

Abstract
The activation of NKG2D on innate and adaptive cytotoxic lymphocytes contributes to immune-mediated tumor destruction. Nonetheless, tumor cell shedding of NKG2D ligands, such as MHC class I chain-related protein A (MICA), results in immune suppression through down-regulation of NKG2D surface expression. Here we show that some patients who respond to antibody-blockade of cytotoxic T lymphocyte-associated antigen 4 or vaccination with lethally irradiated, autologous tumor cells engineered to secrete granulocyte–macrophage colony-stimulating factor generate high titer antibodies against MICA. These humoral reactions are associated with a reduction of circulating soluble MICA (sMICA) and an augmentation of natural killer (NK) cell and CD8+ T lymphocyte cytotoxicity. The immunotherapy-induced anti-MICA antibodies efficiently opsonize cancer cells for dendritic cell cross-presentation, which is correlated with a diversification of tumor antigen recognition. The anti-MICA antibodies also accomplish tumor cell lysis through complement fixation. Together, these findings establish a key role for the NKG2D pathway in the clinical activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade and granulocyte–macrophage colony-stimulating factor secreting tumor cell vaccines. Moreover, these results highlight the therapeutic potential of anti-MICA antibodies to overcome immune suppression and effectuate tumor destruction in patients.
Keywords: NKG2D, cytotoxic T lymphocyte-associated antigen 4, granulocyte-macrophage colony-stimulating factor, antibody
Tumor cells express a variety of gene products that provoke innate and adaptive immune recognition. In murine models, impaired host responses are associated with increased susceptibility to carcinogen-induced and spontaneous tumors (1). In patients, brisk intratumoral lymphocyte infiltrates, particularly those enriched in CD8+ cytotoxic T cells and decreased in regulatory T cells, are correlated with reduced disease recurrence and prolonged survival after standard oncologic treatment (2). Nonetheless, the formation of clinically evident tumors indicates a failure of host defense, and elucidating the pathways that underlie tumor escape is critical to crafting therapeutic strategies that consistently stimulate protective immunity.
One mechanism that facilitates tumor progression is insufficient tumor antigen presentation (3). Tumor cells usually fail to express costimulatory molecules required for efficient lymphocyte priming, and immunosuppressive factors in the tumor microenvironment impede the maturation of infiltrating dendritic cells. Together, these conditions favor the production of regulatory T cells and functionally impaired effector T cells. To ameliorate these defects, several groups have devised therapeutic strategies that enhance dendritic cell-mediated tumor antigen presentation. Among these approaches, vaccination with irradiated tumor cells engineered to secrete granulocyte–macrophage colony-stimulating factor (GM-CSF) improves the ability of CD11b+ dendritic cells to capture and present cancer antigens to tumor-reactive CD4+ and CD8+ T cells, CD1d-restricted invariant natural killer (NK) T cells, and B cells (4). Several early-stage clinical trials of this immunization scheme revealed the induction of a coordinated humoral and cellular antitumor response in patients with advanced melanomas or carcinomas of the lung, kidney, prostate, ovary, or pancreas.
Although a minority of vaccinated subjects manifested prolonged survival, most eventually succumbed to progressive disease, implying the existence of additional immune defects. In this context, an important role has emerged for cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) in attenuating antitumor responses (5). Whereas the triggering of CD28 by B7-1 or B7-2 contributes to T cell activation, the engagement of CTLA-4 by these ligands results in cell cycle arrest and diminished cytokine production. In murine models, antibody blockade of CTLA-4 enhances tumor rejection, especially in conjunction with cancer vaccines. Moreover, we showed that the administration of a fully human CTLA-4 blocking antibody (MDX-010) to advanced melanoma and ovarian carcinoma patients previously immunized with irradiated, autologous, GM-CSF secreting tumor cells effectuated significant tumor destruction, albeit with a modest compromise of tolerance to normal tissues (6). Similar to the gene-modified whole tumor cell vaccines, effective CTLA-4 antibody blockade elicited dense T and B cell infiltrates in metastatic lesions.
The detailed analysis of subjects achieving durable responses on these clinical trials should yield insights into the targets and mechanisms of protective tumor immunity. Here, we used antibody-based screening of tumor-derived cDNA expression libraries to examine an advanced melanoma patient who achieved a sustained clinical response to CTLA-4 antibody blockade after vaccination with irradiated, autologous, GM-CSF secreting tumor cells.
Results
Clinical Course.
MEL15 is a 48-year-old female who had a primary melanoma removed in 2000. Four years later, she developed abdominal pain and was found to harbor multiple lung and pleural-based nodules, primarily left-sided, that were biopsy proved as metastatic disease. She underwent thoracotomy on protocol to harvest tissue for autologous, GM-CSF secreting tumor cell vaccine manufacture and received six immunizations (the first three at weekly intervals and the last three at every 2 weeks) during May and June 2004. Vaccination evoked strong local reactions and delayed-type hypersensitivity responses to injections of irradiated, autologous, unmodified melanoma cells, but thoracic computerized tomography (CT) scans at restaging disclosed slightly enlarged pulmonary metastases.
In August 2004, MEL15 complained of significant left-sided chest pain radiating to the left shoulder and neck, likely referred from the pleural metastases, and narcotic analgesia was instituted. Treatment on protocol with the fully human anti-CTLA-4 monoclonal antibody (MDX-010) at 3 mg/kg was begun, and 1 month later MEL15 reported a marked improvement in the referred pain. Repeat computerized tomography (CT) scans in September 2004, 2 months after the initiation of CTLA-4 antibody blockade, demonstrated a mixed response with a slight increase in some lung masses (<10%) but a reduction in the pleural-based lesions. MEL15 received additional MDX-010 infusions at 2-month intervals (for a total of nine treatments as of March 2006), with complete resolution of the pain and requirement for analgesia. Her lung disease steadily improved, with the most recent CT scans documenting a >50% reduction in the size of all lesions. Toxicities of therapy were limited to a mild erythematous rash, reflecting the development of lymphocyte infiltrates in the superficial dermis with some T cell apposition to normal melanocytes.
Serologic Response to MHC Class I Chain-Related Protein A (MICA).
Sera obtained from MEL15 after MDX-010 infusion were used to screen two melanoma cDNA expression libraries previously constructed from heavily infiltrated metastases of patients achieving long-term responses to autologous, GM-CSF secreting tumor vaccines. This investigation yielded 16 gene products, including CD63, macrophage migration inhibitory factor (MIF), and galectin-3, which likely contribute to melanoma biology and/or host defense (Table 1, which is published as supporting information on the PNAS web site). Nonetheless, MICA was selected for detailed study, given the importance of NKG2D in tumor suppression (7). MICA is detected in some normal gastrointestinal epithelial cells and thymocytes (8), but double-stranded DNA breaks trigger high-level expression in a broad range of human cancers (9).
To examine the humoral response to MICA more fully, we established an ELISA with recombinant protein (Fig. 1). Unexpectedly, MEL15 harbored anti-MICA antibodies before initiating vaccination, likely indicative of a nascent host reaction, because sera from 20 healthy controls failed to recognize the protein (data not shown). Vaccination modestly influenced anti-MICA antibody titers, but CTLA-4 blockade intensified the humoral immunity, which was sustained with continued treatment. Additional analysis revealed that IgG2 antibodies constituted the dominant anti-MICA Ig subclass (data not shown).
Fig. 1.

CTLA-4 blockade after autologous tumor vaccination elicited a potent humoral reaction to MICA that was temporally associated with a reduction in sMICA. (Upper) Longitudinal sera samples from MEL15 were diluted 1:500 and analyzed by ELISA with recombinant MICA protein. Reactivity was determined with a pan-IgG secondary. Downward arrows denote tumor cell vaccinations and upward arrows depict infusions of MDX-010. (Lower) Serial sMICA levels were measured with a sandwich ELISA.
Because tumor cell shedding of soluble MICA (sMICA) impairs host defense (10), we wondered whether the increase in anti-MICA antibodies might be correlated with alterations in sMICA levels. Although high sMICA was present on study entry and during vaccination, CTLA-4 antibody blockade resulted in a sharp decrease in sMICA that was temporally linked to the rise in anti-MICA antibodies (Fig. 1). The reduction in sMICA was confirmed with Western blot analysis of MEL15 sera by using an anti-MICA monoclonal antibody (data not shown). Although these findings highlight an association between anti-MICA antibodies and sMICA, further investigations are required to clarify whether immune complexes were formed in vivo and cleared from the circulation.
Enhanced Innate Antitumor Immunity.
Because sMICA compromises NK cell function (11), we examined whether CTLA-4 blockade modulated innate antitumor responses. CD56+ NK cells were purified from healthy donors, cultured for 48 h in various sera, and analyzed with flow cytometry (Fig. 2A). Sera from normal donors or vaccinated melanoma patients (M2 and M9) without detectable sMICA (limits of detection 90 pg/ml) did not influence NKG2D expression. However, sera obtained from MEL15 during vaccination diminished NKG2D surface levels, whereas sera collected after MDX-010 infusion did not. The addition of anti-MICA monoclonal antibodies to MEL15 early sera blocked the decrease in NKG2D expression, suggesting that sMICA and not TGF-β (or other factors) was the dominant suppressive mechanism. In accordance with this finding, late MEL15 sera also antagonized the NKG2D down-regulation triggered with early sera (data not shown).
Fig. 2.

Therapy-induced anti-MICA antibodies antagonized sMICA suppression of innate immune responses. (A) Normal donor PBMCs were incubated for 48 h in sera, and NKG2D expression on gated NK cells (CD56+ and CD3−) was determined with flow cytometry. Sera samples were obtained from a healthy donor, vaccinated melanoma patients without sMICA (M2 and M9), and MEL15 during immunization or after CTLA-4 antibody blockade. Anti-MICA monoclonal antibodies or isotype controls were added where indicated. (B) Healthy donor PBMCs were incubated in normal or MEL15 sera for 48 h and washed; NK cells were then purified with magnetic bead selection and tested for lytic activity against 51Cr-labeled K562 targets. The NKG2D-dependent lysis was determined with the addition of the mAb 1D11 (anti-NKG2D) or isotype control (IgG).
Donor NK cells exposed to normal sera efficiently lysed K562 cells, which was substantially blocked with anti-NKG2D antibodies, indicating a major role for NKG2D in target cell recognition (Fig. 2B). However, NK cells incubated in MEL15 early sera showed impaired K562 killing, whereas NK cells cultured in MEL15 late sera manifested NKG2D-dependent lysis that was comparable with healthy controls. NK cells incubated in a mixture of MEL15 early and late sera also displayed robust NKG2D-dependent killing, illustrating that high titer anti-MICA antibodies neutralized the deleterious effects of sMICA.
Consistent with these studies, NKG2D levels were reduced on CD56+ cells obtained from MEL15 during vaccination compared with those collected after CTLA-4 antibody blockade (Fig. 3A). Moreover, MEL15 NK cells collected during vaccination displayed decreased killing, whereas NK cells obtained after MDX-010 infusion mediated NKG2D-dependent lysis at levels that were nearly equivalent to normal controls (Fig. 3B). MEL15 CD56+ cells collected after CTLA-4 blockade efficiently killed K562 cells in the presence of late sera (Fig. 7, which is published as supporting information on the PNAS web site), indicating that high titer anti-MICA antibodies did not interfere with target cell lysis.
Fig. 3.

Immunotherapy restored protective antitumor innate responses in MEL15. (A) PBMCs were obtained from MEL15 during vaccination or after CTLA-4 blockade, and NKG2D expression on gated NK cells was determined with flow cytometry. (B) Magnetic bead-purified healthy donor or MEL15 NK cells obtained at different times were tested for lytic activity against 51Cr-labeled K562 targets.
Enhanced Adaptive Antitumor Immunity.
Whereas sMICA inhibits CD8+ T lymphocyte cytotoxicity through down-regulation of NKG2D (10), anti-MICA antibodies promote dendritic cell-mediated cross-presentation of tumor cells (12). To explore these mechanisms in MEL15, we purified CD8+ T cells from healthy donors, incubated them for 48 h in various sera, and then performed flow cytometry (Fig. 4A). Compared with freshly isolated cells, NKG2D expression was modestly decreased on CD8+ T lymphocytes incubated in sera from healthy donors or vaccinated melanoma patients without detectable sMICA. However, sera obtained from MEL15 during vaccination evoked significantly greater reductions in NKG2D levels, whereas sera collected after MDX-010 infusion proved equivalent to controls. The addition of anti-MICA monoclonal antibodies or MEL15 late sera (data not shown) to MEL15 early sera blocked the decrease in NKG2D expression, establishing sMICA as the primary suppressive factor.
Fig. 4.

Therapy-induced anti-MICA antibodies antagonized sMICA suppression of adaptive immune responses and enhanced MICA-dependent cross-presentation. (A) Normal donor PBMCs were incubated for 48 h in sera, and NKG2D expression on gated CD8+ T cells (CD8+ and CD3+) was determined with flow cytometry. Sera samples were obtained from a healthy donor, vaccinated melanoma patients without sMICA (M2 and M9), and MEL15 during immunization or after CTLA-4 antibody blockade. Anti-MICA monoclonal antibodies or isotype controls were added where indicated. (B) Dendritic cells were generated from adherent PBMCs of HLA-A2.1+ donors, loaded with MEL15 early or late sera-coated MEL15-T or MEL15-T-MICA tumor cells, matured with LPS, and used to stimulate autologous purified CD8+ T cells for 7 days. IFN-γ production was measured by ELISPOT against the indicated targets. No reactivity against unpulsed dendritic cells or dendritic cells loaded with unopsonized tumors was observed (data not shown). (C) HLA-A2.1+ dendritic cells were loaded with MEL15 late sera-coated K008-T melanoma cells (MICA+), LPS matured, and used to stimulate autologous CD8+ T cells in the presence of early or late MEL15 sera. IFN-γ production was measured by ELISPOT against the indicated targets.
To evaluate the ability of therapy-induced anti-MICA antibodies to enhance cross-presentation, we established an autologous melanoma cell line (MEL15-T) from the pulmonary metastasis resected for vaccine manufacture. Although MEL15-T cells displayed only low levels of MICA during routine culture, γ irradiation augmented expression (data not shown), consistent with previous work linking MICA to the DNA damage response (9). To optimize the analysis, however, we used retroviral-mediated gene transfer to engineer MEL15-T cells with stable, high-level MICA expression (MEL15-T-MICA).
Because MEL15 is HLA-A2−, we tested the ability of HLA-A2+ healthy donors to cross-present opsonized MEL15-T and MEL15-T-MICA cells. Dendritic cells were generated from HLA-A2+ normal donors by culturing peripheral blood monocytes in GM-CSF and IL-4. The expanded dendritic cells were pulsed with sera-coated tumor cells, matured with LPS, and used to stimulate purified donor CD8+ T cells. Melanoma-specific IFN-γ production was assessed by ELISPOT. MEL15 sera obtained after MDX-010 infusion mediated more efficient MICA-dependent cross-presentation of melanoma antigens than MEL15 sera collected during vaccination (Fig. 4B). This stimulation resulted in HLA-A2-restricted CD8+ T cell responses to peptides derived from MART-1, gp100, and tyrosinase, and low-level recognition of dendritic cells loaded with MEL15 late sera-coated M34-T melanoma cells (HLA-A2+). In contrast, minimal reactivity was induced against K562 cells or dendritic cells loaded with unopsonized tumor cells (data not shown). MEL15 sera obtained after CTLA-4 blockade also enhanced the cross-presentation of MEL15-T cells, albeit to a lesser extent.
This cross-presentation scheme was then used to evaluate whether the sMICA-induced down-regulation of NKG2D impaired the generation of tumor-specific CD8+ T lymphocytes. For these studies, K008-T melanoma cells, which constitutively express high levels of MICA (data not shown), were opsonized with MEL15 sera obtained after CTLA-4 blockade and loaded onto dendritic cells. After maturation with LPS, the dendritic cells and CD8+ T cells were cocultured in the presence of either MEL15 sera obtained during vaccination or MEL15 sera collected after CTLA-4 blockade. MEL15 early, but not late, sera markedly inhibited the development of CD8+ T cell responses to opsonized K008-T cells and melanosomal differentiation antigens (Fig. 4C), illustrating the potent suppressive effects of sMICA on adaptive cellular immunity.
Consistent with these studies, NKG2D levels were substantially decreased on CD8+ T cells collected from MEL15 during vaccination relative to those obtained after MDX-010 infusion (Fig. 5A). Moreover, MEL15 samples collected after CTLA-4 blockade manifested much greater cross-presentation of K008-T cells compared with samples obtained during vaccination (Fig. 5B), resulting in enhanced melanoma inhibitor of apoptosis protein (ML-IAP)-specific reactions (13). Augmented CD8+ T cell responses after MDX-010 infusion were also evident without in vitro stimulation (Fig. 5C).
Fig. 5.

Immunotherapy restored protective antitumor innate responses and enhanced cross-presentation in MEL15. (A) PBMCs were obtained from MEL15 during vaccination or after CTLA-4 blockade, and NKG2D expression on gated CD8+ T cells was determined with flow cytometry. (B) PBMCs were obtained from MEL15 during vaccination or after CTLA-4 blockade and used to generate dendritic cells. These cells were loaded with K008-T melanoma cells coated in early or late MEL15 sera and then matured with LPS. Purified CD8+ T cells from the same time points were then stimulated in vitro with the respective tumor-loaded dendritic cells for 7 days. IFN-γ production was measured by ELISPOT against the indicated targets. (C) MEL15 CD8+ T cells were purified from PBMCs collected during vaccination or after CTLA-4 blockade and tested for IFN-γ production against the indicated targets without prior in vitro stimulation. Melanoma inhibitor of apoptosis protein (ML-IAP) is a previously characterized tumor rejection antigen (13).
The increased cross-presentation and CD8+ T cell function in MEL15 raised the possibility that CTLA-4 blockade might evoke a diversification of antigen recognition. To explore this idea, we rescreened the K008 melanoma cDNA expression library with sera obtained at study enrollment and after vaccination but before MDX-010 administration. In contrast to the 12 gene products identified in this library with sera collected after CTLA-4 blockade, these screens yielded only four targets each (Table 1). The greater number of antigens that elicited IgG antibody responses after MDX-010 infusion is consistent with a spreading of T cell reactivity, although further studies are required to characterize the CD4+ and CD8+ T cell responses to these antigens.
Vaccine-Induced Anti-MICA Antibodies.
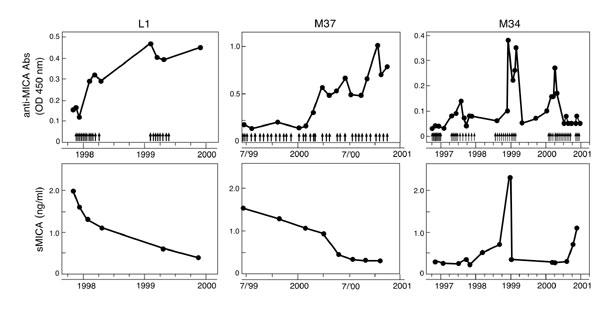
Although CTLA-4 blockade triggered the development of potent anti-MICA humoral immunity in MEL15, we wondered whether vaccination alone might evoke a similar response in some patients. We thus examined 14 additional metastatic melanoma or nonsmall cell lung carcinoma patients who were immunized with irradiated, autologous, GM-CSF secreting tumor cells. On study entry, ten patients harbored anti-MICA antibodies, and nine of these patients manifested circulating sMICA (data not shown). Longitudinal analysis disclosed that vaccination augmented anti-MICA antibody titers that were temporally associated with decreases in sMICA in three cases, each of whom demonstrated pathologic and/or clinical evidence of antitumor activity (Fig. 8, which is published as supporting information on the PNAS web site). In contrast, sMICA levels remained constant or rose in those subjects that did not display increased anti-MICA antibodies.
The vaccine-induced anti-MICA antibodies and decreased sMICA manifested similar biologic activities as were observed in MEL15. In particular, prevaccination sera down-regulated NKG2D levels and inhibited lytic activity of purified CD56+ cells from healthy donors, whereas late sera antagonized these suppressive effects (Fig. 9, which is published as supporting information on the PNAS web site). Further, early sera diminished NKG2D levels on healthy donor CD8+ T cells, whereas postvaccination sera promoted the efficient cross-presentation of MICA expressing melanoma cells (Fig. 10, which is published as supporting information on the PNAS web site).
Lastly, we examined whether the immunotherapy-induced anti-MICA antibodies might accomplish tumor lysis through complement fixation. For these studies, 293 embryonic kidney cells were engineered to express high levels of MICA (wild-type cells show minimal expression). Using this matched set of targets, we found that anti-MICA antibodies stimulated by autologous, GM-CSF secreting tumor cell vaccinations or CTLA-4 blockade mediated MICA-specific cytotoxicity (Fig. 6). In contrast, sera from an immunized melanoma patient without anti-MICA antibodies (M35) failed to induce MICA-dependent lysis.
Fig. 6.

Immunotherapy-induced anti-MICA antibodies mediate complement-dependent lysis. 293T and 293T-MICA embryonic kidney cells were incubated in patient sera and complement, and cell viability was determined with a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay after 6 h. (Upper) Prevaccination sera. (Lower) Postvaccination sera. M35 was a vaccinated, long-term surviving metastatic melanoma patient (7 years) without anti-MICA antibodies. The differences were abrogated by heat inactivation of the complement.
Discussion
The analysis of patients who manifest clinically significant benefits from current immunotherapies provides one informative approach to delineating the requirements for effective tumor immunity. Here we studied an advanced melanoma patient who achieved a sustained tumor response to CTLA-4 antibody blockade after vaccination with irradiated, GM-CSF secreting melanoma cells. By using sera collected after MDX-010 infusion to screen two melanoma cDNA expression libraries, we identified the NKG2D ligand MICA as a target of high titer antibodies. This humoral reaction contributed to the antitumor effects by antagonizing immune suppression and promoting antitumor cytotoxicity. Additional investigations revealed that some patients who responded to vaccination with irradiated, GM-CSF secreting autologous tumor cells alone also developed anti-MICA antibodies with similar functional properties.
MICA is a MHC class I-related molecule that does not bind CD8 and exhibits stable surface expression without peptide loading or β2-microglobulin association (14). Along with MHC class I-related protein B (MICB) and four UL16-binding proteins, MICA activates NKG2D signaling through DNAX-activation protein 10 (DAP10) in human NK cells, γδ T cells, and CD8+ αβ T cells (14–16). Substantial evidence implicates a central role for the NKG2D pathway in cancer suppression (7). Experimental tumors engineered to express the murine NKG2D ligand Rae1 are rejected in wild-type animals through the coordinated activities of NK cells, CD8+ T lymphocytes, and perforin (17–20), whereas the administration of anti-NKG2D blocking antibodies increases susceptibility to chemical carcinogens (21). Although human cancers frequently exhibit MICA, immune destruction is subverted, at least in part, through ligand shedding, which promotes NKG2D internalization in NK cells and CD8+ T lymphocytes (10, 22). Persistent NKG2D ligand expression rather than transformation per se appears to underlie shedding, because transgenic mice engineered for constitutive production similarly generate soluble ligand and manifest immune dysfunction in the absence of cancer (23, 24).
The presence of anti-MICA antibodies proved uncommon among healthy donors, but a majority of patients with advanced melanoma or nonsmall cell lung carcinoma displayed humoral reactions. Almost all of these subjects also harbored sMICA, suggesting that ligand shedding might elicit antibody production. Although these endogenous responses were insufficient to overcome immune suppression or impede disease progression, we identified four patients who mounted increased anti-MICA antibodies as a consequence of CTLA-4 blockade or vaccination with irradiated, GM-CSF secreting tumor cells. In each case, the intensified humoral reaction was correlated with a decrease in sMICA, a restoration of NK cell and CD8+ T lymphocyte function, and clinically significant tumor destruction.
Not all patients who responded to CTLA-4 blockade or vaccination with irradiated, GM-CSF secreting tumor cells, however, developed augmented humoral reactions to MICA (data not shown). As sMICA was present in some of these subjects, other mechanisms might override immune suppression. Indeed, in rheumatoid arthritis or celiac disease, sMICA fails to down-regulate NKG2D, perhaps reflecting the activities of TNF-α or IL-15 (25, 26). The delineation of alternative pathways for antagonizing the deleterious effects of sMICA in cancer patients remains an important issue for further investigation.
In addition to countering immune suppression, the therapy-induced anti-MICA antibodies enhanced the dendritic cell cross-presentation of tumor antigens, yielding more robust antimelanoma CD8+ T lymphocyte responses. The anti-MICA humoral reactions also mediated efficient complement-dependent tumor lysis, which could provide another source of antigen for dendritic cell capture. The improvement in cross-presentation was temporally associated with a diversification of target recognition. Because preclinical experiments suggest that the most potent cancer rejection antigens might be tumor-specific, mutated proteins (27), the ability to immunize against autologous cancer cells is likely to prove advantageous for clinical immunotherapy. Our findings thus raise the intriguing possibility that anti-MICA antibodies might contribute to dendritic cell priming of tumor-specific T cells in vivo. In this scenario, a gene product induced on the tumor cell surface in response to DNA damage might function as a link to the adaptive immune recognition of a large repertoire of neoantigens arising from genomic instability.
Finally, our demonstration that the clinical activity of CTLA-4 blockade and irradiated, GM-CSF secreting tumor cell vaccines involves, at least in part, the reversal of sMICA-induced immune suppression and the stimulation of antitumor cytotoxicity should advance the development of anti-MICA monoclonal antibodies as cancer therapy. These reagents might be effectively used in combination with dendritic cell activating signals and/or treatments that up-regulate NKG2D ligand expression consequent to DNA damage. Moreover, anti-MICA monoclonal antibodies might increase the proportion of patients who benefit from CTLA-4 inhibition or cancer vaccination.
Materials and Methods
Clinical Protocols.
Sera, lymphocytes, and tumor samples were obtained from patients enrolled on Institutional Review Board/Food and Drug Administration/Recombinant DNA Advisory Committee-approved Dana-Farber Partners Cancer Care clinical protocols.
Serology.
The K008 and M34 melanoma cDNA expression libraries were generated and screened as described (28). Anti-MICA antibodies and sMICA levels were measured with ELISAs by using recombinant MICA protein (ProSpec-Tany; TechnoGene, Rehovot, Israel) and anti-human MICA monoclonal antibodies (R & D Systems). For complement assays, 293T and 293T MICA cells, generated by retroviral mediated gene transfer (13), were incubated with patient sera and human complement (Sigma-Aldrich) and lysis quantified by using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) (Roche Diagnostics).
Flow Cytometry.
Peripheral blood mononuclear cells (PBMCs) from healthy donors or patients were incubated for 48 h in patient or control sera (10%) in complete media stained with phycoerythrin (PE)-conjugated anti-NKG2D mAb (Pharmingen), FITC-conjugated anti-CD3 mAb (BD Biosciences Pharmingen), and either PC5-conjugated anti-CD8 mAb or PC5-conjugared anti-CD56 mAb (Beckman-Coulter), and analyzed with a FW501 flow cytometer (Beckman-Coulter) and flowjo software (Tree Star, San Carlos, CA). Cells were gated for CD3+CD8+ T cells and CD3−CD56+ NK cells.
Cellular Assays.
Donor PBMCs were incubated in 10% patient sera as described earlier, and NK cells, isolated by magnetic cell sorting (Miltenyi Biotec, Auburn, CA), were tested in 4-h lysis assays against 51Cr-labeled K562 target cells. Dendritic cells (generated from adherent PBMCs with GM-CSF and IL-4) were cocultured with opsonized tumor cells (1:1 ratio) for 20 h, matured with LPS (Sigma-Aldrich) for 24 h, and then used to stimulate autologous CD8+ T cells, isolated by magnetic cell sorting, for 7 days. Antigen-specific IFN-γ production was then determined by ELISPOT, as described (13). HLA-A2-restricted peptides were derived from MART-1 (M27; AAGIGILTV), gp100 (G154; KTWGQYWQV), and tyrosinase (368D: YMDGTMSQV) (12).
Supplementary Material
Acknowledgments
We thank the members of the Cell Manipulation Core Facility (CMCF) laboratory for sample processing and Jerry Ritz and Ellis Reinherz for reviewing the manuscript. This work was supported by National Institutes of Health Grants CA111506, CA78378, CA092625, and CA66996.
Abbreviations
- CTLA-4
cytotoxic T lymphocyte-associated antigen 4
- GM-CSF
granulocyte-macrophage colony-stimulating factor
- MICA
MHC class I chain-related protein A
- sMICA
soluble MICA
- NK
natural killer
- PBMC
peripheral blood mononuclear cell.
Footnotes
Conflict of interest statement: No conflicts declared.
References
- 1.Dunn G. P., Old L. J., Schreiber R. D. Immunity. 2004;21:137–148. doi: 10.1016/j.immuni.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 2.Sato E., Olson S. H., Ahn J., Bundy B., Nishikawa H., Qian F., Jungbluth A. A., Frosina D., Gnjatic S., Ambrosone C., et al. Proc. Natl. Acad. Sci. USA. 2005;102:18538–18543. doi: 10.1073/pnas.0509182102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dranoff G. Nat. Rev. Cancer. 2004;4:11–22. doi: 10.1038/nrc1252. [DOI] [PubMed] [Google Scholar]
- 4.Hodi F. S., Dranoff G. Adv. Immunol. 2006;90:337–360. doi: 10.1016/S0065-2776(06)90009-1. [DOI] [PubMed] [Google Scholar]
- 5.Korman A., Peggs K., Allison J. P. Adv. Immunol. 2006;90:293–335. doi: 10.1016/S0065-2776(06)90008-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hodi F. S., Mihm M. C., Soiffer R. J., Haluska F. G., Butler M., Seiden M. V., Davis T., Henry-Spires R., MacRae S., Willman A., et al. Proc. Natl. Acad. Sci. USA. 2003;100:4712–4717. doi: 10.1073/pnas.0830997100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diefenbach A., Raulet D. Immunol. Rev. 2002;188:9–21. doi: 10.1034/j.1600-065x.2002.18802.x. [DOI] [PubMed] [Google Scholar]
- 8.Groh V., Bahram S., Bauer S., Herman A., Beauchamp M., Spies T. Proc. Natl. Acad. Sci. USA. 1996;93:12445–12450. doi: 10.1073/pnas.93.22.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gasser S., Orsulic S., Brown E. J., Raulet D. H. Nature. 2005;436:1186–1190. doi: 10.1038/nature03884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Groh V., Wu J., Yee C., Spies T. Nature. 2002;419:734–738. doi: 10.1038/nature01112. [DOI] [PubMed] [Google Scholar]
- 11.Doubrovina E. S., Doubrovin M. M., Vider E., Sisson R. B., O'Reilly R. J., Dupont B., Vyas Y. M. J. Immunol. 2003;171:6891–6899. doi: 10.4049/jimmunol.171.12.6891. [DOI] [PubMed] [Google Scholar]
- 12.Groh V., Li Y. Q., Cioca D., Hunder N. N., Wang W., Riddell S. R., Yee C., Spies T. Proc. Natl. Acad. Sci. USA. 2005;102:6461–6466. doi: 10.1073/pnas.0501953102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmollinger J. C., Vonderheide R. H., Hoar K. M., Maecker B., Schultze J. L., Hodi F. S., Soiffer R. J., Jung K., Kuroda M. J., Letvin N. L., et al. Proc. Natl. Acad. Sci. USA. 2003;100:3398–3403. doi: 10.1073/pnas.0530311100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Groh V., Steinle A., Bauer S., Spies T. Science. 1998;279:1737–1740. doi: 10.1126/science.279.5357.1737. [DOI] [PubMed] [Google Scholar]
- 15.Bauer S., Groh V., Wu J., Steinle A., Phillips J. H., Lanier L. L., Spies T. Science. 1999;285:727–729. [PubMed] [Google Scholar]
- 16.Wu J., Song Y., Bakker A. B., Bauer S., Spies T., Lanier L. L., Phillips J. H. Science. 1999;285:730–732. doi: 10.1126/science.285.5428.730. [DOI] [PubMed] [Google Scholar]
- 17.Jamieson A. M., Diefenbach A., McMahon C. W., Xiong N., Carlyle J. R., Raulet D. H. Immunity. 2002;17:19–29. doi: 10.1016/s1074-7613(02)00333-3. [DOI] [PubMed] [Google Scholar]
- 18.Diefenbach A., Jamieson A. M., Liu S. D., Shastri N., Raulet D. H. Nat. Immunol. 2000;1:119–126. doi: 10.1038/77793. [DOI] [PubMed] [Google Scholar]
- 19.Diefenbach A., Jensen E. R., Jamieson A. M., Raulet D. H. Nature. 2001;413:165–171. doi: 10.1038/35093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cerwenka A., Baron J. L., Lanier L. L. Proc. Natl. Acad. Sci. USA. 2001;98:11521–11526. doi: 10.1073/pnas.201238598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smyth M. J., Swann J., Cretney E., Zerafa N., Yokoyama W. M., Hayakawa Y. J. Exp. Med. 2005;202:583–588. doi: 10.1084/jem.20050994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salih H. R., Rammensee H. G., Steinle A. J. Immunol. 2002;169:4098–4102. doi: 10.4049/jimmunol.169.8.4098. [DOI] [PubMed] [Google Scholar]
- 23.Oppenheim D. E., Roberts S. J., Clarke S. L., Filler R., Lewis J. M., Tigelaar R. E., Girardi M., Hayday A. C. Nat. Immunol. 2005;6:928–937. doi: 10.1038/ni1239. [DOI] [PubMed] [Google Scholar]
- 24.Wiemann K., Mittrucker H. W., Feger U., Welte S. A., Yokoyama W. M., Spies T., Rammensee H. G., Steinle A. J. Immunol. 2005;175:720–729. doi: 10.4049/jimmunol.175.2.720. [DOI] [PubMed] [Google Scholar]
- 25.Groh V., Bruhl A., El-Gabalawy H., Nelson J. L., Spies T. Proc. Natl. Acad. Sci. USA. 2003;100:9452–9457. doi: 10.1073/pnas.1632807100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meresse B., Chen Z., Ciszewski C., Tretiakova M., Bhagat G., Krausz T. N., Raulet D. H., Lanier L. L., Groh V., Spies T., et al. Immunity. 2004;21:357–366. doi: 10.1016/j.immuni.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 27.Basombrio M. A. Cancer Res. 1970;30:2458–2462. [PubMed] [Google Scholar]
- 28.Hodi F. S., Schmollinger J. C., Soiffer R. J., Salgia R., Lynch T., Ritz J., Alyea E. P., Yang J. C., Neuberg D., Mihm M., et al. Proc. Natl. Acad. Sci. USA. 2002;99:6919–6924. doi: 10.1073/pnas.102025999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}