

Abstract
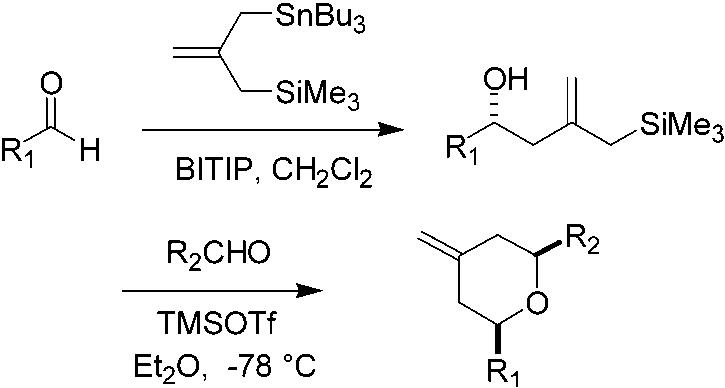
A reaction process for the asymmetric construction of a variety of cis or trans disubstituted pyrans is described. This sequences allows for the asymmetric convergent union of two aldehydes with silyl-stannane reagent 1 in a two-step process: catalytic asymmetric allylation of the first aldehyde using 1 with a BITIP catalyst, followed by reaction of the alcohol so obtained with a second aldehyde and TMSOTf.
An ever increasing number of biologically significant natural products, especially those of marine origin, possess functionalized tetrahydropyran rings as critical substructural motifs. Prominent examples of considerable current interest would include bryostatin 1 and phorboxazole (Figure 1).1-2 Our interest in these structures, particularly bryostatin 1 and other potential therapeutic agents based upon this structural platform,3 has led us to investigate new methods for the asymmetric synthesis of pyrans bearing the substitution patterns commonly encountered in these natural products. The occurrence of such pyran motifs in a number of biologically active natural products has inspired the development of many clever methods for obtaining these oxacycles. The intramolecular attack of an olefin on an oxocarbenium ion, or intramolecular Prins reaction, although one of the established methods, has been underutilized in this capacity. This is in part due to the paucity of methods for obtaining suitable substrates to provide appropriately functionalized nonracemic tetrahydropyrans.
Figure 1.
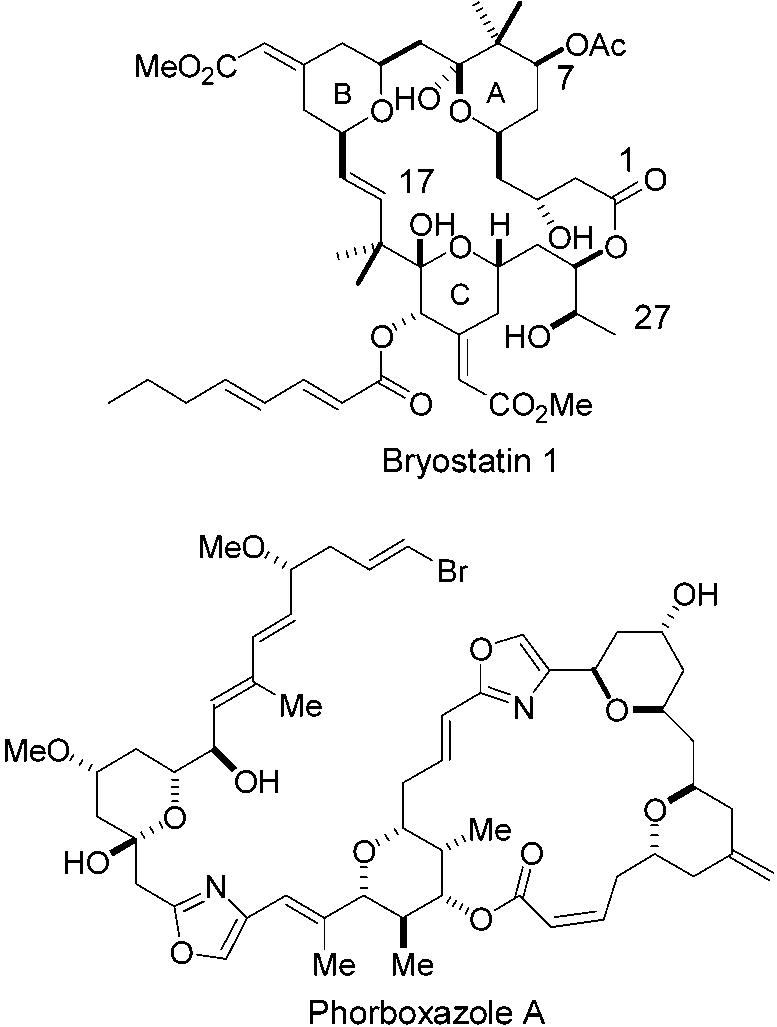
Structures of bryostatin 1 and phorboxazole.
There exists extensive precedent for the construction of oxygen-containing heterocycles, including tetrahydropyrans, by approaches based upon intramolecular versions of the basic Prins reaction.4 Previous work has also documented the utility and enhanced reactivity of allylsilanes as intramolecular traps for oxocarbenium ions generated in various ways.5 Kang has described reactions of racemic hydroxy allylsilanes of general structure 2 with vinyl ethers and α-halo acetals to give acetals that were subsequently cyclized to afford pyrans.6 Marko has developed an efficient pyran synthesis that combines the Noyori method7 for generation of oxocarbenium ions with intramolecular allylsilane trapping and has coined the term ISMS (intramolecular silyl modified Sakurai) to describe this reaction (eq 1, Scheme 1).8 Studies by Marko have also documented the use of racemic hydroxy allylsilanes in sequences leading to pyrans via an initial ene reaction; Marko has suggested the term “IMSC” (intramolecular Sakurai cyclization) to refer to the cyclization step of the process (eq 2, Scheme 1).9 Very recently, during the preparation of our manuscript, Leroy and Marko reported on the use of a functionalized allylstannane in BF3•OEt2-promoted reactions with aldehydes, followed by Bi(OTf)3-promoted cyclocondensation with a second aldehyde, to yield differentially substituted pyrans as shown in Scheme 1 below (eq 3).10
Scheme 1.
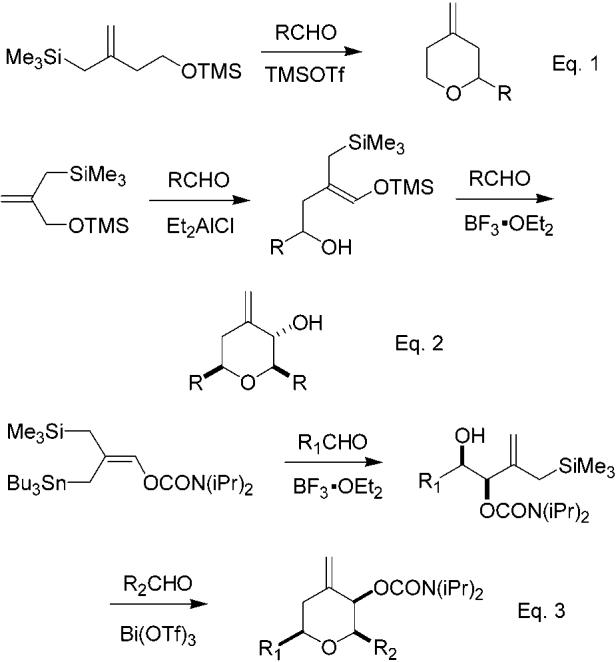
Related Racemic Pyran Syntheses
Rychnovsky has also developed a process termed “segment coupling Prins cyclization”11 and applied it to an extremely concise construction of the C20–C27 pyran subunit of phorboxazole.12 Rychnovsky and Kopecky have also recently reported a tandem Mukaiyama aldol–Prins sequence utilizing allylsilanes, which bears similarity to the process described herein.13 Both of these methods access nonracemic pyrans. Smith has utilized the Petasis–Ferrier rearrangement in efficient syntheses of nonracemic tetrahydropyrans, and Panek has employed nonracemic allylsilanes to access dihydropyrans.14
Herein we report an exceptionally facile enantioselective synthesis of 2,6-cis-disubstituted-4-methylenetetrahydropyran systems. These are obtained in a very brief three steps from commercially available starting materials. As indicated in Scheme 2, the overall two-step reaction process results in the convergent union of two aldehyde components with a four-carbon unit derived from the known 2-(trimethylsilylmethyl)allyltri-n-butylstannane reagent 1.15 For purposes of discussion, it is convenient to refer to these as the first aldehyde, R1CHO, and the second aldehyde, R2CHO.
Scheme 2.
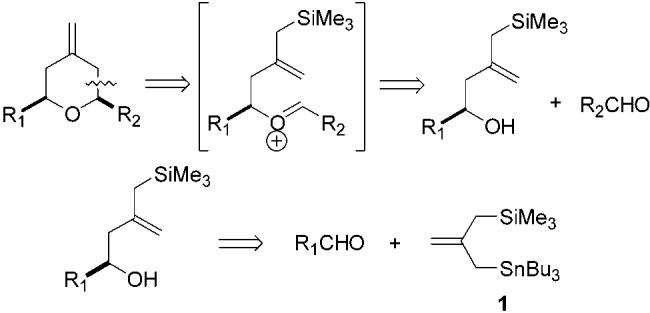
Overall Pyran Annulation Process
The first step in the overall process is the preparation of the requisite hydroxy allylsilanes in nonracemic form. This was achieved for the present examples by catalytic asymmetric allylation of the first aldehyde R1CHO with stannane 1.16 These reactions were found to proceed well using our previously described BINOL titanium tetraisopropoxide (BITIP) catalyst;17 in general, the hydroxy allylsilanes were obtained in high yields and with high levels of enantioselectivity. (Table 1).
Table 1.
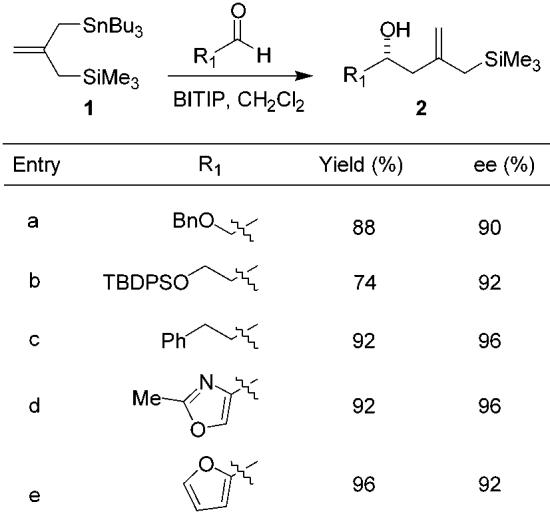
Isolated Yields and Enantiomeric Excesses for the Reaction of 1 with Aldehydes
The second step of the process, the TMSOTf-promoted annulation of such silanes with the second aldehyde R2CHO, was found to occur rapidly (generally within 15 min at −78°C) to provide the 2,6-cis-tetrahydropyran containing an exo-methylene in the 4-position. The trans diastereomer was not detected. The stereochemical outcome is in accord with previous observations and with the expectation that cyclization should occur via a chairlike transition state with R1 and R2 equatorially disposed. The results for several examples are provided in Table 2.
Table 2.
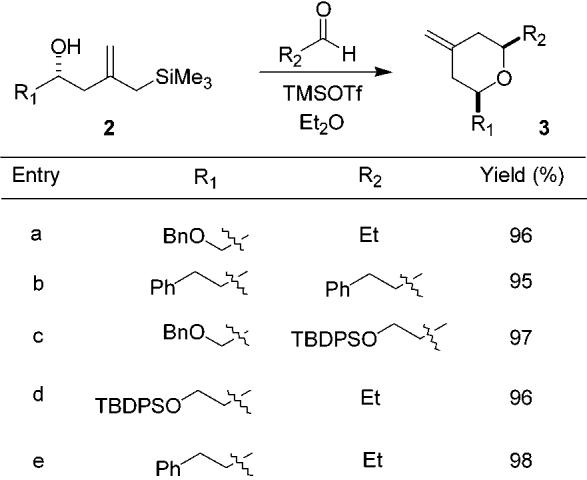
Isolated Yields for TMSOTf-Promoted Annulations
We have noticed an interesting combination of solvent effects and substrate dependence with respect to this annulation reaction. The reaction generally proceeded well in CH2-Cl2, but in all cases at least a trace amount of an endocyclic olefin that was inseparable from the desired product was obtained. In the cases where the cyclization substrate is 2c, this problem was greatly magnified. Previous attempts by Marko to cyclize similar preformed TMS ethers also resulted in mixtures of endo and exo olefins, which were very difficult to separate.8a,18 However, noting that the substrates that performed best in this reaction contained α or β-alkoxy substituents, diethyl ether was also examined as solvent. A dramatic improvement was found with this oxygenated solvent as no endocyclic olefins were obtained regardless of the substrate employed.
Another problem observed in these reactions is related to the use of substrates 2d and 2e (Table 1). In CH2Cl2, these substrates decomposed immediately upon exposure to TMSOTf. In ether at −78 °C no decomposition was observed; however, no cyclized product was obtained either. Under these conditions the alcohol is rapidly converted to the TMS ether, but the reaction stalls at this juncture.19 We are currently still examining this issue.
It is also possible to use this pyran annulation to obtain access to 2,6-trans-disubstituted pyrans. As shown in eq 4, 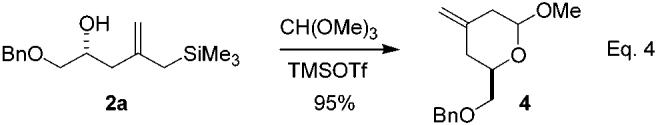 the annulation reaction works very well when an ortho ester is used as the electrophile.20 Thus, the reaction of 2a with trimethylorthoformate smoothly provides methylacetal 4 in excellent yield (eq 4). Although 4 is obtained as an epimeric mixture, this is of little consequence. Reaction of a storable nucleophile (allylsilane, allylstannane, or enol silane) with the oxocarbenium ion generated upon treatment of such acetals with a Lewis acid is well-known to afford the 2,6-trans isomer with high selectivity as a consequence of stereoelectronically preferred axial addition to the intermediate oxocarbenium ion.21
the annulation reaction works very well when an ortho ester is used as the electrophile.20 Thus, the reaction of 2a with trimethylorthoformate smoothly provides methylacetal 4 in excellent yield (eq 4). Although 4 is obtained as an epimeric mixture, this is of little consequence. Reaction of a storable nucleophile (allylsilane, allylstannane, or enol silane) with the oxocarbenium ion generated upon treatment of such acetals with a Lewis acid is well-known to afford the 2,6-trans isomer with high selectivity as a consequence of stereoelectronically preferred axial addition to the intermediate oxocarbenium ion.21
This annulation process also lends itself to iterative applications that allow for the rapid assembly of structures containing multiple pyran rings, such as the bis pyran subunit of phorboxazole. Thus, if a pyran constructed using this process contains a latent or protected aldehyde, exposure of the aldehyde function and reaction with an appropriate hydroxy allylsilane in a second annulation affords a bis pyran. An example of this iterative annulation is shown in Scheme 3. In this case, bis pyran 6 is assembled from stannane 1, α-benzyloxyacetaldehyde, and β-OTBDPS propionaldehyde in five linear steps. The compatibility of both α- and β-alkoxy aldehydes in these reactions thus easily provides pyrans containing three differentiated and highly malleable functional groups on the THP ring. Finally, this pyran synthesis is quite flexible since the annulation can be approached from two different directions for any given pyran. This should increase its utility and help to promote its application.
Scheme 3.
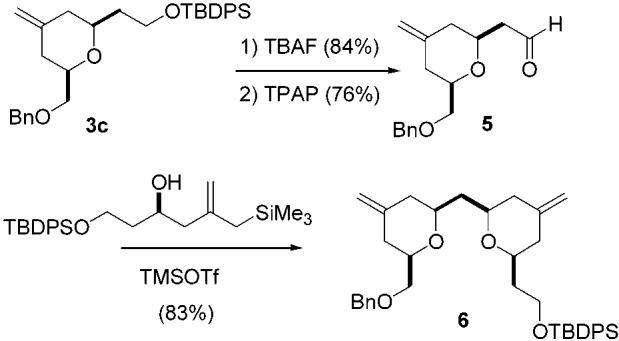
Iterative Pyran Annulation
Supplementary Material
Acknowledgment
Financial support from the National Institutes of Health (through GM-28961) and by Pfizer, Inc. is gratefully acknowledged.
Footnotes
Supporting Information Available: Spectral and analytical data and experimental procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Kageyama M, Tamura T, Nantz MH, Roberts JC, Somfai P, Whritenour DC, Masamune S. J. Am. Chem. Soc. 1990;112:7407. For total syntheses of bryostatin and leading references to other work, see: [Google Scholar]; (b) Evans DA, Carter PH, Carreira EM, Charette AB, Prunet JA, Lautens M. J. Am. Chem. Soc. 1999;121:7540. [Google Scholar]; (c) Ohmori K, Ogawa Y, Obitsu T, Ishikawa Y, Nishiyama S, Yamamura S. Angew. Chem., Int. Ed. 2000;39:2290. [PubMed] [Google Scholar]
- 2.(a) Forsyth CJ, Ahmed F, Cink RD, Lee CS. J. Am. Chem. Soc. 1998;120:5597. For total syntheses of phorboxazole and leading references to other work, see: [Google Scholar]; (b) Evans DA, Fitch DM, Smith TE, Cee VJ. J. Am. Chem. Soc. 2000;122:10033. doi: 10.1002/1521-3773(20000717)39:14<2533::aid-anie2533>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]; (c) Smith AB, III, Verhoest PR, Minbiole KP, Schelhaas M. J. Am. Chem. Soc. 2001;123:4834. doi: 10.1021/ja0105055. [DOI] [PubMed] [Google Scholar]
- 3.(a) Wender PA, De Brabander J, Harran PG, Jimenez J-M, Koehler MFT, Lippa B, Park C-M, Shiozaki S. J. Am. Chem. Soc. 1998;120:4534. [Google Scholar]; (b) Wender PA, De Brabander J, Harran PG, Jimenez JM, Koehler MF, Lippa B, Park CM, Siedenbiedel C, Pettit GR. Proc. Natl. Acad. Sci. U.S.A. 1998;95:6624. doi: 10.1073/pnas.95.12.6624. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wender PA, Blaise L. Tetrahedron Lett. 2000;41:1007. [Google Scholar]; (d) Wender PA, Hinkle KW, Koehler MFT, Lippa B. Med. Res. Rev. 1999;19:388. doi: 10.1002/(sici)1098-1128(199909)19:5<388::aid-med6>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 4.(a) Adams DR, Bhaynagar SD. Synthesis. 1977:661. [Google Scholar]; (b) Snider BB. In: Comprehensive Organic Synthesis. Trost BM, editor. Vol. 2. Pergamon Press; Oxford, U.K.: 1991. p. 527. [Google Scholar]; (c) Cloninger MJ, Overman LE. J. Am. Chem. Soc. 1999;121:1092. [Google Scholar]; (d) Kozmin SA. Org. Lett. 2001;3:755. doi: 10.1021/ol015514x. [DOI] [PubMed] [Google Scholar]
- 5.(a) Mohr P. Tetrahedron Lett. 1993;34:6251. [Google Scholar]; (b) Paquette LA, Tae J. J. Org. Chem. 1996;61:7860. doi: 10.1021/jo961306k. [DOI] [PubMed] [Google Scholar]; (c) Sano T, Oriyama T. Synlett. 1997:716. [Google Scholar]; (d) Suginome M, Iwanami T, Ito Y. J. Org. Chem. 1998;63:6096. doi: 10.1021/jo981173y. [DOI] [PubMed] [Google Scholar]; (e) Chen C, Mariano PS. J. Org. Chem. 2000;65:3252. doi: 10.1021/jo0000832. [DOI] [PubMed] [Google Scholar]
- 6.Sung TM, Kwak WY, Kang K-T. Bull. Korean Chem. Soc. 1998;19:862. [Google Scholar]
- 7.Murata S, Suzuki M, Noyori R. Tetrahedron. 1988;44:4259. [Google Scholar]
- 8.(a) Mekhalfia A, Marko IE. Tetrahedron Lett. 1991;32:4779. [Google Scholar]; (b) Mekhalfia A, Marko IE, Adams H. Tetrahedron Lett. 1991;32:4783. [Google Scholar]
- 9.(a) Marko IE, Bayston DJ. Tetrahedron Lett. 1993;34:6595. [Google Scholar]; (b) Marko IE, Bayston DJ. Tetrahedron. 1994;50:7141. [Google Scholar]; (c) Marko IE, Mekhalfia A, Murphy F, Bayston DJ, Bailey M, Janousek Z, Dolan S. Pure Appl. Chem. 1997;69:565. [Google Scholar]; (d) Marko IE, Plancher J-M. Tetrahedron Lett. 1999;40:5259. [Google Scholar]
- 10.Leroy B, Marko IE. Tetrahedron Lett. 2001;41:8685. [Google Scholar]
- 11.Jaber JJ, Mitsui K, Rychnovsky SD. J. Org. Chem. 2001;66:4679. doi: 10.1021/jo010232w. [DOI] [PubMed] [Google Scholar]
- 12.Rychnovsky SD, Thomas CR. Org. Lett. 2000;2:1217. doi: 10.1021/ol005646a. [DOI] [PubMed] [Google Scholar]
- 13.Kopecky DJ, Rychnovsky SD. J. Am. Chem. Soc. 2001;123:8420. doi: 10.1021/ja011377n. [DOI] [PubMed] [Google Scholar]
- 14.(a) Petasis NA, Lu S-P. Tetrahedron Lett. 1996;36:141. For other recent asymmetric approaches to pyrans, see: [Google Scholar]; (b) Smith AB, III, Verhoest PR, Minbiole KP, Lim JJ. Org. Lett. 1999;1:909. doi: 10.1021/ol990830l. [DOI] [PubMed] [Google Scholar]; (c) Smith AB, III, Minbiole KP, Verhoest PR, Beauchamp TJ. Org. Lett. 1999;1:913. doi: 10.1021/ol990829m. [DOI] [PubMed] [Google Scholar]; (d) Huang H, Panek JS. J. Am. Chem. Soc. 2000;122:9836. [Google Scholar]
- 15.(a) Kang K-T, Hwang SS, Kwak WY, Yoon UC. Bull. Korean Chem. Soc. 1999;20:801. [Google Scholar]; (b) Clive DLJ, Paul CC, Wang Z. J. Org. Chem. 1997;62:7028. [Google Scholar]; (c) The 2-(chloromethyl)allylsilane precursor to 1 is commercially available (Aldrich)
- 16. For an alternative preparation of such hydroxy silanes, by Noyori reduction of β-ketoesters followed by application of the Brunelle allylsilane synthesis, see ref 13.
- 17.Keck GE, Tarbet KH, Geraci LS. J. Am. Chem. Soc. 1993;115:8467. The “method B” catalyst preparation was found to be the most suitable for the reaction of stannane 1 with aldehydes. Experimental procedures may be found in Supporting Information. [Google Scholar]
- 18.Marko IE, Bayston DJ, Mekhalfia A, Adams H. Bull. Soc. Chim. Belg. 1993;102:655. Marko subsequently reported a solution to this problem through the use of trimethylsilyl ethers of simple alcohols as an additive: [Google Scholar]
- 19. Both Kang and Marko have also noticed greatly diminished yields using certain aromatic or heteroaromatic materials in their acetal cyclizations and ene reactions. Note refs 6 and 9a.
- 20.(a) Marko IE, Mekhalfia A. Tetrahedron Lett. 1992;33:1799. For previous uses of acetals, ketals, and ortho esters in this context, note: [Google Scholar]; (b) Marko IE, Mekhalfia A, Bayston DJ, Adams H. J. Org. Chem. 1992;57:2211. Note also ref 8b. [Google Scholar]
- 21. For two recent examples, see refs 2b and 14b.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.