

Abstract
SV40 vectors packaged in vitro (pseudovirions) are an efficient delivery system for plasmids up to 17.7 kb, with or without SV40 sequences. A truncated Pseudomonas exotoxin gene (PE38) was delivered into various human cells (HeLa, KB-3-1, human lymphoblastoids, and erythroleukemia cells) in vitro using pseudovirions. The number of viable cells was reduced significantly in the PE38-transduced cells. Human KB adenocarcinomas growing in mice were treated with intratumoral injection of PE38 packaged in vitro and tumor size decreased significantly. Intraperitoneal treatments were as effective in reducing tumor size as intratumoral treatments. To check the viability of mock- or PE38-treated mice, every four days they were weighed, their blood was tested, and various tissues were screened for pathology. All parameters showed that the in vitro-packaged vectors, injected into tumors or intraperitoneally, caused no abnormalities in mice. The combined treatment of doxorubicin with in vitro-packaged PE38 reduced tumor size only slightly more than each of the treatments separately. However, the combined treatment did not cause the weight loss seen with doxorubicin alone. These results indicate that SV40 in vitro packaging is an effective system for cancer gene delivery using two different routes of injection and in combination with chemotherapy.
Keywords: gene delivery, SV40 pseudovirions, in vitro packaging, Pseudomonas exotoxin, doxorubicin, adenocarcinoma
INTRODUCTION
The need to develop new approaches for cancer therapy led us to test a new strategy for gene delivery using SV40 pseudovirions carrying a lethal gene. The use of Pseudomonas exotoxin A (PE) to kill cancer cells was demonstrated successfully by Pastan and colleagues.1 Pseudomonas Exotoxin A is a 66-kDa pathogenic protein secreted by Pseudomonas aeruginosa as a proenzyme, under the selective pressure of a low iron environment. This toxin assists bacteria in the invasion of animal tissues, including human tissues. As such, it enters cells via the low-density lipoprotein receptor-related protein (LRP). Within the cell, PE is cleaved into two peptide fragments linked by a disulfide bond. When this bond is broken, the enzymatically active C-terminal fragment translocates to the cytosol, where it inactivates elongation factor 2, thereby preventing protein synthesis and promoting apoptotic cell death.2,3 PE is comprised of three major domains: domain Ia is the cell binding domain and domain Ib may serve to enhance toxin stability; domain II harbors the protease processing site and mediates translocation; and domain III has ADP-ribosylating activity, and therefore is responsible for the toxic activity of the protein.2
Pastan, Fitzgerald and colleagues created recombinant immunotoxins (RIT) in E.coli.1 They fused the Fv portion of a monoclonal antibody (MAb) in a single chain form directly to mutants of PE missing domain 1a. The advantage of this method is that it combines the higher specificity of the antibody with the killing power of the toxin. In order to achieve stability of the RIT at 37°C, Brinkman and colleagues4,5 designed a new molecule in which the light and heavy chains of the Fv portion are held together by a disulfide bond. Trials of RIT have shown that one of them, BL22, can produce complete remissions in many patients with drug resistant leukemia.6 Suicide gene therapy is one of several strategies7 used to deliver genes to cancer cells by converting non-toxic prodrugs into active chemotherapeutic agents capable of killing cancer cells. This approach enables selective killing of cells; a maximal therapeutic effect is achieved while systemic toxicity is limited.8 Despite the so-called “bystander effect”, studies show that the efficacy of suicide gene therapy of tumors is limited by the efficiency of delivery of the vector to the tumor and the less than 100% efficiency of killing by the activated prodrug.9 However, some have found that it is possible to improve cytotoxicity by combining suicide gene therapy with conventional chemotherapy. There are certain drawbacks that need to be taken into account when dealing with viral gene delivery systems, such as the bloodstream's rapid clearance of the virus-based gene transfer system. Consequently, the development of synthetic gene delivery vectors (non-viral gene delivery systems) is required10 in order to achieve the goal of delivering the suicide gene to its target.
Simian virus 40 (SV40) is an attractive potential vector for gene transfer to kill cancer cells. The vectors are prepared with nuclear extract of SF9 insect cells containing the main viral capsid protein of the SV40 wild-type virus, VP1. For the transfer of circular DNA encapcidated in VP1, the SV40 major capsid protein demonstrates high transduction efficiency and can be introduced into a wide variety of human, murine and monkey tissues. In vitro packaging of DNA with SV40 capsid protein enables efficient delivery of plasmids with a length of up to 17.7 kb. Moreover, it does not require any SV40 sequence, thus providing efficient gene delivery at the same level of safety when using nonviral vectors.11-13
In this study, we demonstrate delivery of PE38 toxin with or without a combination of the chemotherapeutic agent doxorubicin. SV40 pseudovirion delivery of PE was found to be effective in the treatment of human adenocarcinomas growing in mice either by direct injection or systemically. Using a combined treatment of PE38 with doxorubicin, we were able to reduce the side effects of chemotherapy.
Materials and Methods
Transduction of Sf9 cells with baculovirus
A baculovirus construct containing the VP1 gene which encodes the SV40 main capsid protein was added to 250 ml Sf9 cells with occasional mixing at a multiplicity of infection (MOI) of 10. These cells were harvested 4-5 days later.
Preparation of nuclear extracts from Sf9 cells
Isolation of the nuclei was achieved by the addition of 10% Nonidet™-40 (NP40) (Sigma, St. Louis, MO) to a buffer comprising 10 mM Hepes pH 7.9 (Cellgro, Herndon, VA,), 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA (all from Digene, Beltsville, MD), 0.5 mM phenylmethylsulfonylfluoride (PMSF; Fluka, Basel, Switzerland), 1 mM Dithiothreitol (DTT) (Sigma) and Protease Inhibitor cocktail tablet (Roche, Mannheim, Germany). The nuclei were then extracted by buffering the cell culture in 20 mM Hepes pH 7.9, 0.4 M NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM PMSF, 1mM DTT and a protease inhibitor cocktail tablet (Roche). DTT, PMSF and the protease inhibitor cocktail were added to the buffer immediately before use.14,15 Nuclear extract concentration was measured using the BCA Protein Assay protocol (Pierce, Rockford, Illinois). The nuclear extract was kept for more than a year at −20°C.
Preparation of in vitro packaging vectors
Packaged DNA in this study included the PE38 construct under the CMV promoter, kindly provided by Dr. Ira Pastan, NCI, NIH, and the pEGFP-C1 construct (4.7Kb; Clontech, Palo Alto, CA).16 Packaging was performed using scaled-up production methods.17 100 μg of nuclear extract from Sf9 cells, along with 100 μg of DNA construct in the presence of 5 mM ATP and 8 mM MgCl2 formed pseudoviral particles during incubation at 37°C for 6 hours in a 600 μL reaction volume (hereafter referred to as 1 reaction volume). The reaction tube was then incubated on ice for 1 hour with 1 mM CaCl2. In vitro-prepared vectors were stored at −20°C.
Cell culture and cell lines
The drug sensitive KB-3-1 cell line, derived from human HeLa epidermal carcinoma cells,18 and the HeLa cells were maintained in DMEM media (Dulbecco's modified Eagle medium, Invitrogen, Carlsbad, CA) with 10% fetal bovine serum, 50 μg/ml penicillin, 50 μg/ml streptomycin and 5 mM L-glutamine (all obtained from Invitrogen, Carlsbad, CA). Cells were incubated at 37°C, in 5% CO2. Splitting of cells was done using 0.25% trypsin-EDTA (Invitrogen). .45 human lymphoblastoid cells with high levels of major histocompatibility complex class I (MHC I), and K562 human erythroleukemia cells were maintained in RPMI media (Invitrogen).
In vitro transductions of a variety of cell lines with in vitro -packaged PE38 plasmid DNA
1-5×105 adherent cells (KB-3-1 and HeLa cells) were plated 24 hours prior to transduction in 60 mm dishes. K562 and .45 cells in suspension were split using their growth media 24 hours prior to transduction, but were counted on the day of transduction. A reaction of in vitro-packaged SV40 vectors carrying the PE38 plasmid DNA or empty SV40 vectors without DNA as a control were suspended in 340 μL of DMEM, supplemented with 5 mM L-glutamine, 50 μg /ml penicillin and 50 μg /ml streptomycin. Cells were transduced and placed on a rotary shaker at 30 rpm for 2.5 hours (at 37°C in 5% CO2). Post-infection, the dishes were supplemented with 4 ml of growth medium. Every two days the cells were counted and replated in new T-25 flasks. Fresh media was added to bring the total to 5 ml.
In Vivo treatment
Athymic nude female mice were supplied by the National Cancer Institute's Fredrick Animal Facility and housed under standard conditions based on the guidelines of the NIH Office of Animal Care and Use. The conditions in mouse cages were uniform concerning feed and water. At the age of 6-10 weeks, each received a subcutaneous injection of 2 × 106 KB-3-1 cells grown in tissue culture in 0.2 ml medium into the right flank. Within two to three days, all of the mice grew subcutaneous adenocarcinomas. From zero to fifteen days subsequent to cell injection, mice were divided into equal groups, so that each group was comprised of 5-10 mice. The treatments administered are shown in Table 1. The animals were regularly monitored for changes in weight and tumor size. Palpable tumors were measured with calipers, and the tumor diameter in two orthogonal dimensions was measured every four days over a period of several months. Tumor size (in mm3) was calculated for each mouse by multiplying the two measured dimensions together and then multiplying by the smaller dimension. Mice were inspected every day for the condition of their health and behavior. Mouse mortality in both experimental and control cages was similar. Tumors were allowed to grow until they reached approximately 2 cm in diameter. At this endpoint, the mice were euthanized according to Standard Operating Procedures.
Table 1.
Treatments to nude mice with human adenocarcinoma tumors
| Treatment | Amounta | Routeb | Frequency |
|---|---|---|---|
| PBS | 200 or 300 μl | SC | 4-5 times a week |
| PBS | 200 or 300 μl | IP | 4-5 times a week |
| Empty vectors | 200 or 300 μl | SC | 4-5 times a week |
| Empty vectors | 200 or 300 μl | IP | 4-5 times a week |
| DNA onlyc | 1 μg/μl | SC | 4-5 times a week |
| DNA onlyc | 1 μg/μl | IP | 4-5 times a week |
| IVP-PE | 200 or 300 μl | SC | 4-5 times a week |
| IVP-PE | 200 or 300 μl | IP | 4-5 times a week |
| Doxorubicin | 3.5-5 mg/kg | SC | Every 4th day |
| Doxorubicin | 3.5-5 mg/kg | IP | Every 4th day |
| IVP-PE with | 200 or 300 μl | SC | 4-5 times a week |
| Doxorubicind | 3.5-5 mg/kg | SC | Every 4th day |
| IVP-PE with | 200 or 300 μl | SC | 4-5 times a week |
| Doxorubicind | 3.5-5 mg/kg | IP | Every 4th day |
| IVP-PE with | 200 or 300 μl | IP | 4-5 times a week |
| Doxorubicind | 3.5-5 mg/kg | SC | Every 4th day |
| IVP-PE with | 200 or 300 μl | IP | 4-5 times a week |
| Doxorubicind | 3.5-5 mg/kg | IP | Every 4th day |
| IVP-EGFP | 200 or 300 μl | IP | 4-5 times a week |
The amounts varied from experiment to experiment and are specified in the Results section.
SC, subcutaneous; IP, intraperitoneal
The amount is based on 1% packaging efficiency estimation
Combined treatment
Histological, pathological and serological tests
Animals were first bled and then immediately euthanized by carbon dioxide inhalation and necropsied. Tissues were examined by a pathologist from the Division of Veterinary Resources at NIH. The following tissues were harvested and fixed in 10% buffered formalin phosphate (Fisher Scientific, Inc., Fairlawn, NJ): skin, liver, spleen, lungs, heart, kidneys, and brain. After fixation, tissues were processed for routine paraffin embedding, sectioned, and H&E-stained. Serum analyses were performed within 2 hours of blood collection for albumin, amylase, blood urea nitrogen, calcium, glucose, inorganic phosphorus, total protein, alkaline phosphatase, -glutamyl transferase, lactate dehydrogenase, alanine aminotransferase, aspartate aminotransferase, and total bilirubin. For GFP expression some tissues were harvested and frozen, and frozen slides were screened using a Leica fluorescent microscope. These experiments were conducted under approved NCI animal protocol LCB-004.
Statistical analysis
Tumor volume as well as mouse weight were plotted individually for each mouse. Average size was calculated for each 5-10 mice that received the same treatment in a specific experiment, and standard deviations were determined and plotted.
Results
Low cell viability after delivery of PE38 using SV40 pseudovirions
In order to test the efficiency and expression of the Pseudomonas exotoxin plasmid delivered by SV40 vectors, we measured the viability of cells when transduced with PE38 DNA encapsidated in VP1. Four different cell lines (HeLa, KB-3-1, K562 and .45 cells) (Figure 1, A, B, C, D panels respectively) were tested. The results shown here demonstrate that this transduction of PE by SV40 inhibits the growth of all four cell lines. The number of cells and the rate of growth of PE38-transduced cells were much lower. The experiment with all four-cell lines was repeated 4 times with similar results.
Figure 1.

Expression of PE38 packaged in vitro in four cell lines. Cells were transduced with a full reaction of PE38 plasmid DNA packaged in vitro (solid lines), and the number of cells was compared to mock transduced cells (dashed lines). Cells were counted every other day in order to monitor their viability. Cell lines were: HeLa cells (a), KB-3-1 cells (b), K562 human erythroleukemia cells (c), and .45 human lymphoblastoid cells (d).
Treating adenocarcinoma tumors in nude mice using PE38 in vitro-packaged vectors starting on the day of inoculation
To determine if the in vitro packaged PE38 is active in vivo, we injected PE38 encapsidated with VP1 or control vector at the same time and site as 2 × 106 KB-3-1 human adenocarcinoma cells.18 Five mice with these tumors were injected 4-5 times a week with 200 μl PBS (Table 1, line 1), or 200 μl empty in vitro-packaged vectors (Table 1, line 3) (Figure 2, panel A) and five mice were injected with 200 μl in vitro-packaged PE38 (IVP-PE38) (Table 1, line 7) (Figure 2, panel B). Figure 2, panel C demonstrates the average tumor size measured for groups shown in panels A and B with standard deviations. Figure 2 (panels A-C) clearly shows a large increase in tumor size 30 days after inoculation when PBS was used for treatment, while only very small tumors appeared in PE38-treated mice. Tumor size and growth rate obtained when using a control consisting of empty SV40 pseudovirions were similar to those using PBS treatment. The weight of the mice was measured every 4-6 days (Figure 2, panels D-F): both PE38-treated mice (Figure 2, panel D) and control-treated mice (Figure 2, panel E) gained weight. Figure 2, panel F demonstrates the average weight of the groups shown in panels A and B with standard deviations. One of the mice died on day 25. Three of the PE38-treated mice (# 342, 343, 345) were followed more than a year and half after 30 days of PE38-in vitro packaged treatment: no tumors appeared and the mice continued to grow and gain weight (data not shown). The results of pathology tests were the same for tissues from PE38-treated mice as for control, PBS-treated animals. Blood tests for both groups were within the normal range.
Figure 2.

Tumor size and weight of PE38 packaged in vitro and control mice treated from the day of tumor challenge. Mice were injected with 2 × 106 KB-3-1 cells along with the treatment. Tumor size as well as mouse weight were followed for 30 days. Figure 2 (a-c) demonstrates tumor size in mm3 of empty vector-treated mice (panel a) or IVP-PE38 (panel b) (five mice in each group, each line represents one mouse), and panel c is the average of each five mice (IVP-PE is shown in dark blue, and control-treated mice in magenta) with standard deviation (z = (−2.6191), one sided p-value=0.004 for day 25 of treatment). Figure 2 (d-f) demonstrates the weight in grams for these groups in this order (z = (−2.252), one sided p-value = 0.0122 for day 30 of treatment, z = (−1.744), one sided p-value = 0.0406 for relative weight gain by day 30 of treatment (wts on day 30/wts on day 4) (panels d, e, and f respectively).
Treatment of tumors using PE38 in vitro-packaged vectors beginning several days after inoculation
An experiment similar to the previous one was carried out with a modification—the treatment started on day 7 post KB-3-1 inoculation, when the average size of tumors was approximately 150 mm3. The tumors of five mice were injected 4-5 times a week directly (SC) with 200 μl IVP-PE38 (Table 1, line 7) (Figure 3, panel A), and the tumors of five mice were injected with DNA only (Table 1, line 5) or 200 μl empty in vitro packaged vectors (Table 1, line 3) (Figure 3, panel B). Figure 3 shows clearly a major growth in tumors 21 days after inoculation when empty vectors were used for treatment, while there was markedly less tumor growth in PE38-treated mice (Figure 3, panel C, average tumor size with standard deviations). Animals maintained their weight through the 21 days of the experiment (Figure 3, panels D and E respectively to Figure 3, panels A and B), although control-treated mice lost 2.5% of their body weight from day 17 to day 21 (Figure 3, panel E). Figure 3, panel F indicates the average weight of the groups shown in panels D and E with standard deviations. This experiment was repeated 3 times with a similar pattern of results. All types of treated mice, i.e. those treated with PBS, empty vectors or IVP-PE38, were sent for pathology and blood tests. Results were all within normal parameters.
Figure 3.

Tumor size and weight of PE38 packaged in vitro and control mice treated subcutaneously several days post tumor challenge. Treatment was followed six days after mice were injected with 2 × 106 KB-3-1 cells. Tumor size as well as mouse weight were followed for 21 days. Figure 3 (a-c) demonstrates tumor size in mm3 of empty vector-treated mice (panel a) or IVP-PE38 (panel b) (five mice in each group, each line represents one mouse), and panel c is the average of each five mice (IVP-PE is shown in dark blue, and control-treated mice in magenta) with standard deviation. Figure 3 (d-f) demonstrates the weight in grams for these groups in this order (panels d, e, and f respectively).
Intraperitoneal (IP) treatment of adenocarcinoma tumors in nude mice using PE38 in vitro-packaged vectors reduced tumor growth
As in the experiments described above, 2 × 106 KB-3-1 cells were injected subcutaneously into mice. Then, three to seven days after inoculation, five to ten mice were injected intraperitoneally with 200 μl PBS 4-5 times a week (Table 1, line 2), and five to ten mice were injected intraperitoneally with 200 μl IVP-PE38 in vitro vectors IP (Table 1, line 8). Weight and tumor size were measured every 4-5 days. Clearly, as seen from Figure 4, panels A and B, or the average with standard deviation in panel C, treatment which started on day 3 using IP injection of in vitro-packaged PE38 vectors led to smaller tumors as compared to IP treatment with PBS (z = 1.7975, one sided p-value = 0.036 on day 25 of treatment). A comparison of the weight of treated animals revealed continuous growth after PBS treatment, or after PE38 treatment using IP injection, with a greater weight gain in the control group (z = 2.0153, one sided p-value = 0.0219 on day 34 of treatment) (Figure 4 panels D-F). The experiment was repeated three times with similar results. IP injections of pEGFP-C1 packaged in vitro revealed similar results. Five tissues were examined from mice treated with PBS and mice treated with pEGFP-C1: kidney, heart, liver, spleen and tumor. All tissues from GFP-treated mice expressed high fluorescence as compared to tissues from PBS-treated mice (Figure 5), except kidney tissue, which expressed similar high background fluorescence in both PBS and EGFP mice. The difference in fluorescence intensity between tumors of mice treated with pEGFP-C1 and those treated with PBS was the most obvious. When the delivery of PE38 by SV40 started later than 7 days after inoculation, and tumor size exceeded 150 mm3 on average, it did not significantly decrease tumor size (data not shown). Pathology and blood tests were also done on animals that received IP treatment, and all results were within normal ranges.
Figure 4.

Tumor size and weight of PE38 packaged in vitro and control mice treated intraperitoneally several days post tumor challenge. Treatment was followed three days after mice were inoculated with 2 × 106 KB-3-1 cells. Tumor size as well as mouse weight were followed for 34 days. Figure 4 (a-c) demonstrates tumor size in mm3 of PBS-treated mice (panel a) or IVP-PE38 (panel b) (five mice in each group, each line represents one mouse), and panel c is the average of each five mice (IVP-PE is shown in dark blue, and control-treated mice in magenta) with standard deviation (z = 1.7975, one sided p-value = 0.0361 for day 25 of treatment). Figure 4 (d-f) demonstrates the weight in grams for these groups in this order (panels d, e, and f respectively) (z = 2.0153, one sided p-value = 0.0219 for day 34 of treatment).
Figure 5.
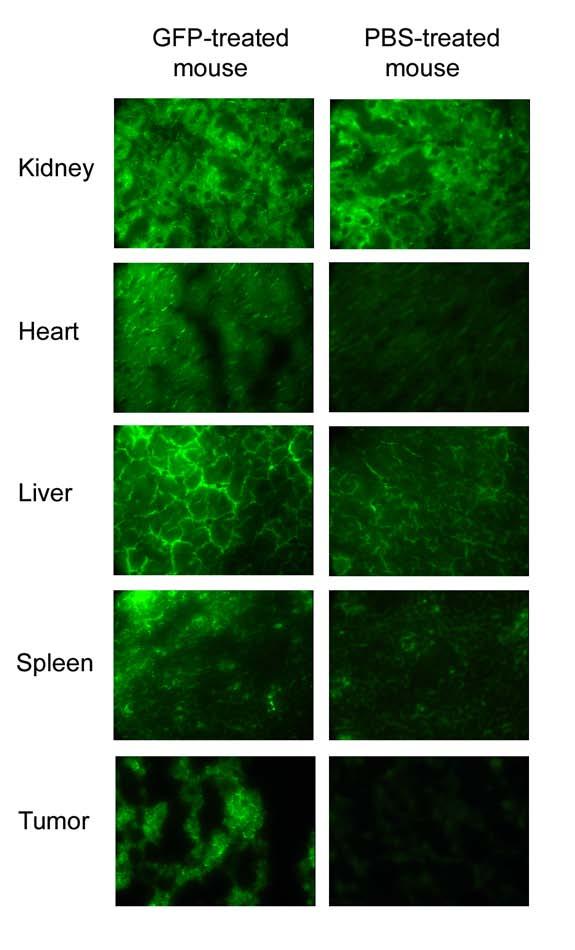
Tissue fluorescence of EGFP packaged in vitro and control mice treated intraperitoneally several days post tumor challenge. Treatment was followed three days after mice were inoculated with 2 × 106 KB-3-1 cells. Tumor size as well as mouse weight were followed for 30 days. Figure 5 shows five different tissues (from the top) kidney, heart, liver, spleen and tumor of IVP-EGFP (left) and PBS-treated mice (right). Figures were taken at 400X magnification.
Combination of PE38 gene delivery and doxorubicin chemotherapy treatment in cultured cells
In order to improve the treatment and to reduce tumor size more effectively, we first tested the combination of in vitro-packaged PE38 together with doxorubicin in cultured cells. Doxorubicin was applied to 5 × 105 PE38-transduced cells in culture immediately after transduction in two different concentrations (6 ng/ml and 12 ng/ml).19 We added the following four controls to the experiment: cells only, cells transduced with in vitro-packaged PE38, cells treated with 6 ng/ml doxorubicin only, and cells treated with 12 ng/ml doxorubicin only. Cell counts were performed every 2-3 days. The viability of cells in the combined treatment was higher than in doxorubicin alone, but lower than the viability of cells transduced with PE38 (Figure 6). The experiment was repeated three times with similar results.
Figure 6.

Viability of KB-3-1 cells treated in tissue culture with PE38 packaged in vitro and doxorubicin. 105 KB-3-1 cells were transduced with PE38 plasmid DNA packaged in vitro (dark blue), and the number of cells was compared to mock transduced cells (magenta). Other treatments on transduced cells were as follows: 105 KB-3-1 cells treated with 6 ng/ml (green), 105 KB-3-1 cells treated with 12 ng/ml (light blue), 105 KB-3-1 cells transduced with PE38 plasmid DNA packaged in vitro (purple), and treated with 6 ng/ml, and 105 KB-3-1 cells transduced with PE38 plasmid DNA packaged in vitro (brown), and treated with 12 ng/ml. Cells were counted every 2-3 days in order to monitor their viability.
Combination of PE38 gene delivery and doxorubicin chemotherapy treatment in vivo
The combination of chemotherapy treatment together with PE38 packaged in vitro was tested using both types of injection – IP and intratumoral. 5 mg/kg doxorubicin was injected every 4 days into either five or ten mice. Treatment started on day 3 after inoculation with KB-3-1 cells, when average tumor size was 1-2 mm3. Controls which were tested in this series of experiments were: 5 mg/kg doxorubicin alone, IVP-PE alone, and PBS only. Four different combined treatments were tested, as shown in Table 1 (last four lines). The first three combinations resulted in tumor size and mice weight similar to treatment with doxorubicin alone (data not shown). However, the combined treatment of IP injection of both PE38 and doxorubicin (Table 1, last line) reduced tumor size slightly more than each of the treatments separately, and kept mice viable for a longer time without weight loss (Figure 7, average tumor size (a) and weight (b) with standard deviations). Mice treated with doxorubicin alone lost weight, while mice treated with the combined treatment maintained their weight or gained weight (z = (−1.8991), one sided p-value = 0.028 on day 40 of treatment). Using less doxorubicin (3.5 mg/kg) did not reduce tumor size or produce differences in weight between the combined treatment and doxorubicin alone. The experiment was repeated four times with similar results.
Figure 7.
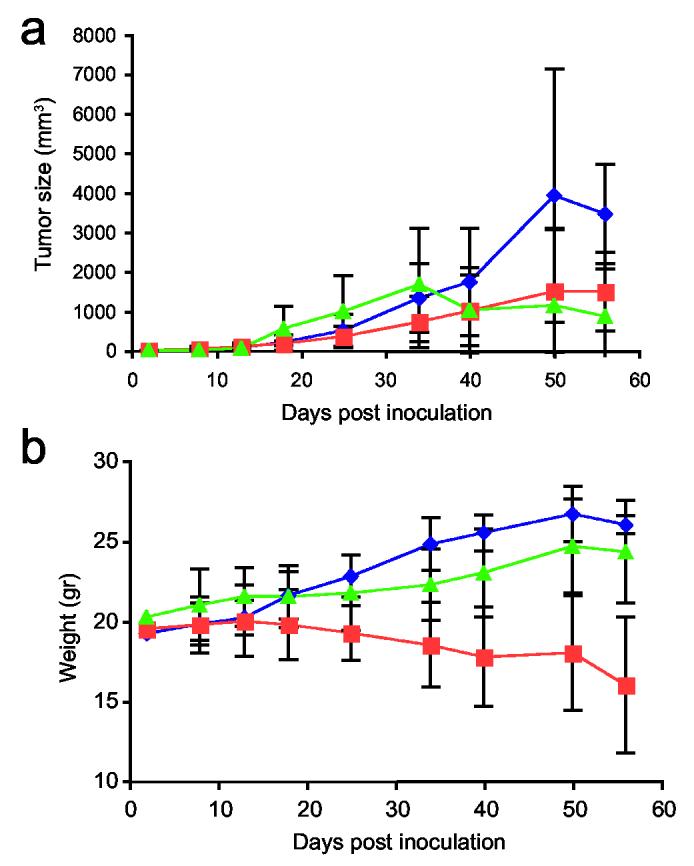
Tumor size and weight of mice treated with a combination of PE38 packaged in vitro and doxorubicin via IP. Treatment was followed three days after inoculation of mice with 2 × 106 KB-3-1 cells. Tumor size as well as the weight of the mice were followed for 56 days. Figure 6a demonstrates average tumor size in mm3 of IVP-PE-treated mice (blue), doxorubicin-treated mice (orange), and the combined treatment of IVP-PE38 with 5 mg/kg doxorubicin-treated mice (green) (five mice in each group). Panel b is the average weight of each of these groups with standard deviations (z = (−1.8991), one sided p-value = 0.0288 on day 40 of treatment with doxorubicin in comparison to the combination of doxorubicin and IVP-PE38)
Discussion
The ability of Pseudomonas exotoxin (PE) to kill cancer cells was demonstrated earlier by Pastan and colleagues1,3,20-23 and by other groups as well.24-26 PE was delivered into cancer cells as an immunotoxin, and successfully reduced tumor size. Other studies have used gene delivery of PE to treat various malignancies. In this study, we deliver a cDNA for Pseudomonas toxin using the SV40 in vitro-packaging delivery system. Our in vitro studies using cells in suspension and adherent cells demonstrate high efficiency of gene delivery of the encapsidated PE vectors. Periodic injections of the PE vectors into adenocarcinoma tumors in nude mice were shown in this study to reduce tumor size significantly. The mice did not lose weight and serological parameters were very similar to those of control-injected mice. Interestingly, here we show that intraperitoneal injection into nude mice with adenocarcinomas also results in significant reduction in tumor size. This delivery mode has not previously been used with SV40 vectors. Animals receiving the treatment did not show any abnormal pathological manifestations as compared to control-treated animals.
PE38 delivery in vitro using SV40 vectors
The Pseudomonas toxin delivered in vitro using the SV40 system very efficiently eradicated various types of cell lines, adherent cells (KB-3-1 cells and HeLa cells) and cells in suspension (K562, human erythroleukemia cells, and .45 human lymphoblastoid cells). The efficiency of delivery was very similar between the different cell lines, which indicates that the PE protein has similar toxicity in cells of various origins. The plasmid DNA contains the C-terminus domain 3, the catalytic domain, which is responsible for the inactivation of eukaryotic elongation factor 2 (eEF2) by catalyzing the ADP-ribosylation of this target protein once in the cytoplasm, thus inducing cell death. These results are in accord with other studies where cancer cells were treated in vitro very efficiently with the Pseudomonas exotoxin.1
Delivery of PE alone, not as a fused protein, directly to adenocarcinoma tumors
Bacterial toxins which have been targeted to cancer cells include Pseudomona exotoxin, ricin, and diphtheria toxin. These can be designed to form recombinant fusion toxins, and delivered by injection of the fused protein or by gene delivery using adenoviral, retroviral and liposome delivery systems.27,28 Therefore, the majority of the research on lethal gene delivery has been done on fused-PE proteins. TGF-α fused with PE, for example, was delivered to bladder carcinoma.29 Xu and colleagues30 reported delivery of PE and caspase-6 into BALB/c athymic mice bearing human breast SK-BR-3 tumors. They designed a fusion gene, immunocasp-6, consisting of an NH2-terminal leader sequence to promote secretion of the recombinant immunocasp-6 protein fused with an anti-HER2 single-chain Ab, the translocation domain (domain II) of Pseudomonas exotoxin A (PEA), and an active caspase-6. IL-4 fused with pseudomonas exotoxin was delivered to brain tumors and destroyed the brain tumor selectively.31,32 Chen and colleagues33 attempted to introduce the membrane translocation domain of Pseudomonas exotoxin A into a DNA delivery vehicle to increase the translocation efficiency of DNA into the cytosol34. That work, however, was cell-type dependent. They also used PE with a highly positively charged DNA binding region of the N-terminal 198 amino acid residues of human DNA Topo I. Their aim was to use DNA Topo I so that PE would bind to DNA without a DNA sequence or topological specificity. Hafkemeyer et al.16 delivered the same PE DNA vector as was used in the current study, but not as a fused construct, to animal models. The liposome delivery system was tested via tail-vein injections to assess the activity of PE, specifically in mouse lungs, using assays which detect apoptosis. Our current study demonstrates high efficiency of delivery to adenocarcinoma tumors of a non-targeted PE using a new delivery system based on SV40. Controls including daily delivery of this DNA as naked DNA, as well as empty capsid proteins or GFP encapcidated using the same pseudovirion system, all failed to inhibit tumor growth.
Treating adenocarcinoma tumors in nude mice using PE38 in vitro-packaged vectors via tumor injection or IP injection
Treating adenocarcinoma tumors using PE38 packaged with VP1, the SV40 main capsid protein, significantly reduced the size of adenocarcinomas. The earlier we started the treatment, shortly after KB-3-1 inoculation, the more tumor reduction we observed. Treatment that started later, on larger tumors, was less effective, as compared to treatment that started only two days after inoculation. A simple explanation of this phenomenon is that the effectiveness of the therapy is related to the ratio of pseudovirions to the number of tumor cells.
Interestingly, both methods of injection—injection of the pseudovirions directly into the tumor, or IP injection—resulted in tumor reduction. In fact, both methods proved to be very efficient in delivery of the virions to the tumor. These results parallel previous findings such as reported by Kawakami et al.31 or Jakubzick et al.35 Those investigators used a chimeric protein comprised of human IL-13 and a truncated version of an exotoxin from Pseudomonas (IL13-PE), and observed that IL13-PE selectively targeted human pulmonary fibroblasts grown from IIP SLBs, and had a minimal effect on fibroblasts grown from biopsies from normal patients.35 These results are also supported by our findings of GFP expressing cells in tumors from IP treated mice. We hypothesize that the SV40 pseudovirions circulate in the blood, but probably because of the proliferation of blood vessels in the tumors they are taken up by the tumors. The PE38 seems to be somewhat specifically taken up or expressed by the tumor, because otherwise, we would have seen tissue abnormalities in the PE-treated mice. We also found that both methods of injection had a minimal effect on other tissues, with similar serological and pathological results in control-treated mice and PE38 pseudovirion-treated mice. Furthermore, mice do not exhibit any adverse effects from prolonged 3-5 times a week in vivo administration of the pseudovirions during tumor treatment, which indicates that the capsid proteins are also harmless.
One of the main advantages of the SV40 in vitro packaging delivery system is its high efficiency. However, following a detailed examination of the pathway of the SV40 pseudovirus particles in human lymphoblastoid cells (which were tested among the four cell lines for cell viability), it is suggested that the low expression provided by the delivery system might be also due to the DNA trapped in the cytoplasm.36 Moreover, expression levels of toxin in cancer cells must be very low since even one molecule in the cytosol may lead to cell death, and these levels could not be quantified. Therefore, the combination of a delivery system that is highly efficient, although with a low-level of expression. and a very potent toxin results in high levels of cell death”.
Doxorubicin chemotherapy treatment and PE lethal gene delivery do not necessarily have an additive effect
A combined treatment of doxorubicin and in vitro packaged PE38 to cultured cells resulted in a major arrest of cell proliferation, but cell numbers were higher than when each of the treatments was applied separately. While doxorubicin acts on DNA synthesis, PE inhibits protein synthesis and induces caspase-dependent programmed cell death. This partially antagonistic effect could result from the need for protein synthesis as part of the doxorubicin killing process.37 Moreover, we have demonstrated successful gene delivery of Pseudomas exotoxin using SV40 pseudovirions in mice that actually continue to gain weight. Treating the same adenocarcinoma tumors using chemotherapy resulted in less tumor growth, but in significant weight loss. The combined treatment in vivo resulted in only slightly less tumor growth than when each of the treatments was applied separately. However, the mice did not lose weight. Thus, it might be possible to combine PE38 therapy with chemotherapy to improve the antitumor effect and reduce the side effects of chemotherapy. As seen in Figure 5, GFP was expressed in normal tissues to a lesser degree, which might suggest that PE targets normal cells as well. It is possible that through this pathway, the metabolism of doxorubicin is increased, which reduces its toxic activity.
Here, we establish a new method of delivery of a lethal gene into cells in vitro and in vivo. We demonstrate efficient delivery of the truncated Pseudomonas exotoxin gene to dramatically reduce the size of human adenocarcinomas using SV40 pseudovirions. Delivery is very effective either by direct injection into the tumor or intraperitoneally. Our results show that this method of treatment could potentially improve the chemotherapy of human adenocarcinomas by reducing tumor size with fewer side effects, as compared to treatment by chemotherapy alone.
Acknowledgments
This research was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research. We thank Ariella Oppenheim (The Hebrew University, Hadassah Medical School and Hadassah University Hospital, Jerusalem, Israel) for fruitful collaboration on the SV40 vectors, and Ira Pastan, LMB, NIH for productive discussions. We thank Georgina F. Miller and Michael Eckhaus, ORS/DIRS/VRP, NIH, for providing us with pathological reports and blood tests results, and George Leiman for insightful editorial assistance.
References
- 1.Pastan I. Immunotoxins containing Pseudomonas exotoxin A: a short history. Cancer Immunol Immunother. 2003;52:338–341. doi: 10.1007/s00262-002-0353-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen TY, Shang HF, Chen TL, Lin CP, Hui CF, Hwang J. Recombinant protein composed of Pseudomonas exotoxin A, outer membrane proteins I and F as a vaccine against P. aeruginosa infection. Biomed Sci. 1999;6:727–32. doi: 10.1007/s002530051555. [DOI] [PubMed] [Google Scholar]
- 3.Fitzgerald D, Pastan I. Pseudomonas exotoxin and recombinant immunotoxins derived from it. Ann N Y Acad Sci. 1993;685:740–745. doi: 10.1111/j.1749-6632.1993.tb35935.x. [DOI] [PubMed] [Google Scholar]
- 4.Brinkmann U, Reiter Y, Jung SH, Lee B, Pastan I. A Recombinant immunotoxin containing a disulfide-stabilized Fv fragment. Proc Natl Acad Sci. USA. 1993;90:7538–42. doi: 10.1073/pnas.90.16.7538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bera TK, Onda M, Brinkmann U, Pastan I. A Bivalent disulfide-stabilized Fv with improved antigen binding to erbB2. J Mol Biol. 1998;281:475–83. doi: 10.1006/jmbi.1998.1948. [DOI] [PubMed] [Google Scholar]
- 6.Kreitman RJ, Wilson WH, Bergeron K, Raggio M, Stetler-Stevenson M, FitzGerald DJ, et al. Efficacy of the anti-CD22 recombinant immunotoxin BL22 in chemotherapy-resistant hairy-cell leukemia. N Eng J Med. 2001;345:241–7. doi: 10.1056/NEJM200107263450402. [DOI] [PubMed] [Google Scholar]
- 7.Vassaux G, Martin-Duque P. Use of suicide genes for cancer gene therapy: study of the different approaches. Expert Opin Biol Ther. 2004;4:519–530. doi: 10.1517/14712598.4.4.519. [DOI] [PubMed] [Google Scholar]
- 8.Yazawa K, Fisher WE, Brunicardi FC. Current progress in suicide gene therapy for cancer. World J Surg. 2002;26:783–789. doi: 10.1007/s00268-002-4053-5. [DOI] [PubMed] [Google Scholar]
- 9.Gottesman MM. Cancer gene therapy: an awkward adolescence. Cancer Gene Ther. 2003;10:501–508. doi: 10.1038/sj.cgt.7700602. [DOI] [PubMed] [Google Scholar]
- 10.Ogris M, Wagner E. Targeting tumors with non-viral gene delivery systems. Drug Discov Today. 2002;7:479–485. doi: 10.1016/s1359-6446(02)02243-2. [DOI] [PubMed] [Google Scholar]
- 11.Kimchi-Sarfaty C, Ben-Nun-Shaul O, Rund D, Oppenheim A, Gottesman MM. In Vitro-Packaged SV40 Pseudovirions as Highly Efficient Vectors for Gene Transfer. Hum Gene Ther. 2002;13:299–310. doi: 10.1089/10430340252769815. [DOI] [PubMed] [Google Scholar]
- 12.Kimchi-Sarfaty C, Arora M, Sandalon Z, Oppenheim A, Gottesman MM. High cloning capacity of in vitro packaged SV40 vectors with no SV40 virus sequences. Hum Gene Ther. 2003;14:167–177. doi: 10.1089/104303403321070865. [DOI] [PubMed] [Google Scholar]
- 13.Kimchi-Sarfaty C, Gottesman MM. SV40 pseudovirions as highly efficient vectors for gene transfer and their potential application in cancer therapy. Curr Pharm Biotechnol. 2004;5:451–458. doi: 10.2174/1389201043376670. [DOI] [PubMed] [Google Scholar]
- 14.Sandalon Z, Dalyot-Herman N, Oppenheim AB, Oppenheim A. In vitro assembly of SV40 virions and pseudovirions: vector development for gene therapy. Hum Gene Ther. 1997;8:843–849. doi: 10.1089/hum.1997.8.7-843. [DOI] [PubMed] [Google Scholar]
- 15.Sandalon Z, Oppenheim A. Self-assembly and protein-protein interactions between the SV40 capsid proteins produced in insect cells. Virology. 1997;237:414–421. doi: 10.1006/viro.1997.8796. [DOI] [PubMed] [Google Scholar]
- 16.Hafkemeyer P, Brinkmann U, Gottesman MM, Pastan I. Apoptosis induced by Pseudomonas exotoxin: a sensitive and rapid marker for gene delivery in vivo. Hum Gene Ther. 1999;10:923–934. doi: 10.1089/10430349950018328. [DOI] [PubMed] [Google Scholar]
- 17.Kimchi-Sarfaty C, Alexander NS, Brittain S, Ali S, Gottesman MM. Transduction of multiple cell types using improved conditions for gene delivery and expression of SV40 pseudovirions packaged in vitro. BioTechniques. 2004;37:270–275. doi: 10.2144/04372RR04. [DOI] [PubMed] [Google Scholar]
- 18.Akiyama S, Fojo A, Hanover JA, Pastan I, Gottesman MM. Isolation and genetic characterization of human KB cell lines resistant to multiple drugs. Somat Cell Mol Genet. 1985;11:117–126. doi: 10.1007/BF01534700. [DOI] [PubMed] [Google Scholar]
- 19.Gottesman MM, Germann UA, Aksentijevich I, Sugimoto Y, Cardarelli CO, Pastan I. Gene transfer of drug resistance genes. Implications for cancer therapy. Ann N Y Acad Sci. 1994;716:126–138. doi: 10.1111/j.1749-6632.1994.tb21708.x. discussion 138-143. [DOI] [PubMed] [Google Scholar]
- 20.Chaudhary VK, Jinno Y, Gallo MG, FitzGerald D, Pastan I. Mutagenesis of Pseudomonas exotoxin in identification of sequences responsible for the animal toxicity. J Biol Chem. 1990;265:16306–16310. [PubMed] [Google Scholar]
- 21.FitzGerald DJ, Kreitman R, Wilson W, Squires D, Pastan I. Recombinant immunotoxins for treating cancer. Int J Med Microbiol. 2004;293:577–582. doi: 10.1078/1438-4221-00302. [DOI] [PubMed] [Google Scholar]
- 22.Kreitman RJ, Bailon P, Chaudhary VK, FitzGerald DJ, Pastan I. Recombinant immunotoxins containing anti-Tac(Fv) and derivatives of Pseudomonas exotoxin produce complete regression in mice of an interleukin-2 receptor-expressing human carcinoma. Blood. 1994;83:426–434. [PubMed] [Google Scholar]
- 23.Kreitman RJ, Chaudhary VK, Waldmann T, Willingham MC, FitzGerald DJ, Pastan I. The recombinant immunotoxin anti-Tac(Fv)-Pseudomonas exotoxin 40 is cytotoxic toward peripheral blood malignant cells from patients with adult T-cell leukemia. Proc Natl Acad Sci USA. 1990;87:8291–8295. doi: 10.1073/pnas.87.21.8291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beseth BD, Cameron RB, Leland P, You L, Varricchio F, Kreitman RJ, et al. Interleukin-4 receptor cytotoxin as therapy for human malignant pleural mesothelioma xenografts. Ann Thorac Surg. 2004;78:436–443. doi: 10.1016/j.athoracsur.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 25.Kawakami M, Kawakami K, Kioi M, Leland P, Puri RK. Hodgkin's lymphoma therapy with interleukin-4 receptor-directed cytotoxin in an infiltrating animal model. Blood. 2004;105:3707–3713. doi: 10.1182/blood-2004-08-3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kioi M, Kawakami K, Puri RK. Analysis of antitumor activity of an interleukin-13 (IL-13) receptor-targeted cytotoxin composed of IL-13 antagonist and Pseudomonas exotoxin. Clin Cancer Res. 2004;10(Pt 1):6231–6238. doi: 10.1158/1078-0432.CCR-04-0700. [DOI] [PubMed] [Google Scholar]
- 27.Vallera DA, Jin N, Shu Y, Panoskaltsis-Mortari A, Kelekar A, Chen W. Retroviral immunotoxin gene therapy of leukemia in mice using leukemia-specific T cells transduced with an interleukin-3/Bax fusion protein gene. Hum Gene Ther. 2003;14:1787–1798. doi: 10.1089/104303403322611791. [DOI] [PubMed] [Google Scholar]
- 28.Jain KK. Use of bacteria as anticancer agents. Expert Opin Biol Ther. 2001;1:291–300. doi: 10.1517/14712598.1.2.291. [DOI] [PubMed] [Google Scholar]
- 29.Theuer CP, FitzGerald DJ, Pastan I. A recombinant form of Pseudomonas exotoxin A containing transforming growth factor alpha near its carboxyl terminus for the treatment of bladder cancer. J Urol. 1993;149:1626–1632. doi: 10.1016/s0022-5347(17)36464-9. [DOI] [PubMed] [Google Scholar]
- 30.Xu YM, Wang LF, Jia LT, Qiu XC, Zhao J, Yu CJ, et al. A caspase-6 and antihuman epidermal growth factor receptor-2 (HER2) antibody chimeric molecule suppresses the growth of HER2-overexpressing tumors. J Immunol. 2004;173:61–67. doi: 10.4049/jimmunol.173.1.61. [DOI] [PubMed] [Google Scholar]
- 31.Kawakami M, Kawakami K, Puri RK. Interleukin-4-Pseudomonas exotoxin chimeric fusion protein for malignant glioma therapy. J Neurooncol. 2003;65:15–25. doi: 10.1023/a:1026294416718. [DOI] [PubMed] [Google Scholar]
- 32.Kreitman RJ, Puri RK, Pastan IA. Circularly permuted recombinant interleukin 4 toxin with increased activity. Proc. Natl. Acad. Sci. USA. 1994;91:6889–6893. doi: 10.1073/pnas.91.15.6889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen TY, Hsu CT, Chang KH, Ting CY, Whang-Peng J, Hui CF, et al. Development of DNA delivery system using Pseudomonas exotoxin A and a DNA binding region of human DNA topoisomerase I. Appl Microbiol Biotechnol. 2000;53:558–567. doi: 10.1007/s002530051657. [DOI] [PubMed] [Google Scholar]
- 34.Yerushalmi N, Brinkmann U, Brinkmann E, Pai L, Pastan I. Attenuating the growth of tumors by intratumoral administration of DNA encoding Pseudomonas exotoxin via cationic liposomes. Cancer Gene Ther. 2000;7:91–96. doi: 10.1038/sj.cgt.7700115. [DOI] [PubMed] [Google Scholar]
- 35.Jakubzick C, Kunkel SL, Puri RK, Hogaboam CM. Therapeutic targeting of IL-4- and IL-13-responsive cells in pulmonary fibrosis. Immunol Res. 2004;30:339–349. doi: 10.1385/IR:30:3:339. [DOI] [PubMed] [Google Scholar]
- 36.Kimchi-Sarfaty C, Garfield S, Alexander NS, Ali S, Cruz C, Chinnasamy D, et al. The pathway of uptake of SV40 pseudovirions packaged in vitro: from MHC class I receptors to the nucleus. Gene Ther Mol Biol. 2004;8:439–450. [Google Scholar]
- 37.Cogan PS, Koch TH. Studies of targeting and intracellular trafficking of an anti-androgen doxorubicin-formaldehyde conjugate in PC-3 prostate cancer cells bearing androgen receptor-GFP chimera. J Med Chem. 2004;47:5690–5699. doi: 10.1021/jm0495226. [DOI] [PubMed] [Google Scholar]