

Abstract
Platelet-derived lysophosphatidic acid (LPA) supports the progression of breast and ovarian cancer metastasis to bone. The mechanisms through which LPA promotes bone metastasis formation are, however, unknown. Here we report that silencing of the type 1 LPA receptor (LPA1) in cancer cells blocks the production of tumor-derived cytokines that are potent activators of osteoclast-mediated bone destruction and significantly reduces the progression of osteolytic bone metastases. Moreover, functional blockade of LPA action on its cognate receptor LPA1 using a pharmacological antagonist mimics the effects of silencing LPA1 in tumor cells in vitro and substantially reduces bone metastasis progression in animals. Overall, these results suggest that inhibition of platelet-derived LPA action on LPA1 expressed by tumor cells may be a promising therapeutic target for patients with bone metastases.
Keywords: breast cancer, platelet, treatment
Bone is a frequent metastatic site for many cancers (1). Bone metastasis formation is associated with a high morbidity rate because of intractable bone pain, pathological fractures, hypercalcemia, and nerve compression (1). Bone-residing metastatic cells are not directly able to destroy bone. Instead, they secrete paracrine factors [IL-6, IL-8, and parathyroid hormone-related peptide (PTHrP)] that stimulate osteoblast-mediated bone resorption, leading to osteolysis (1). In this respect, bisphosphonates (as inhibitors of osteoclast activity) are the standard of care in the treatment of patients with bone metastases. Unfortunately, these treatments are only palliative and do not provide a life-prolonging benefit to metastatic patients (2), indicating that more efficacious therapies are required.
We have recently demonstrated that the naturally occurring bioactive lipid, lysophosphatidic acid (LPA), produced by activated blood platelets (3), is coopted by breast and ovarian cancer cells as a tumor mitogen and an inducer of tumor-derived cytokine (IL-6 and IL-8) that, altogether, promote the progression of bone metastases (4). Endogenous receptors through which LPA promotes breast cancer progression and bone metastasis are, however, unknown. LPA binds to three GTP-binding protein-coupled receptors, LPA1 (5), LPA2 (6), and LPA3 (7), that mediate the growth factor-like activity of LPA in a large variety of cell types in culture, including cancer cells (8). Interestingly, mRNA levels for LPA receptors are up-regulated in various cancers (9–11). Yet, the clinical significance of such observations remains to be determined. Using both genetic and pharmacological approaches in vitro and in vivo, we demonstrate here that inhibition of LPA action on its receptor LPA1 is a promising therapeutic target in cancer, especially for metastasis to bone.
Results
Silencing of LPA1 Expression in Cancer Cells Markedly Impairs Bone Metastasis Progression.
We have shown previously that CHOβ3wt ovarian cancer cells express only LPA1 (4). To analyze the role of LPA1 in bone metastasis formation, we generated a CHOβ3wt cell line in which the expression of LPA1 was stably down-regulated with specific small interference RNAs (siRNAs; see Fig. 1A). In vivo experiments indicated that the down-regulation of LPA1 did not alter the propensity of these cells to induce bone metastases in nude mice (Fig. 1B and Table 1). However, the silencing of LPA1 expression markedly reduced (77% inhibition) the progression of osteolytic bone lesions in animals at the time of death (Fig. 1B and Table 1). The extent of osteolytic lesions was associated with an increase of the bone volume (BV)/tissue volume (TV) ratio, which indicated a prevention of bone loss, and with a decrease of the tumor burden (TB)/TV ratio, which indicated a decrease in skeletal TB (Fig. 1B and Table 1). We next focused our attention on the human MDA-BO2 breast cancer cell line, which endogenously expresses all three LPA receptors (LPA1, LPA2, and LPA3) and induces bone metastases in nude mice (4). Human MDA-BO2 cells have been previously stably transfected to express GFP (MDA-BO2/GFP) to detect fluorescent bone metastases in animals (12). The silencing of LPA1 expression in MDA-BO2/GFP cells was achieved by using a siRNA strategy similar to that described for CHOβ3wt cells (Fig. 1A). Radiographic analysis indicated that all MDA-BO2/GFP transfectants had the same bone metastasis incidence in animals (Table 1). However, animals injected with MDA-BO2/GFP-SiLPA1 cells displayed an 80% decrease in the extent of osteolytic lesions (Fig. 1C and Table 1). Moreover, there was a 50% decrease in the extent of fluorescent lesions in metastatic animals inoculated with MDA-BO2/GFP-SiLPA1 cells (Fig. 1C and Table 1). Histological examination revealed that the silencing of LPA1 expression in MDA-BO2/GFP cells was associated with a decrease of bone destruction (increased BV/TV ratio) and reduced skeletal TB (decreased TB/TV ratio; see Fig. 1C and Table 1).
Fig. 1.

Effect of silencing of LPA1 expression in CHOβ3wt and MDA-BO2/GFP cells on osteolytic lesions and skeletal TB. (A) Expression levels of LPA1 mRNA expression were quantified by real-time RT-PCR. Data were presented as the mean ± SD of LPA1:GAPDH mRNA ratio of three independent experiments (Upper). ∗, P < 0.001, parental cells vs. SiLPA1 transfectant. Lysates of tumor cells were resolved by SDS/PAGE and immunoblotted with an anti-LPA1 polyclonal antibody (Lower). (B) CHOβ3wt cells and stable transfectants (clones Sbl#1 and SiLPA1#6) were assayed for their ability to induce osteolytic bone metastases. Representative radiographs (x-rays) of hind limbs from mice, 21 days after tumor cell inoculation. There was a marked reduction in the extent of osteolytic lesions (arrows) in CHO-SiLPA1#6 cell-bearing mice. Representative bone histology (Histo) of Goldner’s trichrome stained tibial metaphysis from metastatic animals. Bone is stained in green; bone marrow and tumor cells (T) are stained in red. Trabecular bone was almost completely preserved in tibial metaphysis from animals bearing CHO-SiLPA1#6 cells. (C) MDA-BO2/GFP cells and stable transfectants were assayed for their ability to induce bone metastases. Animals were analyzed 30 days after tumor cell injection. Osteolytic lesions were detected by radiography (x-rays), and bone destruction and TB were examined by histology (Histo), as described in B. Fluorescent tumor lesions were identified by fluorescence scanning (Fluo). There was a marked reduction in the extent of osteolytic (arrows) and fluorescent lesions, and trabecular bone was almost completely preserved in tibial metaphysis from animals bearing MDA-BO2/GFP-SiLPA1 cells.
Table 1.
Effect of LPA1 expression on CHOβ3wt or MDA-BO2/GFP cell-induced osteolytic lesions, skeletal tumor lesions, BV, and TB in metastatic animals
| Cell lines | Osteolytic lesions |
Fluorescent lesions |
Histomorphometry |
||||
|---|---|---|---|---|---|---|---|
| Incidence | Area, mm2 | Incidence | Area, mm2 | Incidence | BV/TV, % | TB/TV, % | |
| CHOβ3wt | 6/6 | 15.5 ± 1.3 | N.A. | N.A. | 6/6 | 1.1 ± 0.8 | 92.5 ± 3.6 |
| CHOβ3wt-Sb1#1 | 6/6 | 15.6 ± 2.5 | N.A. | N.A. | 6/6 | 1.1 ± 0.6 | 91.6 ± 2.3 |
| CHOβ3wt-SiLPA1#6 | 5/6 | 3.5 ± 1.7* | N.A. | N.A. | 5/6 | 10.9 ± 1.1* | 10.0 ± 2.5* |
| BO2/GFP | 4/4 | 23.6 ± 2.3 | 4/4 | 21.7 ± 2.7 | 4/4 | 1.4 ± 0.4 | 96.0 ± 2.0 |
| BO2/GFP-Sb1 #9 and #10 | 5/5, 8/8 | 20.3 ± 1.2 | 5/5, 8/8 | 23.2 ± 3.1 | 5/5, 8/8 | 1.1 ± 0.2 | 93.2 ± 1.2 |
| BO2/GFP-SiLPA1 #122, #123, and #132 | 7/7, 6/6, 8/8 | 4.1 ± 0.5* | 2/7, 2/7, 3/8 | 10.9 ± 2.8** | 7/7, 6/6, 8/8 | 11.3 ± 0.2* | 9.5 ± 0.6* |
Animals were analyzed by noninvasive radiography and fluorescence imaging 21 or 30 days after intravenous inoculation of CHOβ3wt or MDA-BO2/GFP cell lines, respectively. Metastatic hind limbs were analyzed by histomorphometry. Incidence indicates the number of metastatic animals over the total number of animals used in each experiment. Data of osteolytic and fluorescent lesions are expressed as the mean ± SE (in mm2) of n metastatic mice. The BV/TV and TB/TV ratios are expressed as the mean +/− SD (in %) of n metastatic mice. N.A., not applicable.
*, P < 0.0001;
**, P < 0.01 using unpaired Student’s t test when comparing animals injected with SiLPA1 transfectants with animals injected with Sb1 transfectants.
LPA1 Controls Tumor Cell Proliferation in Vitro and in Vivo.
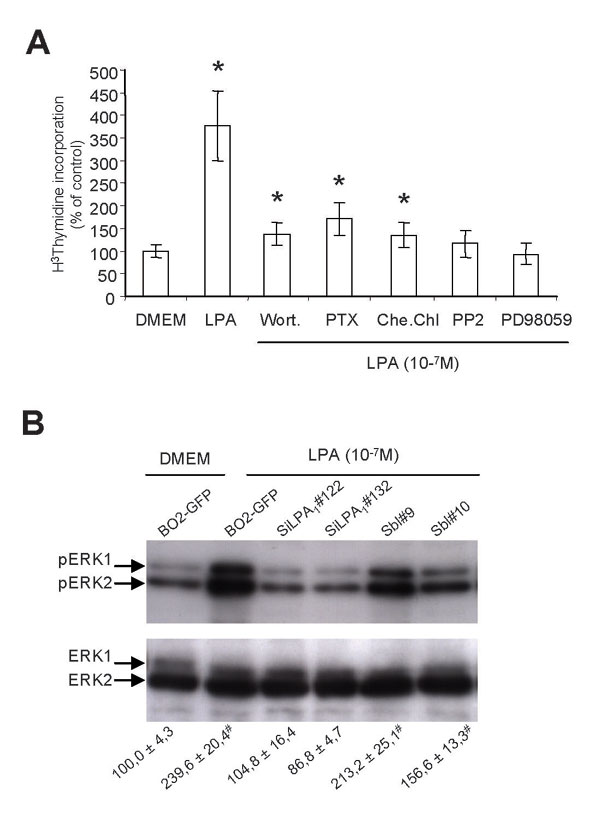
To assess whether LPA1 played a role in tumor cell proliferation, we introduced wild-type cDNA for LPA1 into the LPA-insensitive human breast cancer SKBr-3 cells (Fig. 2A). As opposed to parental cells, LPA1-expressing SKBr-3 cells (clones #3.1 and #4.1) dose-dependently responded to the mitogenic activity of LPA (Fig. 2B). Conversely, the silencing of LPA1 expression in CHOβ3wt cells almost totally abolished the mitogenic activity of LPA on these cells (Fig. 2C). The silencing of LPA1 expression also markedly altered the proliferation of MDA-BO2/GFP cells in response to LPA (Fig. 2D). In this respect, the residual LPA-dependent proliferation of MDA-BO2/GFP-SiLPA1 cells indicated that among LPA receptors, LPA1 was the main transducer of LPA mitogenic activity on parental cells (Fig. 2D). We also observed that LPA-dependent MDA-BO2/GFP cell proliferation was markedly inhibited in the presence of inhibitors for Pi3Kinase (Wortmannin), Gi protein (pertussis toxin), and PKC (chelerythrine chloride) and was totally blocked with inhibitors for mitogen-activated protein kinase (MAPK) kinase kinase, MEK (PD98059), and the Src kinase family (PP2; see Fig. 8A, which is published as supporting information on the PNAS web site). These results suggested that the mitogenic activity of LPA on these cells depended on Src kinase family MAPK extracellular signal-regulated kinase (ERK)1/2 activation. We therefore investigated the role of LPA1 on the LPA-dependent activation of ERK1/2 on MDA-BO2/GFP-SiLPA1 and -Sbl clones (Sbl, scrambled). Silencing of LPA1 expression totally blocked the LPA-dependent phosphorylation of ERK1/2 (Fig. 8B). These results, combined with previous observations (Fig. 2D), demonstrated that the mitogenic activity of LPA, at a concentration of 10−7 M, on MDA-BO2/GFP cells was mediated through a LPA1-Src kinase-ERK1/2-dependent signaling pathway. In vivo, the silencing of LPA1 markedly inhibited the growth of MDA-BO2/GFP tumors (80% reduction at day 55; see Fig. 3A). As judged by Ki67 nuclear antigen staining, the silencing of LPA1 expression in MDA-BO2/GFP cells led to a decreased proliferation of both s.c. and skeletal tumors (Fig. 3B). Thus, we demonstrated that LPA1 mediates the proliferation of cancer cells both in vitro and in vivo.
Fig. 2.

Effect of LPA1 on tumor cell proliferation in vitro. (A) Total RNA extracts of parental and SKBr-3 cells stably transfected with pCi/HA-LPA1 (clones LPA1#3.1 and #4.1) were subjected to real-time RT-PCR (Upper) and cell lysates were analyzed by Western blotting for LPA1 detection (Lower), as described in Fig. 1A. SKBr-3 cells (B), CHOβ3wt (C), and MDA-BO2/GFP (D) cell proliferation assays. Quiescent tumor cells plated in 96-well tissue culture plates were incubated overnight with 1-oleoyl LPA (0–1 μM) in serum-free medium containing 0.1% (wt/vol) BSA fatty acid-free then pulsed with [H3]thymidine for 8 h. Data of [H3]thymidine incorporation were expressed in cpm, are the mean ± SD of six replicates, and are representative of at least three separate experiments. ∗, P < 0.001, parental cells vs. transfectants.
Fig. 3.

Effect of LPA1 on tumor growth and cancer cell proliferation in vivo. (A) Animals were injected s.c. into the flank with parental cells or MDA-BO2/GFP transfectants. Tumor size was assessed by external measurement of the tumors by using a Vernier caliper. Tumor volume was calculated at the indicated time point and expressed as the mean ± SD (in mm3) of 10 animals per group. ∗, P < 0.001, MDA-BO2/GFP cells vs. SiLPA1 clones. (B) s.c. tumor (xenograft) and decalcified metastatic bone tissues (skeletal) were fixed and embedded in paraffin. Six-micrometer tissue sections were subjected to immunohistochemistry by using a monoclonal antibody against the nuclear Ki67 antigen. The mitotic index (numbers are indicated on each image) was calculated as the percentage of nuclei positive for Ki67. Results are expressed as the mean ± SD (in %) of six independent tumor sections with 10 animals per group. #, P < 0.0001, MDA-BO2/GFP tumor-bearing animals vs. transfectant SiLPA1#122 tumor-bearing animals. (Scale bars: 100 μm.)
LPA1 Mediates Tumor Cell-Induced Osteoclast Activity Through the Production of Specific Cytokines.
LPA has recently been shown to stimulate the production of IL-6 and IL-8 in ovarian and breast cancer cells because of the activation of each LPA receptor (LPA1, LPA2, and LPA3; see ref. 13). Artificial overexpression of LPA1 in MDA-BO2 cells increases IL-6 and IL-8 production in response to LPA (4). Among the cytokines, chemokines, and growth factors naturally secreted by MDA-BO2/GFP cells and detected by RayBio (Norcross, GA) human cytokine antibody arrays, LPA up-regulated the secretion of growth-related oncogene (Gro), granulocyte/macrophage colony-stimulating factor (GM-CSF), IL-6, IL-8, angiogenin, macrophage chemoattractant protein 1 (MCP-1), and tissue inhibitor of metalloproteinase 1 (TIMP-1) (Fig. 9, which is published as supporting information on the PNAS web site). Cytokines play a significant role during bone remodeling (1, 14). Therefore, we examined the effect of LPA1 expression in tumor cells on the production of IL-6, IL-8, GM-CSF, Groα, and MCP-1 (Table 2). As opposed to the parental SKBr-3 cell line, which is refractory to LPA-induced cytokine secretion, LPA strongly stimulated the secretion of IL-6, IL-8, GM-CSF, Groα, and MCP-1 by LPA1-expressing SKBr-3 transfectants (Table 2). Conversely, the silencing of LPA1 expression in MDA-BO2/GFP cells inhibited partially the LPA-dependent secretion of all these cytokines. The residual LPA-dependent cytokine secretion of MDA-BO2/GFP-SiLPA1 cells indicated that LPA1 activity accounted for partial secretion of IL-6 (42%), IL-8 (37%), and GM-CSF (66%), and for almost all of the secretion of Groα (90%) and MCP-1 (95%) by parental cells. Thus, these results indicated that LPA1 plays a significant role in the LPA-dependent production of proosteoclastic cytokines IL-6, IL-8, GM-CSF, Groα, and MCP-1 by tumor cells.
Table 2.
Effect of LPA1 expression by tumor cells on LPA-induced cytokine secretion
| Cell lines | IL-6 | IL-8 | GM-CSF | GROα | MCP-1 |
|---|---|---|---|---|---|
| SKBRr-3 | 4 ± 0 | 0 | 0 | 0 | 1 ± 1 |
| SKBr/LPA1#3.1 | 454 ± 64 | 572 ± 42 | 1,033 ± 133 | 577 ± 62 | 281 ± 40 |
| SKBr/LPA1#4.1 | 491 ± 54 | 578 ± 91 | 1,013 ± 144 | 576 ± 72 | 282 ± 38 |
| BO2/GFP | 467 ± 71 | 987 ± 10 | 2,313 ± 267 | 1,161 ± 55 | 407 ± 45 |
| BO2/GFP-Sb1#9 | 524 ± 47 | 1,048 ± 182 | 2,241 ± 228 | 1,142 ± 124 | 382 ± 113 |
| BO2/GFP-Sb1#10 | 570 ± 25 | 1,079 ± 68 | 2,219 ± 368 | 1,117 ± 72 | 380 ± 132 |
| BO2/GFP-SiLPA1#122 | 236 ± 45 | 491 ± 7 | 665 ± 143 | 10 ± 17 | 6 ± 4 |
| BO2/GFP-SiLPA1#123 | 217 ± 13 | 446 ± 16 | 597 ± 160 | 14 ± 4 | 5 ± 4 |
| BO2/GFP-SiLPA1#132 | 209 ± 6 | 439 ± 11 | 679 ± 139 | 9 ± 4 | 7 ± 4 |
The specific production of cytokines secreted in the culture media from cells treated with 1-oleoyl LPA (1 μM) for 48 h was quantified by ELISA. Data are expressed as mean ± SD (in pg/ml per 106 cells) of three replicates and are representative of two separate experiments.
Consistent with this observation, conditioned media collected from LPA1-expressing MDA-BO2/GFP parental mock-transfectant (clone Sbl#9) and SKBr-3/HA-LPA1 cells treated with LPA markedly enhanced the differentiation of osteoclasts from bone marrow cells in vitro (Fig. 4A). In contrast, the conditioned medium collected from LPA-treated MDA-BO2/GFP-SiLPA1 cells did not stimulate osteoclastogenesis nor did the conditioned medium from LPA-treated SKBr-3 cells that lack LPA1 (Fig. 4A). These data do not show the functional role of each individual cytokine. However, consistent with an important role of the secretion of IL-6 and IL-8 by tumor cells in osteoclast-mediated bone destruction in vivo (15, 16), the recruitment of mature osteoclasts located at the bone/tumor interface of metastatic long bones in animals bearing tumor cells (MDA-BO2/GFP-SiLPA1 and CHOβ3wt/SiLPA1 cells) that lack LPA1 was also markedly decreased, as judged by tartrate-resistant acid phosphatase (TRAP) staining of bone resorption surfaces (Fig. 4B). Altogether, these results indicate that LPA1 expressed by tumor cells mediates LPA action that leads to osteoclast activation in vitro and in vivo.
Fig. 4.

Effect of LPA1 expression on tumor cell-mediated osteoclast activity in vitro and in vivo. (A) Mononuclear cells isolated from bone marrow were incubated with mouse M-CSF, RANK-L, and conditioned medium collected from indicated tumor cell lines, treated with 1-oleoyl LPA (1 μM). Differentiated osteoclasts were enumerated under a light microscope as a function of multinucleation (more than three nuclei) and TRAP staining. Results were expressed as the mean ± SD of osteoclast number per mm2. #, P < 0.0001 for conditioned media from MDA-BO2/GFP cells vs. transfectant clone SiLPA1#122, and from SKBr-3 cells vs. transfectant clone HA-LPA1#3.1. (Scale bars: 100 μm.) (B) Representative histological examination of TRAP-stained proximal tibia sections from metastatic animals, 30 days after tumor cell inoculation of parental BO2/GFP cells, mock transfectants (clones Sbl#9), or BO2/GFP-SiLPA1 cells (clones SiLPA1#132) (Left). Bone is stained in dark blue, and osteoclasts are stained in red. (Scale bar: 200 μm.) The percentage of active-osteoclast resorption surface per bone surface (Oc.S/BS) was quantified. Results are expressed as the mean ± SD (in %) of five to eight animals per group. ∗, P < 0.001, BO2/GFP (Cont.), or CHOβ3wt (Cont) vs. SiLPA1 tumor-bearing animals (Right).
Pharmacological Blockade of LPA1 Activity Inhibits Bone Metastasis Progression.
3-(4-[4-([1-(2-chlorophenyl)ethoxy]carbonyl amino)-3-methyl-5-isoxazolyl]benzylsulfanyl)propanoic acid (Ki16425) is a potent antagonist of LPA activity on LPA1 and LPA3 (17). Ki16425 blocks the LPA-induced motility of various LPA1-expressing carcinoma cell lines in vitro (18). Ki16425 also dose-dependently inhibited LPA-induced proliferation of LPA1-expressing MDA-BO2/GFP and CHOβ3wt cell lines in vitro (Fig. 5) and specifically blocked the LPA-mediated secretion of cytokines (IL-6, IL-8, GM-CSF, Groα, and MCP-1) in LPA1-expressing breast cancer cell lines (data not shown). We therefore examined the effect of Ki16425 on the progression of established bone metastases using our fluorescent animal model. Ki16425 inhibited by 90% the progression of osteolytic lesions and totally blocked the formation of fluorescent tumor lesions (Fig. 6). Histological examination of bone tissue sections showed that bone destruction and skeletal TB were markedly decreased upon Ki16425 treatment (Fig. 6). Consistent with in vitro observations, Ki16425 treatment substantially inhibited the recruitment of mature osteoclasts at the bone/tumor interface and also decreased the in vivo proliferation of tumor cells (80% decrease of the Ki67 cell mitotic index; see Fig. 6). Plasma of vehicle-treated metastatic mice was as equipotent as purified 1-oleoyl LPA (0.1 μM) to stimulate proliferation of tumor cells (Fig. 7A). In contrast, the mitogenic activity of plasma from Ki16425-treated animals was markedly reduced (80% inhibition; see Fig. 7A). As indicated in Fig. 7A Inset, the circulating concentration of Ki16425 was 0.1 μM in animals treated for 16 days with a daily dose of 20 mg/kg. In addition, Ki16425 did not affect platelet count after a 16-day treatment of metastatic animals, because the mean ± SD of platelet number per nanoliter was 253.8 ± 32.7 and 261.7 ± 40.7 for vehicle- and Ki16425-treated animals, respectively. Moreover, platelets isolated from Ki16425-treated animals aggregated in response to collagen with the same kinetic and magnitude as platelets isolated from vehicle-treated animals (Fig. 7B). This rules out a possible antiplatelet activity of this compound. In addition to an absence of side effect of Ki16425 on hemostasis, no alterations of animal weight and behavior have been observed along with treatment. Altogether, these results strongly suggest that the inhibitory activity of Ki16425 on bone metastasis progression is associated with a blockade of platelet-derived LPA action on bone residing-tumor cells.
Fig. 5.

Effect of Ki16425 on LPA-induced tumor cell proliferation. (A) Quiescent BO2/GFP parental cells, scramble and SiLPA1 transfectants, and cells sensitized to the mitogenic action of LPA (MDA-BO2/HA-LPA1 cells) or (B) CHOβ3wt parental cells, scramble, and SiLPA1 transfectants, were treated overnight with increasing concentrations of Ki16425 (0–10 μM) in the presence of 1-oleoyl LPA (0.1 μM). Cell proliferation was monitored as described in Fig. 2B. Data are expressed as the mean ± SD (in cpm) of six replicates and are representative of at least three separate experiments.
Fig. 6.

Effect of Ki16425 on the progression of breast cancer bone metastases. Representative radiographs, fluorescence imaging, bone histology, TRAP, and Ki67 staining of hind limbs from metastatic mice treated daily by s.c. injection with PBS (vehicle) or Ki16425. Quantification values, indicated below each image, were the mean ± SD of osteolytic lesion area (x-ray, in mm2), fluorescent lesion area (Fluo, in mm2; ND, not detectable), active-osteoclast resorption surface per bone surface (TRAP, in %), and mitotic index (Ki67, in %) of 10 animals per group. ∗, P < 0.001, vehicle-treated vs. Ki16425-treated animals. Osteolytic lesions are indicated by arrows and TB (T).
Fig. 7.

Quantification of circulating Ki16425 in the plasma of metastatic animals and effect of a Ki16425 treatment on platelet aggregation. (A) Quiescent MDA-BO2 cells and cells sensitized to the mitogenic action of LPA (MDA-BO2/HA-LPA1 cells) were incubated overnight with 1-oleoyl LPA (0.1 μM) in serum-free medium in presence of 0.1% (wt/vol) BSA fatty acid–free (Cont.) or with plasma collected from vehicle- or Ki16425-treated animals for 16 days. Cell proliferation was quantified as described in Fig. 2B. Data were expressed as the mean ± SD (in cpm) of six replicates. #, P < 0.0001 plasma-vehicle vs. plasma-Ki16425 for each cell line. (Inset) Standard curve of Ki16425 inhibition on the mitogenic activity of LPA (0.1 μM) for MDA-BO2 and MDA-BO2/HA-LPA1 cells. Arrows indicate the relation between the concentration of Ki16425 and the mitogenic activity of the plasma from Ki16425-treated animals (in cpm). (B) Platelet aggregation experiments were carried out from blood collected from vehicle- and Ki16425-treated animals as described in Methods. Pl, platelets; Col, collagen.
Discussion
Recent studies suggest that LPA plays a significant role in the development of cancers (8). In breast and ovarian cancer, we have recently demonstrated that LPA produced by blood platelets is a major factor involved in the progression of bone metastases (4). The signaling pathways through which platelet-derived LPA promotes bone metastasis are, however, unknown. LPA mediates its activity by binding to G protein-coupled receptors (LPA1, LPA2, and LPA3; see refs. 5–8), and these receptors share signaling pathways, suggesting that their activities might be redundant (19). The silencing of LPA1 expression in breast and ovarian cancer cells markedly altered the progression of bone metastases because of inhibition of TB and bone resorption. Our results do not exclude a possible contribution of LPA2 and LPA3 in mediating LPA action on bone metastasis progression. Although an artificial overexpression of LPA1 in human MDA-BO2 cells enhances bone metastasis progression (4), such experimental strategy remains inconclusive on the physiopathological role of endogenous LPA1. This LPA receptor was consistently detected at primary and secondary sites of human breast cancers but with no significant variation of expression (9–11). Overexpression of LPA2 and/or LPA3 was observed in several cancers (9–11), and these receptors are involved in the LPA-dependent proliferation of colon cancer HCT116 and LS174T cells in vitro (20), which would suggest that up-regulation of these receptors could play a role in carcinogenesis. However, as shown here, using both genetic and pharmacological approaches, most of the LPA activities on human breast cancer MDA-BO2 cell proliferation and production of proosteoclastic cytokines were LPA1-dependent. Silencing of LPA1 expression decreased TB both in bone and soft tissue, suggesting that the LPA1-dependent promoting effect of LPA on tumor growth was independent of the metastatic site. However, bone destruction is mediated by osteoclasts under the control of growth factors and cytokines. Therefore, factors such as LPA that increased tumor cell cytokine secretion might have a more marked influence on the progression of bone metastases than that of metastases located in other organs.
Clinical trials using antiplatelet agents such as aspirin or heparin have yielded inconclusive results (21, 22). Moreover, bleedings that are frequently encountered in cancer patients treated with cytotoxic chemotherapies because of platelet toxicity often require life-saving transfusions with platelets from healthy donors (23). We demonstrated here that a specific LPA1 antagonist exhibited specificity for tumor cell–platelet interaction without abrogating normal platelet functions. Ki16425 blocked in vivo tumor cell proliferation and inhibited the production by tumor cells of proosteoclastic cytokines, whereas normal platelet functions were unaffected. Different antagonists targeting LPA1 and, to a lesser extent, LPA3 have been described (17, 24–26). In vitro, the concentration of Ki16425 (10 μM) used in this study was suitable with a complete inhibition of LPA1 (Ki, 0.35 μM) and LPA3 (Ki, 0.93 μM; see ref. 17). In contrast, the concentration of Ki16425 (0.1 μM) in the plasma of animals treated for 16 days with a daily dose of 20 mg/kg suggested that the main target in vivo was LPA1. In accordance with an involvement of LPA receptors in pathophysiological processes, the treatment of mice with Ki16425 had no obvious deleterious effects, because the animals behaved normally and had normal platelet functions. Altogether, our results demonstrate that LPA1 expressed by tumor cells plays an essential role in bone metastasis progression and indicate that antagonists blocking LPA1-dependent effects of LPA may be promising in the treatment of bone metastasis.
Methods
Cell Culture.
Characteristics of SKBr-3, MDA-MB-231/BO2 (MDA-BO2), MDA-BO2/GFP, MDA-BO2/HA-LPA1, and CHOβ3wt cell lines were described previously (4, 12, 27). SKBr-3/HA-LPA1 cell lines were established as followed. The cDNA encoding the HA-LPA1 was removed from the pBiL/HA-LPA1 (4) using NheI and HindIII restriction enzymes. The 5′ end of the cDNA was blunted by using the T4 DNA polymerase before digestion with HindIII. The vector pCi/HA-LPA1 was constructed by inserting the blunt/HindIII fragment encoding for HA-LPA1 into the pCi-neo plasmid (Promega), previously digested with SmaI and HindIII restriction enzymes. SKBr-3 cells were transfected with pCi/HA-LPA1 using Lipofectamine 2000 (Invitrogen). Cells were cultured for 4 weeks in the presence of G418 (1 mg/ml), and stable SKBr-3/HA-LPA1 clones were isolated by using cloning cylinders. LPA1 expression was assessed at the mRNA level by real-time quantitative RT-PCR, as described (4), and at the protein level by Western blot analysis with a polyclonal antibody to LPA1 (Abgent, San Diego). Oleoyl C18:1 LPA (Avanti Polar Lipids) proliferation assays and production of cell conditioned media, as described (4). Ki16425 is a competitive inhibitor of in vitro LPA-dependent signaling pathways through LPA1 (Ki, 0.35 μM), and to a lesser extent, through LPA3 (Ki, 0.93 μM) and LPA2 (Ki, 6.5 μM; see ref. 23). Increasing concentrations of Ki16425 (from 10−10 to 10−5 M) were used in cell proliferation assay in the presence of LPA (10−7 M).
Stable Silencing of LPA1 mRNA Expression in Hamster Ovarian and Human Breast Cancer Cells.
We designed small hairpin RNAs (SiRNA) and corresponding SblRNA sequences directed to LPA1 mRNA target sites based on the human and hamster sequences (GenBank accession nos., NM_001401 and AY522544, respectively), using the online sirna target designer 1.51 software (Promega). Pairwise oligonucleotides for hamster SiRNA-LPA1 5′-ACCGCCTCTGTCGCCAATCTGTAAGTTCTCTACAGATTGGCGACAGAGGCTTTTTC3′, 5′-TGCAGAAAAAGCCTCTGTCGCCAATCTGTAGAGAACTTACAGATTGGCGACAGAGG-3′ (target nucleotide site: 343–361), and for human SiRNA-LPA1 5′-ACCGAATGTCTCGGCATAGTTCAAGTTCTCTGAACTATGCCGAGACATTCTTTTTC-3′, 5′-TGCAGAAAAAGAATGTCTCGGCATAGTTCAGAGAACTTGAACTATGCCGAGACATT-3′ (target nucleotide site, 769–786), as well as sbl control sequences were cloned into the psiSTRIKE puromycin vector containing the U6 promoter (Promega). Plasmids were transfected into tumor cells by using the Transfast reagent (Promega). Cells were cultured for 2 weeks in the presence of puromycin (2 μg/ml), and CHOβ3wt-siLPA1, CHOβ3wt-sbl, MDA-BO2/GFP-siLPA1, and MDA-BO2/GFP-sbl clones were isolated by using cloning cylinders. LPA1 expression was assessed by real-time RT-PCR and Western blot analysis, as described above.
Cytokine Quantification.
Cytokines produced in cell-culture-conditioned media were quantified by ELISA following the manufacturer’s instructions (Bender MedSystems, Vienna).
Animal Studies.
All procedures were performed on female NMRI nu/nu mice 4 weeks of age (Janvier, Le Genest St.-Isle, France). Studies involving animals, including housing and care, method of euthanasia, and experimental protocols, were conducted in accordance with a code of practice established by the Experimentation Review Board from the Laënnec School of Medicine. These studies were routinely inspected by the attending veterinarian to ensure continued compliance with the proposed protocols. Bone metastasis and tumorigenesis experiments conducted with MDA-BO2/GFP cells were as described (4, 28). Metastatic animals bearing MDA-BO2/GFP tumors were also treated daily from day 14 to day 30 with Ki16425 by s.c. injection (20 mg/kg per day). Bone metastasis experiments with CHOβ3wt cells were carried out as described (27).
Bone Histology and Immunohistochemistry.
Quantification of BV/TV, TB/TV, and in situ detection of osteoclasts (TRAP cells) was carried out on undecalcified bone tissue sections, as described (4). The mitotic index of tumor cells in vivo was quantified by immunohistochemistry by using a mouse anti-human Ki67 monoclonal antibody (Dakocytomation, Dako), as described (4).
Osteoclastogenesis Assay in Vitro.
Bone marrow cells from mice were collected, and mononuclear cells were isolated by using lymphocyte separation media (ICN), then seeded in a 48-well tissue culture plate at a density of 2.5 × 103 cells per well in α-MEM medium (Invitrogen) supplemented with FCS, macrophage–CSF (PreproTech, Rocky Hill, NJ) and receptor-activated nuclear receptor factor κB ligand (RANK-L, generous gift of Pr. Jurdic, CNRS, Lyon, France). After incubation for 6 days, differentiated osteoclasts were enumerated under a light microscope as a function of multinucleation (more than three nuclei) and TRAP activity. Results were expressed as the number of osteoclasts per mm2.
Platelet Experiments.
Blood samples were collected from metastatic mice in the presence of ACD-A as an anticoagulant. Platelets were counted, and aggregation experiments were performed by using washed platelets under stirring conditions at 37°C in the absence or presence of collagen (2.5 μg/ml). Platelet aggregation was monitored over time as the percentage of light transmission, as described (4).
Statistical Analysis.
Unpaired Student’s t test analyses were carried out by using stat-view 5.0 software. P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank Kirin Brewery for providing Ki16425. This study was supported by grants from Institut National de la Santé et de la Recherche Médicale (to O.P. and P.C.), the Comité Départemental de la Loire de la Ligue Nationale Contre le Cancer (to O.P.), and the European Commission (contract LSHC-CT-2004-503049, to P.C.). A.B. was a recipient of fellowships from the Ligue Nationale Contre le Cancer and the Fondation pour la Recherche Médicale.
Abbreviations
- LPA
lysophosphatidic acid
- LPA1–3
LPA receptor type 1–3
- GM-CSF
granulocyte/macrophage colony-stimulating factor
- Gro
growth-related oncogene
- BV
bone volume
- TV
tissue volume
- TB
tumor burden
- TRAP
tartrate-resistant acid phosphatase
- sbl
scrambled
- Ki16425
3-(4-[4-([1-(2-chlorophenyl)ethoxy]carbonyl amino)-3-methyl-5-isoxazolyl]benzylsulfanyl)propanoic acid.
Footnotes
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Mundy G. R. Nat. Rev. Cancer. 2002;2:584–593. doi: 10.1038/nrc867. [DOI] [PubMed] [Google Scholar]
- 2.Hillner B. E., Ingle J. N., Berenson J. R., Janjan N. A., Albain K. S., Lipton A., Yee G., Biermann J. S., Chlebowski R. T., Pfister D. G. J. Clin. Oncol. 2000;18:1378–1391. doi: 10.1200/JCO.2000.18.6.1378. [DOI] [PubMed] [Google Scholar]
- 3.Aoki J., Taira A., Takanezawa Y., Kishi Y., Hama K., Kishimoto T., Mizuno K., Saku K., Taguchi R., Arai H. J. Biol. Chem. 2002;277:48737–48744. doi: 10.1074/jbc.M206812200. [DOI] [PubMed] [Google Scholar]
- 4.Boucharaba A., Serre C.-M., Gres S., Saulnier-Blache J. S., Bordet J.-C., Guglielmi J., Clezardin P., Peyruchaud O. J. Clin. Invest. 2004;114:1714–1725. doi: 10.1172/JCI22123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.An S., Dickens M. A., Bleu T., Hallmark O. G., Goetzl E. J. Biochem. Biophys. Res. Commun. 1997;231:619–622. doi: 10.1006/bbrc.1997.6150. [DOI] [PubMed] [Google Scholar]
- 6.An S., Bleu T., Hallmark O. G., Goetzl E. J. J. Biol. Chem. 1998;273:7906–7910. doi: 10.1074/jbc.273.14.7906. [DOI] [PubMed] [Google Scholar]
- 7.Bandoh K., Aoki J., Hosono H., Kobayashi S., Kobayashi T., Murakami-Murofushi K., Tsujimoto M., Arai H., Inoue K. J. Biol. Chem. 1999;274:27776–27785. doi: 10.1074/jbc.274.39.27776. [DOI] [PubMed] [Google Scholar]
- 8.Mills G. B., Moolenaar W. H. Nat. Rev. Cancer. 2003;3:582–591. doi: 10.1038/nrc1143. [DOI] [PubMed] [Google Scholar]
- 9.Kitayama J., Shida D., Sako A., Ishikawa M., Hama K., Aoki J., Arai H., Nagawa H. Breast Cancer Res. 2004;6:640–646. doi: 10.1186/bcr935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fang X., Schummer M., Mao M., Yu S., Tabassam F. H., Swaby R., Hasegawa Y., Tanyi J. L., LaPushin R., Eder A., et al. Biochim. Biophys. Acta. 2002;1582:257–264. doi: 10.1016/s1388-1981(02)00179-8. [DOI] [PubMed] [Google Scholar]
- 11.Schulte K. M., Beyer A., Kohrer K., Oberhauser S., Roher H. D. Int. J. Cancer. 2001;92:249–256. doi: 10.1002/1097-0215(200102)9999:9999<::aid-ijc1166>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 12.Peyruchaud O., Winding B., Pecheur I., Serre C. M., Delmas P., Clezardin P. J. Bone Miner. Res. 2001;16:2027–2034. doi: 10.1359/jbmr.2001.16.11.2027. [DOI] [PubMed] [Google Scholar]
- 13.Fang X., Yu S., Bast R. C., Liu S., Xu H. J., Hu S. X., LaPushin R., Claret F. X., Aggarwal B. B., Lu Y., Mills G. B. J. Biol. Chem. 2004;279:9653–9661. doi: 10.1074/jbc.M306662200. [DOI] [PubMed] [Google Scholar]
- 14.Teitelbaum S. L. Science. 2000;289:1504–1508. doi: 10.1126/science.289.5484.1504. [DOI] [PubMed] [Google Scholar]
- 15.de la Mata J., Uy H. L., Guise T. A., Story B., Boyce B. F., Mundy G. R., Roodman G. D. J. Clin. Invest. 1995;95:2846–2852. doi: 10.1172/JCI117990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bendre M. S., Gaddy-Kurten D., Mon-Foote T., Akel N. S., Skinner R. A., Nicholas R. W., Suva L. J. Cancer Res. 2002;62:5571–5579. [PubMed] [Google Scholar]
- 17.Ohta H., Sato K., Murata N., Damirin A., Malchinkhuu E., Kon J., Kimura T., Tobo M., Yamazaki Y., Watanabe T., et al. Mol. Pharmacol. 2003;64:994–1005. doi: 10.1124/mol.64.4.994. [DOI] [PubMed] [Google Scholar]
- 18.Hama K., Aoki J., Fukaya M., Kishi Y., Sakai T., Suzuki R., Ohta H., Yamori T., Watanabe M., Chun J., Arai H. J. Biol. Chem. 2004;279:17634–17639. doi: 10.1074/jbc.M313927200. [DOI] [PubMed] [Google Scholar]
- 19.Anliker B., Chun J. J. Biol. Chem. 2004;279:20555–20558. doi: 10.1074/jbc.R400013200. [DOI] [PubMed] [Google Scholar]
- 20.Yang M., Zhong W. W., Srivastava N., Slavin A., Yang J., Hoey T., An S. Proc. Natl. Acad. Sci. USA. 2005;102:6027–6032. doi: 10.1073/pnas.0501535102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Falanga A. J. Thromb. Haemost. 2004;2:1263–1265. doi: 10.1111/j.1538-7836.2004.00868.x. [DOI] [PubMed] [Google Scholar]
- 22.Varki N. M., Varki A. Semin. Thromb. Hemost. 2002;28:53–66. doi: 10.1055/s-2002-20564. [DOI] [PubMed] [Google Scholar]
- 23.Nash G. F., Turner L. F., Scully M. F., Kakkar A. K. Lancet Oncol. 2002;3:425–430. doi: 10.1016/s1470-2045(02)00789-1. [DOI] [PubMed] [Google Scholar]
- 24.Fischer D. J., Nusser N., Virag T., Yokoyama K., Wang D., Baker D. L., Bautista D., Parrill A. L., Tigyi G. Mol. Pharmacol. 2001;60:776–784. [PubMed] [Google Scholar]
- 25.Heise C. E., Santos W. L., Schreihofer A. M., Heasley B. H., Mukhin Y. V., Macdonald T. L., Lynch K. R. Mol. Pharmacol. 2001;60:1173–1180. doi: 10.1124/mol.60.6.1173. [DOI] [PubMed] [Google Scholar]
- 26.Sardar V. M., Bautista D. L., Fischer D. J., Yokoyama K., Nusser N., Virag T., Wang D. A., Baker D. L., Tigyi G., Parrill A. L. Biochim. Biophys. Acta. 2002;1582:309–317. doi: 10.1016/s1388-1981(02)00185-3. [DOI] [PubMed] [Google Scholar]
- 27.Pecheur I., Peyruchaud O., Serre C. M., Guglielmi J., Voland C., Bourre F., Margue C., Cohen-Solal M., Buffet A., Kieffer N., Clezardin P. FASEB J. 2002;16:1266–1268. doi: 10.1096/fj.01-0911fje. [DOI] [PubMed] [Google Scholar]
- 28.Peyruchaud O., Serre C.-M., NicAmhlaoibh R., Fournier P., Clezardin P. J. Biol. Chem. 2003;278:45826–45832. doi: 10.1074/jbc.M309024200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}