

Abstract
Receptor-mediated endocytosis of ligands, such as transferrin and LDL, is suppressed when clathrin synthesis is blocked by RNA interference in HeLa cells. We have found that domains containing the adapter complex 2 (AP2)-coated vesicle adapter and the endocytic accessory proteins CALM (clathrin assembly lymphoid myeloid leukemia protein), epsin, and eps15/eps15R (EGF receptor pathway substrate 15-related) nevertheless persist at the plasma membrane. They are similar in size and number to those seen in clathrin-expressing cells. Here we characterize these membrane domains by fluorescence and electron microscopy in detail. Fluorescence recovery after photobleaching measurements suggest that the exchange between membrane-bound and free cytosolic AP2 molecules is not significantly influenced by the depletion of clathrin. The AP2 membrane domains are dispersed upon interfering with protein–protein interactions that involve the α appendage domain of AP2. Electron microscopy of cellular cortices revealed that the AP2 membrane domains lack any curvature, suggesting that clathrin is essential for driving coated pit invagination. A model for coated vesicle formation, incorporating a mechanism commonly referred to as a “Brownian ratchet,” is consistent with our observations.
Keywords: adapter complex 2, Brownian ratchet, electron microscopy, endocytosis, RNA interference
Possible mechanisms leading to the deformation of membranes have been recently widely discussed (1–6). The archetypal example is the formation of clathrin-coated vesicles, the principal agents of endocytic membrane transport, at the plasma membranes of eukaryotic cells. The life cycle of such vesicles begins with the recruitment of adapter complex 2 (AP2) adapters, accessory proteins, and clathrin to phosphatidylinositol-4,5-bisphosphate (PtdIns-4,5-P2)-enriched plasma membrane regions. This step is presumed to be coordinated with cargo selection and binding (7). Numerous accessory proteins that interact with AP2 and clathrin have been identified in recent years (8). Like AP2, most of them are also capable of binding to PtdIns-4,5-P2. Clathrin assembly lymphoid myeloid leukemia protein (CALM), epsin, and eps15/eps15R (EGF receptor pathway substrate 15-related) are the ones most likely to be involved in early stages of coat formation. All three bind to the appendage domains of the α- and β-subunits of AP2, and the first two also associate with clathrin. Epsin and CALM possess an N-terminally located PtdIns-4,5-P2 binding module, referred to as epsin N-terminal homology and AP180 N-terminal homology domain, respectively, next to which are flexible segments, containing multiple short binding motifs for clathrin, AP2, and eps15/eps15R. In addition, epsin contains ubiquitin-binding motifs (9). Because CALM and epsin will also bind to PtdIns-4,5-P2-containing monolayers, these proteins may also associate with the plasma membrane independent of AP2 (10, 11). By means of their unstructured segments, epsin and CALM have been conjectured to capture additional AP2 adapters and clathrin from the cytosol and may coordinate their assembly into membrane coats (12). Recent studies on HeLa cells depleted of CALM by RNA interference, in which abnormally shaped coated buds were observed at the plasma membrane, support a role of CALM in the coordination of coat assembly (13). Whether the depletion of epsin has similar consequences is not as yet known, but there is evidence that the interaction of the epsin N-terminal homology domain with membranes induces membrane curvature, suggesting that epsin might aid plasma membrane invagination and bud formation (11). Similarly, the respective Bin1/Amphiphysin/Rvs167 (BAR) domains of amphiphysin and endophilin bind preferentially to convexly curved membranes, and at high concentration these proteins are even capable of inducing membrane tube formation in vivo and in vitro (14–16). The family of BAR domain proteins was recently extended to include membrane tubule-forming proteins that possess an N-terminal BAR-related module dubbed F-BAR domain (5). At least three of them appear to be required for efficient transferrin uptake (5). The function of amphiphysin and endophilin is probably linked to the recruitment of dynamin to invaginated pits as a prelude to the scission event that releases a coated vesicle from the plasma membrane.
Electron microscopy has captured images of clathrin lattices at the plasma membrane, showing intermediates of the lattice that range from flat to deeply invaginated (17). Clathrin triskelia will readily polymerize in vitro under physiological conditions in the presence of accessory proteins into closed baskets, implying that this structure constitutes its energetically most stable state. Thus, clathrin could drive membrane deformation, or it could equally well serve as a flexible or polymorphic scaffold for adapters and accessory proteins that constitute the agents of deformation (1). The highly dynamic behavior of the molecular components of clathrin-coated structures revealed by fluorescence recovery after photobleaching (FRAP) measurements in living cells is consistent with both models (18, 19). A functional actin cytoskeleton was shown to be essential for endocytosis in yeast (20), but a coherent view of the role of actin in clathrin-dependent endocytosis in mammalian cells has not yet emerged. However, there is general agreement that actin polymerization at the neck of constricted coated vesicles pushes coated pits and nascent coated vesicles away from the plasma membrane and into the cytosol (21). Moreover, there is recent evidence that actin is also involved in the formation of new coated pits, in their lateral movement in the plane of the plasma membrane, and in constricting the neck of budding coated vesicles (22). On the other hand, the transition from flat to invaginated coated pits does not seem to implicate actin (22). Here we have set out to clarify the processes that lead to assembly of the coated pit and, in particular, govern the critical invagination step.
Results and Discussion
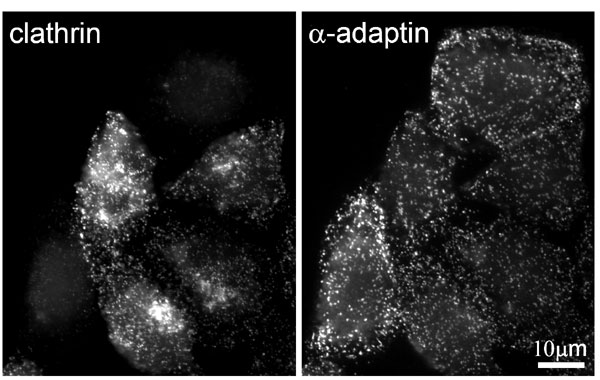
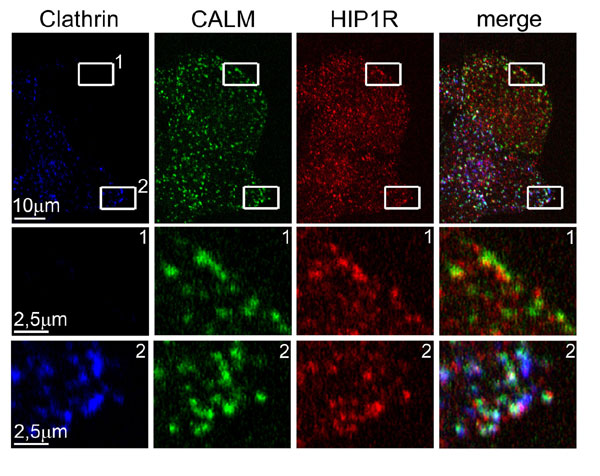
Transfection of HeLa cells with small interfering RNA (siRNA) specific for clathrin heavy chains acts within 72 h to suppress the cellular concentration of the heavy chains by >80% (Fig. 1A) (23, 24). Whereas the clathrin light chains are also depleted (23, 25), the cellular concentrations of AP2 and the accessory proteins epsin, CALM, and eps15 were hardly affected (23–25). Moreover, the AP2 adapter, together with its accessory proteins, remained associated with the plasma membrane in puncta that, when viewed by fluorescent microscopy, are similar to those seen in cells with normal amounts of clathrin (23–25) (Fig. 6, which is published as supporting information on the PNAS web site). Another example is the protein eps15R, which colocalized precisely with AP2 and plasma membrane-associated clathrin (26), and, like epsin, it remained concentrated in puncta in the clathrin-depleted HeLa cells (Fig. 1 B and C). The proteins Hip1 and Hip1R are known to potentially link the clathrin coat to actin. Both proteins associate with membrane surfaces through their respective epsin N-terminal homology domains, and with a central helical domain they engage in homodimerization and heterodimerization (27, 28). In addition, Hip1 is capable of associating directly with the clathrin heavy chain, with AP2, and with actin, whereas Hip1R binds to clathrin light chains (28–31). The numerous potential interactions of Hip1 and Hip1R with clathrin coat components readily explain the partial overlap of Hip1R with coated structures at the plasma membrane (32) (Fig. 7, which is published as supporting information on the PNAS web site). In clathrin-depleted cells we did not observe any significant changes in the extent of the overlap between Hip1R and coat components, probably because Hip1R is still indirectly connected with AP2 through its interaction with Hip1 (Fig. 7).
Fig. 1.

Effect of clathrin depletion on the plasma membrane association of eps15R and epsin. HeLa cells were transfected with siRNA specific for the clathrin heavy chain. (A) Western blot of clathrin heavy chain-depleted HeLa cells 3 days after transfection. (B) eps15R-containing puncta at the plasma membrane after clathrin depletion. (C) Punctate epsin staining in clathrin-depleted cells. Forty-eight hours after transfection with siRNA the HeLa cells were replated on glass coverslips together with mock-transfected control cells and cultivated for another 24 h. The cells were subsequently processed for immunofluorescence by using X22 monoclonal antibody for clathrin and rabbit antisera for eps15R (26) and epsin (46). Note that the clathrin knockdown does not affect the punctate distribution of eps15R and epsin. (Scale bars: 10 μm.)
To determine whether the removal of clathrin affects the dynamics of the association of AP2 with the plasma membrane we used the FRAP technique, which was previously used to show that clathrin and AP2 constantly exchange between their membrane-associated states and the free cytosolic pool. Under physiological conditions the half-time for this exchange is 14–18 s (18, 19). We confirmed this exchange rate in cells transiently transfected with a β2 adaptin–yellow fluorescent protein (YFP) construct (data not shown). Next HeLa cells were transfected with clathrin heavy chain siRNA and 48 h later with the β2 adaptin–YFP construct. After another 24 h the exchange of β2 adaptin–YFP was analyzed by FRAP. Unexpectedly, we did not observe any significant differences in the exchange rate of AP2 (t1/2 for recovery 17 ± 3 s) upon clathrin depletion (Fig. 2). It follows that the stabilizing effect of clathrin on membrane-bound AP2 is too small to be discerned by the FRAP technique or that the rate of clathrin exchange is largely dictated by that of AP2.
Fig. 2.

AP2 exchange in clathrin-depleted HeLa cells. (A) Transiently expressed β2 adaptin colocalizes with clathrin at the plasma membrane, clathrin heavy chain clathrin (cla), and β2 adaptin–YFP (β-YFP). (B) Forty-eight hours after transfection with clathrin heavy chain siRNA the cells were mixed with control cells, replated, and then transfected with β2 adaptin (64). After 24 h the cells were immunostained for clathrin with X22 monoclonal antibody. The β2 adaptin is found in the clathrin-depleted cell in AP2 puncta at the plasma membrane. (Scale bars: 10 μm.) (C) Time course of recovery of β adaptin–YFP after photobleaching at 37°C. The data from the knockdown cells are the average of 15 scans obtained from seven cells. For the control, the data of eight scans from four different cells were averaged. Note that the recovery of the fluorescence was not affected by the clathrin knockdown. The half-time for the recovery (t1/2 = 17 ± 3 s) is similar to the one determined previously by others (19).
The observation that AP2 and its accessory proteins cluster at the plasma membrane in puncta raises the question of what holds them together and what determines the size of these domains. An obvious candidate is PtdIns-4,5-P2, which was shown in a perforated cell assay to be required for early and late events in coated vesicle formation (33). Moreover, the amount of AP2 associated with the plasma membrane showed a correlation with cellular levels of phosphatidylinositol-4-phosphate 5-kinase I (34). The observed increase in the association of AP2 with the plasma membrane upon overexpression of this kinase can thus be inferred to accompany the increased PtdIns-4,5-P2 concentration. The importance of PtdIns-4,5-P2 for clathrin-dependent endocytosis was also highlighted when the prenylated phosphatase domain of synaptojanin 1 was used to hydrolyze PtdIns-4,5-P2 specifically at the plasma membrane (35). The expression of this phosphatase domain in COS-7 cells reduced AP2 binding to the plasma membrane and consequently the uptake of transferrin and EGF (35). These observations strongly suggest that PtdIns-4,5-P2 is one necessary determinant for the formation of AP2 domains, but interprotein interactions would also be expected to contribute to the stability of these domains. The endocytic accessory proteins AP180, epsin, amphiphysin, and eps15 possess multiple short peptide motifs known to engage the appendage domains of both the α- and β-subunits of the AP2 adapter (36) and therefore are suited for crosslinking AP2 molecules at the plasma membrane. In addition, earlier work on purified AP2 showed that this adapter can undergo reversible self-association in physiological buffer solutions (37) and even causes the aggregation of membrane vesicles (38). This aggregation was shown to involve the α appendage domain of AP2 (38). To test the idea that the α appendage domain is involved in organizing the AP2 domains we microinjected recombinant appendage domain of the α-subunit of AP2 into control HeLa cells and into cells that had been depleted of clathrin by RNA interference. Introduction of the α appendage domain into control cells and clathrin-depleted cells caused a drastic reduction in the amount of membrane-associated AP2 (Fig. 3). We also examined epsin in the microinjected cells and noted that this protein clustered in what appeared to be large aggregates (Fig. 3). The sensitivity of AP2 domains to high cellular concentrations of the microinjected α appendage domain implies that lateral protein–protein interactions between AP2 molecules and/or AP2 and endocytic accessory proteins are required in addition to PtdIns-4,5-P2 for maintaining stable AP2 membrane domains.
Fig. 3.

Microinjection of α appendage domain interferes with protein–protein interactions in AP2 membrane domains. (A and B) Four HeLa cells were microinjected with GST α appendage (bright fluorescent cells in A and arrowheads in B), and after 3 h they were stained for AP2 with anti-α adaptin (A) or anti-β adaptin (B). Note that the microinjected cells are characterized by reduced membrane-associated AP2 when compared with control cells. (C and D) Displacement of AP2 from the plasma membrane upon clathrin knockdown and microinjection of GST α appendage. HeLa cells were microinjected with GST α appendage after they had been depleted of clathrin by transfection with siRNA targeting its heavy chain. Clathrin-depleted cells were identified by immunostaining in C and are labeled by asterisks in D. Cells microinjected with GST α appendage are labeled with arrowheads in D. Note that microinjected and clathrin-depleted cells (labeled with arrowheads and asterisks) show very little AP2 at the plasma membrane. (E and F) Aggregation of epsin in clathrin-depleted cells after microinjection of GST α appendage. (Scale bar: 10 μm.)
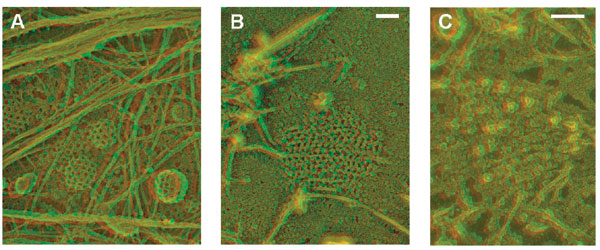
We next examined the AP2 membrane domains in clathrin-depleted HeLa cells by electron microscopy using the surface replication techniques for visualizing cell cortices of “unroofed” cells devised by Nermut et al. (39) and optimized by Heuser (40). This technique has already provided spectacular views of clathrin lattices and cytoskeletal structures on the inner surface of the plasma membrane. Clathrin coats are readily recognized by electron microscopy in such preparations (Fig. 4A and B), but the identification of AP2 membrane domains presented problems, because their appearance in the electron microscope is unknown. To resolve this question we first examined by electron microscopy the plasma membranes of HeLa cells where the clathrin was only partially reduced (Fig. 4 C–F and Fig. 8, which is published as supporting information on the PNAS web site). In these preparations we noted fragmented lattices and few clathrin-coated buds within distinct plasma membrane areas that appeared to be covered by small particles (Fig. 4, arrow). The average size of these particles was 10–15 nm, which is within the size range reported previously for isolated AP2 particles (41). These structures were also recognized in cells in which the expression of clathrin was reduced by >80% (Fig. 4 I and J). To unequivocally prove the presence of AP2 in these membrane domains we used ImmunoGold labeling. To this end the cell cortices were first mildly fixed with formaldehyde and then labeled successively with a monoclonal antibody directed against the α-subunit of AP2 and gold-labeled anti-mouse IgG. In control cells that expressed normal amounts of clathrin 10-nm gold grains were detected on clathrin-coated lattices and coated buds (Fig. 4H). Because of the rotary shadowing procedure, the gold particles appear as dots surrounded by a halo (arrow in Fig. 4 H–J). In cells depleted of >80% of the clathrin heavy chains the gold was found on particle-covered membrane domains (Fig. 4I) with an appearance similar to those shown in Fig. 4 C–G. Close inspection of the AP2 membrane domains revealed that they were always almost completely flat. This is best seen in 3D images of the plasma membrane from clathrin-depleted cells (Fig. 8). On 7 of 39 randomly chosen AP2 membrane domains we observed residual clathrin, and only these parts of the AP2 membrane domains revealed occasionally some curvature (data not shown).
Fig. 4.

AP2 forms membrane microdomains in clathrin-depleted HeLa cells. Shown are views of the inner surface of unroofed HeLa cells that expressed their normal content of clathrin (A, B, and H), ≈40% clathrin (C–F), and <20% clathrin (G and I). Control cells (A, B, and H) are characterized by mainly hexagonal flat clathrin lattices and coated buds. Cells with reduced clathrin content reveal fewer buds (see asterisk in C) and incomplete lattices (e.g., arrow in C and arrowheads in D–F). Arrows in D–F point at clustered particles that are uncovered by the removal of clathrin. A cluster apparently devoid of clathrin is shown in G. They were identified as AP2 molecules by ImmunoGold labeling with AP.6 antibody followed by 10-nm gold-labeled anti-mouse IgG [arrows in control (H) and knockdown cells (I and J)]. The arrowhead in I denotes residual clathrin lattice structures. Anaglyph stereo views reveal that the AP2 membrane domains are almost completely flat (see Fig. 8). (Scale bars: 100 nm.)
Taken together our findings show that, in the absence of clathrin, AP2 and accessory proteins are able to form membrane subdomains that do not differ significantly in size from clathrin-coated structures but are characterized by a lack of curvature. This observation clearly demonstrates that the deformation of the plasma membrane in AP2 patches is strictly dependent on clathrin. This finding confirms the long-held conjecture that clathrin is required for budding (42), and it is also consistent with very recent observations made by live-cell imaging showing that the membrane budding frequency is cooperatively related to the intracellular clathrin concentration (25). Clathrin lattices that cover flat or shallow coated structures show a smaller proportion of pentagonal facets than deeply invaginated structures (17, 42). Whether the transition of hexagonal into pentagonal facets leads to membrane deformation or merely follows membrane curvature driven by other forces remained a matter of debate, but our data now imply that clathrin is indeed required directly or indirectly for membrane bending. How can this be mechanistically accomplished? One possibility is that the clathrin lattice provides a scaffold to concentrate membrane bending proteins such as epsin, which upon binding to PtdIns-4,5-P2 inserts an amphipathic helix into the cytoplasmic leaflet of the plasma membrane and thereby could initiate or support membrane invaginations (11). It is also conceivable that the interaction of clathrin with adapters and accessory proteins induces conformational changes in those proteins that result in membrane bending. In both scenarios the clathrin lattice is also required to adapt to the changing membrane curvature by structural rearrangements within the lattice. These are energetically not too costly, because the lattice is predominantly held together by weak hydrophobic clathrin–clathrin interactions (43). The plasticity of the clathrin lattice was also demonstrated by FRAP experiments in living cells that demonstrated very rapid exchange between lattice-bound and free cytosolic clathrin (18, 19).
However, we do not think that epsin N-terminal homology domain proteins such as epsin have more than a supporting role in membrane bending. First, the AP2 domains in clathrin-depleted cells still contain epsin (Fig. 1) (23, 24), but this does not in itself engender any sustained membrane curvature in living cells, nor does depletion of epsin in HeLa cells inhibit clathrin-dependent endocytosis as observed by us (data not shown) and other investigators (44, 45). Moreover, recent electron micrographs demonstrated the presence of epsin in flat clathrin-coated lattices (46, 47). Taken together these observations showed that the presence of epsin in coats does not automatically bend the underlying membranes.
F-actin dynamics were recently shown to participate in clathrin-mediated endocytosis in mammalian cells (22). However, it is important to note that the disruption of actin function seemed to have little effect on coat invagination, whereas lattice assembly, coat motility in the plane of the membrane, constriction, and the movement of nascent coated vesicles into the cell were affected (22).
How then might clathrin affect the invagination of invariantly flat AP2 domains enriched in CALM, epsin, eps15, and also Hip1R/Hip1? It should be recalled that under in vitro conditions pure clathrin readily polymerizes into closed baskets with diameters ranging from 60 to 150 nm (48–50). This observation suggests that clathrin polymers are most stable in the form of highly curved closed structures. This property, then, should result in a higher affinity of clathrin polymers for curved surfaces. Based on molecular dynamics simulations on red blood cells, spontaneous membrane ripples with amplitudes on the order of 10 nm and wavelengths of ≈100 nm have been predicted under physiological conditions (51). It is now tempting to speculate that transient membrane invaginations resulting from such spontaneous fluctuations are trapped by the dynamic clathrin lattice, which preferentially adopts the curved form. An increasing affinity of the clathrin lattice for higher curvature thus imposes directionality on an inherent random process (Fig. 5). This model for clathrin-coated bud formation conforms loosely to the “Brownian ratchet” principle previously offered as an explanation for the translocation of proteins through membranes (52, 53), lamellipodia formation (54, 55), and as a matter of fact for clathrin pentagon assembly on membranes (3). Based on theoretical considerations it was previously concluded that coat assembly can only stabilize membrane invaginations but not drive them (56). This notion is also consistent with the Brownian ratchet model as outlined above. In lamellipodia formation thermal motion of the plasma membrane temporarily distances the fast growing ends of actin filaments from the membrane and allows their elongation and thus the extension of the cell’s leading edge. The invagination of clathrin-coated structures at the plasma membrane is critically dependent on the presence of cholesterol (57, 58). It may be required for adjusting the difference in area of the inner and outer bilayer leaflets to accommodate changes in curvature that accompany clathrin-dependent budding.
Fig. 5.

A model for the function of clathrin in coated bud formation. An endocytic cycle starts with the recruitment of adapters (red), endocytic accessory proteins (red), and clathrin (blue) to the plasma membrane. The initially flat coated membrane curves because of random thermal fluctuations (A). The clathrin lattice can adjust to the new curvature because both clathrin and adapters are known to exchange readily between their cytosolic and membrane-bound states (18, 19). Clathrin polymerizes in vitro spontaneously into stable 60- to 150-nm closed baskets, which suggests that a highly curved lattice corresponds to the energetically most stable conformation of the polymer. Considering this, the clathrin lattice will stabilize inwardly protruding plasma membrane deformations until the optimal membrane curvature has been reached. Upon vesicle scission the detached vesicle is uncoated because both AP2 and clathrin are losing their affinity for the membrane, which most likely is because of the hydrolysis of PtdIns-4,5-P2 to PtdIns-4-P by synaptojanin. In clathrin-depleted cells the AP2 membrane domains are also subject to thermal fluctuations (B), but these lack the directionality that is given to this process when clathrin is present.
In sum, our results clearly emphasize the role of clathrin in the invagination of coated structures at the plasma membrane. Mechanistically, we favor a model akin to a Brownian ratchet for the function of clathrin in bud formation. This model does not exclude the contributions of membrane-bending accessory proteins that we think are likely to have a supporting function.
Materials and Methods
Antibodies.
Mouse monoclonal antibodies used for immunofluorescence were as follows: AP.6, anti-α-adaptin; X22, anti-clathrin heavy chain (59, 60); monoclonal antibody 100/3 anti-γ-adaptin (61); and anti-Hip1R (BD Bioscience). Mouse monoclonal antibodies used for Western blotting were anti-clathrin heavy chain mouse monoclonal antibody (BD Biosciences) and anti-α-adaptin mouse monoclonal antibody (sc-17771, Santa Cruz Biotechnology). Rabbit polyclonal antibodies used for immunofluorescence and Western blotting were affinity-purified R461 (anti-clathrin light chains) (61) and goat anti-CALM (sc-6433, Santa Cruz Biotechnology). Antiserum directed against eps15R was kindly provided by P. Di Fiore (Istituto Europeo di Oncologia, Milan). Antiserum GD/1 raised against a conserved sequence in the hinge region of all known β-type adaptins (GDLLNLDLGPPV) was kindly supplied by L. Traub (University of Pittsburgh, Pittsburgh) (62). Fluoresceine- or rhodamine-labeled anti-mouse or anti-rabbit antibodies were from Molecular Probes. Fluorescine donkey anti-goat antibody was from Jackson ImmunoResearch. Secondary horseradish peroxidase conjugated anti-mouse, anti-goat, and anti-rabbit antibodies were from ICN. Texas red-labeled transferrin was from Molecular Probes. R. Anderson (University of Texas Southwestern Medical Center, Dallas) provided the construct for the GST α appendage domain of murine αc (residues 701–938) (63).
siRNA and Transfection.
The siRNAs that target the clathrin heavy chain (identical to HC oligo I from ref. 23) were purchased from Dharmacon (Lafayette, CO). HeLaSS6 cells were cultured as described previously (23) and transfected with siRNAs by using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s recommendations. The effects of siRNA-mediated knockdowns were assayed 3 days after transfection unless stated otherwise. For FRAP experiments siRNA transfected cells (≈300,000) were additionally transfected with 0.3 μg of β2 adaptin–YFP construct (64) 2 days after transfection with siRNA.
Fluorescence Microscopy.
Immunostaining of cells grown on glass coverslips was executed exactly as described in ref. 23. Stained cells were viewed with an Axiovert 200M microscope and documented with an AxioCam MRm digital camera controlled by axiovision (rel. 4.2) software (Zeiss). Final images were arranged and labeled by using a Macintosh G4 computer (Apple Computer, Cupertino, CA) with photoshop 7.0 software (Adobe Systems, San Jose, CA). For FRAP experiments cells were grown on 30-mm coverslips and mounted for life cell imaging in a POCmini chamber (Zeiss). The cells were imaged in Leibovitz medium supplemented with 0.1% BSA (Invitrogen) using the Zeiss LSM 510 Meta confocal microscope with a ×63/1.4 oil DIC objective. The microscope was equipped with a heating stage, incubator, and objective heater to keep the cells at 37°C throughout the measurements. Areas within β2 adaptin–YFP transfected cells were bleached by using the 488-nm Ar-laser line of the microscope at 100% intensity for 20 iterations, which reduced the fluorescence intensity by 50–80%. Recovery was monitored by using the laser at 25% of the maximum intensity. Images were analyzed with lsm 510 software and excel (Microsoft).
Microinjection of HeLa Cells.
GST α appendage protein was isolated as described previously (23), dialyzed against buffer G (25 mM Hepes/125 mM K acetate/50 mM Mg acetate, pH 7.3), and concentrated to 3 mg/ml. HeLa cells were seeded on gridded glass-bottom culture dishes (MatTec, Ashland, MA), and after 24 h GST α appendage protein was microinjected with 180 hPa for 0.4 s under microscopic control using an InjectMan NI2 apparatus and Femtotips (Eppendorf). The cells were allowed to recover for 3 h at 37°C and then fixed and processed for immunofluorescence. The grid aided the localization of microinjected cells. For clathrin depletion HeLa cells were transfected with clathrin siRNA as described previously (23), and after 48 h they were seeded on gridded glass-bottom culture dishes. Twenty-four hours later they were microinjected with GST α appendage protein and processed for immunofluorescence as described above.
Electron Microscopy.
HeLa cells grown on glass coverslips were unroofed based on a procedure described by Heuser (40) and modified as detailed recently (13). For ImmunoGold labeling the unroofed cells were fixed with freshly prepared 2% formaldehyde in buffer G (25 mM Hepes/125 mM K acetate/50 mM Mg acetate, pH 7.3) for 30 min. The fixative was quenched with buffer G supplemented with 50 mM NH4Cl and 50 mM glycine for an additional 30 min. Subsequently nonspecific binding sites were blocked with 1% BSA in buffer G for 30 min. AP2 was labeled by using the AP.6 antibody diluted in 1% BSA in buffer G (pH 7.4) and an anti-mouse antibody labeled with 10 nm of gold (British BioCell International, Cardiff, U.K.) diluted in buffer G. Afterward the samples were fixed with 2% glutaraldehyde in G for 30 min and prepared for electron microscopy as described above.
Supplementary Material
Acknowledgments
We are grateful to Drs. L. Traub, A. Sorkin, and P. Di Fiore for generously providing important reagents. We thank Dr. R. Bauerfeind for his advice on microinjection; Dr. B. Sodeik for access to the InjectMan NI2 microinjection apparatus; and B. Grossmann, A. Hundt, G. Preiss, and H. Ungewickell for expert technical assistance. This study was supported by Grants UN 43/4 and UN 43/5 from the German Research Foundation (to E.J.U.) and by a grant from Hannover Medical School (Hochschulinterne Leistungsförderung) (to A.M.).
Abbreviations
- AP2
adapter complex 2
- CALM
clathrin assembly lymphoid myeloid leukemia protein
- FRAP
fluorescence recovery after photobleaching
- PtdIns-4,5-P2
phosphatidylinositol-4,5-bisphosphate
- siRNA
small interfering RNA
- YFP
yellow fluorescent protein.
Footnotes
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.McMahon H. T., Gallop J. L. Nature. 2005;438:590–596. doi: 10.1038/nature04396. [DOI] [PubMed] [Google Scholar]
- 2.Zimmerberg J., McLaughlin S. Curr. Biol. 2004;14:R250–R252. doi: 10.1016/j.cub.2004.02.060. [DOI] [PubMed] [Google Scholar]
- 3.Shraiman B. Biophys. J. 1997;72:953–957. doi: 10.1016/s0006-3495(97)78729-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mashl R. J., Bruinsma R. F. Biophys. J. 1998;74:2862–2875. doi: 10.1016/S0006-3495(98)77993-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Itoh T., Erdmann K. S., Roux A., Habermann B., Werner H., De Camilli P. Dev. Cell. 2005;9:791–804. doi: 10.1016/j.devcel.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 6.Zimmerberg J., Kozlov M. M. Nat. Rev. Mol. Cell Biol. 2006;7:9–19. doi: 10.1038/nrm1784. [DOI] [PubMed] [Google Scholar]
- 7.Ehrlich M., Boll W., Van Oijen A., Hariharan R., Chandran K., Nibert M. L., Kirchhausen T. Cell. 2004;118:591–605. doi: 10.1016/j.cell.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 8.Traub L. M. Biochim. Biophys. Acta. 2005;1744:415–437. doi: 10.1016/j.bbamcr.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 9.Polo S., Sigismund S., Faretta M., Guidi M., Capua M. R., Bossi G., Chen H., De Camilli P., Di Fiore P. P. Nature. 2002;416:451–455. doi: 10.1038/416451a. [DOI] [PubMed] [Google Scholar]
- 10.Ford M. G., Pearse B. M., Higgins M. K., Vallis Y., Owen D. J., Gibson A., Hopkins C. R., Evans P. R., McMahon H. T. Science. 2001;291:1051–1055. doi: 10.1126/science.291.5506.1051. [DOI] [PubMed] [Google Scholar]
- 11.Ford M. G., Mills I. G., Peter B. J., Vallis Y., Praefcke G. J., Evans P. R., McMahon H. T. Nature. 2002;419:361–366. doi: 10.1038/nature01020. [DOI] [PubMed] [Google Scholar]
- 12.Kalthoff C., Alves J., Urbanke C., Knorr R., Ungewickell E. J. J. Biol. Chem. 2002;277:8209–8216. doi: 10.1074/jbc.M111587200. [DOI] [PubMed] [Google Scholar]
- 13.Meyerholz A., Hinrichsen L., Groos S., Esk P. C., Brandes G., Ungewickell E. J. Traffic. 2005;6:1225–1234. doi: 10.1111/j.1600-0854.2005.00355.x. [DOI] [PubMed] [Google Scholar]
- 14.Takei K., Slepnev V. I., Haucke V., De Camilli P. Nat. Cell Biol. 1999;1:33–39. doi: 10.1038/9004. [DOI] [PubMed] [Google Scholar]
- 15.Farsad K., Ringstad N., Takei K., Floyd S. R., Rose K., De Camilli P. J. Cell Biol. 2001;155:193–200. doi: 10.1083/jcb.200107075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peter B. J., Kent H. M., Mills I. G., Vallis Y., Butler P. J., Evans P. R., McMahon H. T. Science. 2004;303:495–499. doi: 10.1126/science.1092586. [DOI] [PubMed] [Google Scholar]
- 17.Heuser J. J. Cell Biol. 1980;84:560–583. doi: 10.1083/jcb.84.3.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu X., Zhao X., Baylor L., Kaushal S., Eisenberg E., Greene L. E. J. Cell Biol. 2001;155:291–300. doi: 10.1083/jcb.200104085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu X., Zhao X., Puertollano R., Bonifacino J. S., Eisenberg E., Greene L. E. Mol. Biol. Cell. 2003;14:516–528. doi: 10.1091/mbc.E02-06-0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Engqvist-Goldstein A. E., Drubin D. G. Annu. Rev. Cell Dev. Biol. 2003;19:287–332. doi: 10.1146/annurev.cellbio.19.111401.093127. [DOI] [PubMed] [Google Scholar]
- 21.Merrifield C. J. Trends Cell Biol. 2004;14:352–358. doi: 10.1016/j.tcb.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 22.Yarar D., Waterman-Storer C. M., Schmid S. L. Mol. Biol. Cell. 2005;16:964–975. doi: 10.1091/mbc.E04-09-0774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hinrichsen L., Harborth J., Andrees L., Weber K., Ungewickell E. J. J. Biol. Chem. 2003;278:45160–45170. doi: 10.1074/jbc.M307290200. [DOI] [PubMed] [Google Scholar]
- 24.Motley A., Bright N. A., Seaman M. N., Robinson M. S. J. Cell Biol. 2003;162:909–918. doi: 10.1083/jcb.200305145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moskowitz H. S., Yokoyama C. T., Ryan T. A. Mol. Biol. Cell. 2005;16:1769–1776. doi: 10.1091/mbc.E04-08-0739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coda L., Salcini A. E., Confalonieri S., Pelicci G., Sorkina T., Sorkin A., Pelicci P. G., Di Fiore P. P. J. Biol. Chem. 1998;273:3003–3012. doi: 10.1074/jbc.273.5.3003. [DOI] [PubMed] [Google Scholar]
- 27.Chopra V. S., Metzler M., Rasper D. M., Engqvist-Goldstein A. E., Singaraja R., Gan L., Fichter K. M., McCutcheon K., Drubin D., Nicholson D. W., Hayden M. R. Mamm. Genome. 2000;11:1006–1015. doi: 10.1007/s003350010195. [DOI] [PubMed] [Google Scholar]
- 28.Chen C. Y., Brodsky F. M. J. Biol. Chem. 2005;280:6109–6117. doi: 10.1074/jbc.M408454200. [DOI] [PubMed] [Google Scholar]
- 29.Mishra S. K., Agostinelli N. R., Brett T. J., Mizukami I., Ross T. S., Traub L. M. J. Biol. Chem. 2001;276:46230–46236. doi: 10.1074/jbc.M108177200. [DOI] [PubMed] [Google Scholar]
- 30.Legendre-Guillemin V., Metzler M., Charbonneau M., Gan L., Chopra V., Philie J., Hayden M. R., McPherson P. S. J. Biol. Chem. 2002;277:19897–19904. doi: 10.1074/jbc.M112310200. [DOI] [PubMed] [Google Scholar]
- 31.Legendre-Guillemin V., Metzler M., Lemaire J. F., Philie J., Gan L., Hayden M. R., McPherson P. S. J. Biol. Chem. 2005;280:6101–6108. doi: 10.1074/jbc.M408430200. [DOI] [PubMed] [Google Scholar]
- 32.Engqvist-Goldstein A. E., Warren R. A., Kessels M. M., Keen J. H., Heuser J., Drubin D. G. J. Cell Biol. 2001;154:1209–1224. doi: 10.1083/jcb.200106089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jost M., Simpson F., Kavran J. M., Lemmon M. A., Schmid S. L. Curr. Biol. 1998;8:1399–1402. doi: 10.1016/s0960-9822(98)00022-0. [DOI] [PubMed] [Google Scholar]
- 34.Padron D., Wang Y. J., Yamamoto M., Yin H., Roth M. G. J. Cell Biol. 2003;162:693–701. doi: 10.1083/jcb.200302051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krauss M., Kinuta M., Wenk M. R., De Camilli P., Takei K., Haucke V. J. Cell Biol. 2003;162:113–124. doi: 10.1083/jcb.200301006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Praefcke G. J., Ford M. G., Schmid E. M., Olesen L. E., Gallop J. L., Peak-Chew S. Y., Vallis Y., Babu M. M., Mills I. G., McMahon H. T. EMBO J. 2004;23:4371–4383. doi: 10.1038/sj.emboj.7600445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beck K. A., Keen J. H. J. Biol. Chem. 1991;266:4437–4441. [PubMed] [Google Scholar]
- 38.Beck K. A., Chang M., Brodsky F. M., Keen J. H. J. Cell Biol. 1992;119:787–796. doi: 10.1083/jcb.119.4.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nermut M. V., Williams L. D., Stamatoglou S. C., Bissell D. M. Eur. J. Cell Biol. 1986;42:35–44. [PubMed] [Google Scholar]
- 40.Heuser J. Traffic. 2000;1:545–552. doi: 10.1034/j.1600-0854.2000.010704.x. [DOI] [PubMed] [Google Scholar]
- 41.Heuser J. E., Keen J. J. Cell Biol. 1988;107:877–886. doi: 10.1083/jcb.107.3.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kanaseki T., Kadota K. J. Cell Biol. 1969;42:202–220. doi: 10.1083/jcb.42.1.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wakeham D. E., Chen C. Y., Greene B., Hwang P. K., Brodsky F. M. EMBO J. 2003;22:4980–4990. doi: 10.1093/emboj/cdg511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sigismund S., Woelk T., Puri C., Maspero E., Tacchetti C., Transidico P., Di Fiore P. P., Polo S. Proc. Natl. Acad. Sci. USA. 2005;102:2760–2765. doi: 10.1073/pnas.0409817102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang F., Khvorova A., Marshall W., Sorkin A. J. Biol. Chem. 2004;279:16657–16661. doi: 10.1074/jbc.C400046200. [DOI] [PubMed] [Google Scholar]
- 46.Hawryluk M. J., Keyel P. A., Mishra S. K., Watkins S. C., Heuser J. E., Traub L. M. Traffic. 2006;7:262–281. doi: 10.1111/j.1600-0854.2006.00383.x. [DOI] [PubMed] [Google Scholar]
- 47.Edeling M. A., Mishra S. K., Keyel P. A., Steinhauser A. L., Collins B. M., Roth R., Heuser J. E., Owen D. J., Traub L. M. Dev. Cell. 2006;10:329–342. doi: 10.1016/j.devcel.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 48.Keen J. H., Willingham M. C., Pastan I. Cell. 1979;16:303–312. doi: 10.1016/0092-8674(79)90007-2. [DOI] [PubMed] [Google Scholar]
- 49.Crowther R. A., Pearse B. M. J. Cell Biol. 1981;91:790–797. doi: 10.1083/jcb.91.3.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ahle S., Ungewickell E. EMBO J. 1986;5:3143–3149. doi: 10.1002/j.1460-2075.1986.tb04621.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brown F. L. Biophys. J. 2003;84:842–853. doi: 10.1016/S0006-3495(03)74903-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Peskin C. S., Odell G. M., Oster G. F. Biophys. J. 1993;65:316–324. doi: 10.1016/S0006-3495(93)81035-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Neupert W., Brunner M. Nat. Rev. Mol. Cell Biol. 2002;3:555–565. doi: 10.1038/nrm878. [DOI] [PubMed] [Google Scholar]
- 54.Theriot J. A. Traffic. 2000;1:19–28. doi: 10.1034/j.1600-0854.2000.010104.x. [DOI] [PubMed] [Google Scholar]
- 55.Vasanji A., Ghosh P. K., Graham L. M., Eppell S. J., Fox P. L. Dev. Cell. 2004;6:29–41. doi: 10.1016/s1534-5807(03)00397-6. [DOI] [PubMed] [Google Scholar]
- 56.Nossal R. Traffic. 2001;2:138–147. doi: 10.1034/j.1600-0854.2001.020208.x. [DOI] [PubMed] [Google Scholar]
- 57.Subtil A., Gaidarov I., Kobylarz K., Lampson M. A., Keen J. H., McGraw T. E. Proc. Natl. Acad. Sci. USA. 1999;96:6775–6780. doi: 10.1073/pnas.96.12.6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rodal S. K., Skretting G., Garred O., Vilhardt F., van Deurs B., Sandvig K. Mol. Biol. Cell. 1999;10:961–974. doi: 10.1091/mbc.10.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brodsky F. M. J. Cell Biol. 1985;101:2055–2062. doi: 10.1083/jcb.101.6.2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chin D. J., Straubinger R. M., Acton S., Nathke I., Brodsky F. M. Proc. Natl. Acad. Sci. USA. 1989;86:9289–9293. doi: 10.1073/pnas.86.23.9289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ahle S., Mann A., Eichelsbacher U., Ungewickell E. EMBO J. 1988;7:919–929. doi: 10.1002/j.1460-2075.1988.tb02897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Traub L. M., Kornfeld S., Ungewickell E. J. Biol. Chem. 1995;270:4933–4942. doi: 10.1074/jbc.270.9.4933. [DOI] [PubMed] [Google Scholar]
- 63.Wang L. H., Sudhof T. C., Anderson R. G. J. Biol. Chem. 1995;270:10079–10083. doi: 10.1074/jbc.270.17.10079. [DOI] [PubMed] [Google Scholar]
- 64.Sorkina T., Huang F., Beguinot L., Sorkin A. J. Biol. Chem. 2002;277:27433–27441. doi: 10.1074/jbc.M201595200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}