koudelka GB, Carlson P. DNA twisting and the effects of non-contacted bases on affinity of 434 operator for 434 repressor. Nature. 1992;355:89–91. doi: 10.1038/355089a0. [DOI] [PubMed] [Google Scholar]
- 8.Koudelka GB, Harbury P, Harrison SC, Ptashne M. DNA twisting and the affinity of bacteriophage 434 operator for bacteriophage 434 repressor. Proc Natl Acad Sci U S A. 1988;85:4633–7. doi: 10.1073/pnas.85.13.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koudelka GB, Harrison SC, Ptashne M. Effect of non-contacted bases on the affinity of 434 operator for 434 repressor and Cro. Nature. 1987;326:886–8. doi: 10.1038/326886a0. [DOI] [PubMed] [Google Scholar]
- 10.Berg OG, von Hippel PH. Selection of DNA binding sites by regulatory proteins , : II. The binding specificity of cyclic AMP receptor protein to recognition sites. Journal of Molecular Biology. 1988;200:709–723. doi: 10.1016/0022-2836(88)90482-2. [DOI] [PubMed] [Google Scholar]
- 11.Ebright RH, Ebright YW, Gunasekera A. Consensus DNA site for the Escherichia coli catabolite gene activator protein (CAP): CAP exhibits a 450-fold higher affinity for the consensus DNA site than for the E. coli lac DNA site. Nucleic Acids Res. 1989;17:10295–305. doi: 10.1093/nar/17.24.10295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gunasekera A, Ebright YW, Ebright RH. DNA sequence determinants for binding of the Escherichia coli catabolite gene activator protein. J Biol Chem. 1992;267:14713–20. [PubMed] [Google Scholar]
- 13.Schultz SC, Shields GC, Steitz TA. Crystal structure of a CAP-DNA complex: the DNA is bent by 90 degrees. Science. 1991;253:1001–7. doi: 10.1126/science.1653449. [DOI] [PubMed] [Google Scholar]
- 14.Parkinson G, Wilson C, Gunasekera A, Ebright YW, Ebright RE, Berman HM. Structure of the CAP-DNA complex at 2.5 angstroms resolution: a complete picture of the protein-DNA interface. J Mol Biol. 1996;260:395–408. doi: 10.1006/jmbi.1996.0409. [DOI] [PubMed] [Google Scholar]
- 15.Parkinson G, Gunasekera A, Vojtechovsky J, Zhang X, Kunkel TA, Berman H, Ebright RH. Aromatic hydrogen bond in sequence-specific protein DNA recognition. Nat Struct Biol. 1996;3:837–41. doi: 10.1038/nsb1096-837. [DOI] [PubMed] [Google Scholar]
- 16.Liu-Johnson HN, Gartenberg MR, Crothers DM. The DNA binding domain and bending angle of E. coli CAP protein. Cell. 1986;47:995–1005. doi: 10.1016/0092-8674(86)90814-7. [DOI] [PubMed] [Google Scholar]
- 17.Porschke D, Hillen W, Takahashi M. The change of DNA structure by specific binding of the cAMP receptor protein from rotation diffusion and dichroism measurements. Embo J. 1984;3:2873–8. doi: 10.1002/j.1460-2075.1984.tb02223.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu HM, Crothers DM. The locus of sequence-directed and protein-induced DNA bending. Nature. 1984;308:509–13. doi: 10.1038/308509a0. [DOI] [PubMed] [Google Scholar]
- 19.Olson WK, Gorin AA, Lu XJ, Hock LM, Zhurkin VB. DNA sequence-dependent deformability deduced from protein-DNA crystal complexes. Proc Natl Acad Sci U S A. 1998;95:11163–8. doi: 10.1073/pnas.95.19.11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bhattacharyya D, Bansal M. Local variability and base sequence effects in DNA crystal structures. J Biomol Struct Dyn. 1990;8:539–72. doi: 10.1080/07391102.1990.10507828. [DOI] [PubMed] [Google Scholar]
- 21.El Hassan MA, Calladine CR. Two distinct modes of protein-induced bending in DNA. J Mol Biol. 1998;282:331–43. doi: 10.1006/jmbi.1998.1994. [DOI] [PubMed] [Google Scholar]
- 22.Hunter CA, Lu XJ. DNA base-stacking interactions: a comparison of theoretical calculations with oligonucleotide X-ray crystal structures. J Mol Biol. 1997;265:603–19. doi: 10.1006/jmbi.1996.0755. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki M, Yagi N. Stereochemical basis of DNA bending by transcription factors. Nucleic Acids Res. 1995;23:2083–91. doi: 10.1093/nar/23.12.2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Packer MJ, Dauncey MP, Hunter CA. Sequence-dependent DNA structure: dinucleotide conformational maps. J Mol Biol. 2000;295:71–83. doi: 10.1006/jmbi.1999.3236. [DOI] [PubMed] [Google Scholar]
- 25.Lyubchenko YL, Shlyakhtenko LS, Appella E, Harrington RE. CA runs increase DNA flexibility in the complex of lambda Cro protein with the OR3 site. Biochemistry. 1993;32:4121–7. doi: 10.1021/bi00066a038. [DOI] [PubMed] [Google Scholar]
- 26.Beutel BA, Gold L. In vitro evolution of intrinsically bent DNA. J Mol Biol. 1992;228:803–12. doi: 10.1016/0022-2836(92)90865-h. [DOI] [PubMed] [Google Scholar]
- 27.McNamara PT, Bolshoy A, Trifonov EN, Harrington RE. Sequence-dependent kinks induced in curved DNA. J Biomol Struct Dyn. 1990;8:529–38. doi: 10.1080/07391102.1990.10507827. [DOI] [PubMed] [Google Scholar]
- 28.Chen S, Vojtechovsky J, Parkinson GN, Ebright RH, Berman HM. Indirect readout of DNA sequence at the primary-kink site in the CAP-DNA complex: DNA binding specificity based on energetics of DNA kinking. J Mol Biol. 2001;314:63–74. doi: 10.1006/jmbi.2001.5089. [DOI] [PubMed] [Google Scholar]
- 29.Okonogi TM, Alley SC, Reese AW, Hopkins PB, Robinson BH. Sequence-dependent dynamics of duplex DNA: the applicability of a dinucleotide model. Biophys J. 2002;83:3446–59. doi: 10.1016/S0006-3495(02)75344-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Passner JM, Steitz TA. The structure of a CAP-DNA complex having two cAMP molecules bound to each monomer. Proc Natl Acad Sci U S A. 1997;94:2843–7. doi: 10.1073/pnas.94.7.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lawson CL, Swigon D, Murakami KS, Darst SA, Berman HM, Ebright RH. Catabolite activator protein: DNA binding and transcription activation. Curr Opin Struct Biol. 2004;14:10–20. doi: 10.1016/j.sbi.2004.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Busby S, Ebright RH. Transcription activation by catabolite activator protein (CAP) J Mol Biol. 1999;293:199–213. doi: 10.1006/jmbi.1999.3161. [DOI] [PubMed] [Google Scholar]
- 33.Zhang XP, Gunasekera A, Ebright YW, Ebright RH. Derivatives of CAP having no solvent-accessible cysteine residues, or having a unique solvent-accessible cysteine residue at amino acid 2 of the helix-turn-helix motif. J Biomol Struct Dyn. 1991;9:463–73. doi: 10.1080/07391102.1991.10507929. [DOI] [PubMed] [Google Scholar]
- 34.Benoff B, Yang H, Lawson CL, Parkinson G, Liu J, Blatter E, Ebright YW, Berman HM, Ebright RH. Structural Basis of Transcription Activation: The CAP-alpha CTD-DNA Complex. Science. 2002;297:1562–6. doi: 10.1126/science.1076376. [DOI] [PubMed] [Google Scholar]
- 35.Schultz SC, Shields GC, Steitz TA. Crystallization of Escherichia coli catabolite gene activator protein with its DNA binding site. The use of modular DNA. J Mol Biol. 1990;213:159–66. doi: 10.1016/S0022-2836(05)80128-7. [DOI] [PubMed] [Google Scholar]
- 36.Chen S, Gunasekera A, Zhang X, Kunkel TA, Ebright RH, Berman HM. Indirect readout of DNA sequence at the primary-kink site in the CAP-DNA complex: alteration of DNA binding specificity through alteration of DNA kinking. J Mol Biol. 2001;314:75–82. doi: 10.1006/jmbi.2001.5090. [DOI] [PubMed] [Google Scholar]
- 37.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 38.Navaza J. AMoRe: an automated package for molecular replacement. Acta Crystallogr A Foundations of Crystallogr. 1994;50:157–163. [Google Scholar]
- 39.Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr D Biol Crystallogr. 2004;60:432–8. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 40.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54(Pt 5):905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 41.Parkinson G, Vojtechovsky J, Clowney L, Brünger AT, Berman HM. New parameters for the refinement of nucleic acid-containing structures. Acta Crystallogr D Biol Crystallogr. 1996;52:57–64. doi: 10.1107/S0907444995011115. [DOI] [PubMed] [Google Scholar]
- 42.Brunger AT, J. K, M. K. Crystallographic R Factor Refinement by Molecular Dynamics. Science. 1987;235:458–460. doi: 10.1126/science.235.4787.458. [DOI] [PubMed] [Google Scholar]
- 43.McRee DE. XtalView/Xfit--A versatile program for manipulating atomic coordinates and electron density. J Struct Biol. 1999;125:156–65. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 44.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993;26:283–291. [Google Scholar]
- 45.Lu XJ, Olson WK. 3DNA: a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003;31:5108–21. doi: 10.1093/nar/gkg680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–42. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]