

Abstract
Maple syrup urine disease (MSUD) is an inborn error of metabolism caused by a deficiency in branched chain α-keto acid dehydrogenase that can result in neurodegenerative sequelae in human infants. In the present study, increased concentrations of MSUD metabolites, in particular α-keto isocaproic acid, specifically induced apoptosis in glial and neuronal cells in culture. Apoptosis was associated with a reduction in cell respiration but without impairment of respiratory chain function, without early changes in mitochondrial membrane potential and without cytochrome c release into the cytosol. Significantly, α-keto isocaproic acid also triggered neuronal apoptosis in vivo after intracerebral injection into the developing rat brain. These findings suggest that MSUD neurodegeneration may result, at least in part, from an accumulation of branched chain amino acids and their α-keto acid derivatives that trigger apoptosis through a cytochrome c-independent pathway.
INTRODUCTION
Maple syrup urine disease (MSUD) is an inborn error of metabolism caused by a deficiency in branched chain α-keto acid dehydrogenase, leading to the accumulation of branched chain amino acids (BCAAs; leucine, valine, and isoleucine) and a corresponding increase in their α-keto acid derivatives (BCKA; α-keto isocaproic acid [KICA], α-keto valeric acid, and α-keto-β-methyl-n-valeric acid [KILE]) levels (Snyderman, 1988). Acute neurological deterioration in children is often associated with increased plasma and cerebrospinal fluid concentrations of BCAA and BCKA (Riviello et al., 1991; Levin et al., 1993). Magnetic resonance imaging studies in children with MSUD have confirmed both white matter and neuronal injury, including extensive brain edema and pathological changes in the basal ganglia (Brismar et al., 1990; Steinlin et al., 1998). At the histopathological level, deficiencies in myelination of major tracts in the pons and spinal cord, widespread areas of spongy change in the white matter, focal areas of astrocytosis, and binucleated neurons have also been reported (Langenbeck, 1984). Because concentrations of BCAA are increased in the cerebrospinal fluid, we hypothesized that pathological changes in the central nervous system may reflect a neurotoxic effect of BCAAs and their keto acids.
Although the underlying mechanisms of cellular toxicity are not known, there is direct evidence that BCKAs affect mitochondrial enzymes, resulting in impaired energy metabolism (Dreyfus, and Prensky, 1967; Walajtys-Rode and Williamson, 1980; Jackson and Singer, 1983; Zielke et al., 1997). Because reduced mitochondrial function can trigger apoptosis, we set out to address the following questions: 1) can BCAAs or BCKAs induce apoptosis in neural cells in vitro and in vivo; and 2) is mitochondrial impairment involved in this toxic effect?
MATERIALS AND METHODS
Materials
Glucose-rich (4.5 g/l) Dulbecco's modified Eagle's medium (DMEM), leucine, valine, isoleucine, KICA, α-keto valeric acid, KILE, 3-(4,5-dimethylthiazol-2-yl)2,5-diphenyl-tetrazolium bromide (MTT), staurosporine (SSP), and cycloheximide were obtained from Sigma (Poole, United Kingdom). Fetal calf serum (FCS) and tissue culture plastics were obtained from Life Technologies (Paisley, United Kingdom). Eight-well chamber slides were obtained from Nunc Laboratories (Naperville, IL). The mitochondrial membrane potential indicators 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzinamidazol carbocyanine iodide (JC-1), rhodamine-123 (Rh-123), and tetramethylrhodamine ethyl ester (TMRE) were purchased from Molecular Probes (Eugene, OR). The cell-permeable caspase inhibitors Boc-Asp-fluoromethylketone (BAF), Z-Asp-Glu-Val-Asp-fluoromethylketone (DEVD-FMK), and Ac-Ile-Glu-Thr-Asp-fluoromethylketone (IETD-FMK) were purchased from Enzyme Systems Products (Livermore, CA).
Cell Culture
Cell Lines.
B104 (rat neuroblastoma), N1E-115 (mouse neuroblastoma/rat glioma hybrid), and C6 (rat glioma) cells were cultured in 10-cm tissue culture dishes, in DMEM containing 10% FCS, supplemented with penicillin and streptomycin. Cells were seeded at 2 × 105 per dish, subcultured twice weekly, and incubated at 37°C in a humidified atmosphere of 10% CO2 and 90% air. Individual cultures were maintained for no more than 6 wk.
Culture and Assay of Cerebellar Granular Neurons.
Cerebellar granule cultures were prepared from the cerebella of 7-d-old rat pups as described previously (Pocock et al., 1993). Cells were plated on poly-d-lysine-coated coverslips at a density of 2.5 × 105 per coverslip and maintained in minimum essential medium with Earle's salts supplemented with 25 mM KCl, 30 mM glucose, 25 mM NaHCO3, 1 mM glutamine, and 10% FCS, incubated at 37°C in a humidified atmosphere of 5% CO2 and 95% air, and used within 8 d.
Hoechst 33342 labeling of granule cell nuclei was carried out as described previously (Yan et al., 1994). Cells were washed in PBS, fixed for 10 min in 4% paraformaldehyde (PFA) at 4°C, washed once in distilled water, and incubated with Hoechst 33342 (5 μg/ml) for 15 min. Nuclear morphology was viewed using an Olympus Optical (London, United Kingdom) IX70 inverted fluorescence microscope with excitation at 365 nm and emission at 490 nm. For quantitative analysis, small, highly fluorescent nuclei were scored as apoptotic, and five separate coverslips (two fields per coverslip, 100–200 cells per field) were counted for each data point.
Preparation of Primary Oligodendrocytes and Astrocytes.
Glial cultures were prepared from newborn Wistar rat brains (Collarini et al., 1992) and enriched for oligodendrocytes and astrocytes by sequential immunopanning (Barres et al., 1992) as described previously.
Survival Assay by the Tetrazolium Salt Method (MTT assay)
The tetrazolium salt assay relies on the conversion of MTT to colored formazan by succinate dehydrogenase in metabolically active cells and provides a measurement of cell viability. For viability experiments, 100-μl aliquots of DMEM/0.5% FCS containing 104 cells (for cell lines) or 50-μl aliquots of Sato's medium containing 5 × 103 primary astrocytes or oligodendrocytes were placed into 96-well tissue culture plates and treated with defined concentrations of BCAA, BCKA, SSP, or cycloheximide. At the end of the experiment, cell viability was measured by MTT assay as previously described (Hansen et al., 1989). Results are expressed as percent viability according to the equation:
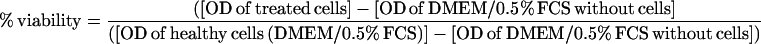 |
To exclude the possibility of misleading readings from cell lysates (e.g., increased metabolism in surviving cells), cell death was also quantified on the basis of apoptotic morphology combined with the absence of MTT metabolism. Both methods yielded comparable results. For all cell survival experiments, either the protein synthesis blocker cycloheximide or the protein kinase inhibitor SSP was used as a positive control for apoptosis. In experiments in which cell-permeable caspase inhibitors were used, these were added to a final concentration of 100 μM at the same time as α-keto isocaproic acid (KICA) or SSP.
Electron Microscopy
C6 cells (105/ml) were cultured in 24-well plates (0.5 ml/well) on sterile coverslips. At the start of the experiment, cells were treated with 600 μl of medium containing 0.5% FCS alone or defined concentrations of BCAA, BCKA, SSP, or cycloheximide. After incubation for 20 h at 37°C, cultures were washed twice in PBS, fixed in 2% glutaraldehyde for 2 h at 4°C, washed, osmicated, and dehydrated before embedding in Taab resin. Coverslips were snapped off with liquid nitrogen, and 1-μm sections were cut and stained with toluidine blue for block selection at the light microscope level. Sections of 100 nm thickness were then cut and collected on nickel grids, stained with uranyl acetate and lead citrate, and examined by electron microscopy (CM-10; Philips, Eindhoven, The Netherlands).
In Situ End Labeling (ISEL)
C6 cells were cultured in eight-well chamber slides at a density of 105/ml (300 μl/well) in DMEM containing 0.5% FCS alone or in the presence of defined concentrations of BCAA, BCKA, SSP, or cycloheximide. After a 20-h incubation at 37°C, ISEL was performed as described previously (Ansari et al., 1993) with minor modifications (Joashi et al., 1999).
DNA Laddering
One of the hallmarks of apoptosis is the endonuclease-mediated degradation of chromatin, giving rise to characteristic DNA laddering (Wyllie et al., 1992). To investigate apoptotic DNA fragmentation, C6 cells were cultured on 5-cm dishes at a density of 4 × 106 cells per dish (5 ml). At the start of the experiment, cultures were washed and treated with 2 ml of DMEM containing 0.5% FCS alone or in the presence of BCAA, BCKA, or SSP for defined times at 37°C. DNA from treated cultures was isolated (Laird et al., 1991) and assayed for oligonucleosomal laddering (Khan et al., 1997) as described previously.
Western Blotting
To measure caspase-dependent cleavage of poly(ADP-ribose)polymerase (PARP), C6 cells were cultured on six-well plates at a density of 106 cells/ml (2.5 ml/well). After 12 h, cultures were washed and treated with 500 μl of DMEM containing 0.5% FCS alone or defined concentrations of BCAA, BCKA, or SSP. At defined times, total cells from each well were lysed in 1% SDS (500 μl/well) and heated for 5 min at 90°C. Protein concentrations were determined by the bicinochinic acid method using a commercial kit from Pierce (Chester, United Kingdom), and lysates were stored at −80°C before use.
Total cellular proteins (50 μg/lane) were separated on a 7.5% polyacrylamide gel and electrotransferred onto nitrocellulose membranes (Hybond ECL; Amersham, Little Chalfont, United Kingdom). Primary incubations were with mouse monoclonal anti-PARP antibody C-2-10 (Transduction Laboratories, Lexington, KY) diluted 1:3000 for 1 h at room temperature. Secondary incubations were with horseradish peroxidase-conjugated anti-mouse antibody (Amersham) diluted 1:1000 under the same conditions. Bound antibodies were visualized with enhanced chemiluminescence reagent (Supersignal horseradish peroxidase; Pierce), and serial exposures were made to radiographic film (Hyperfilm ECL; Amersham). The resulting blots were scanned by densitometry for band quantitation.
Caspase Activity Assays
C6 cells (1 × 106 per well) were grown on six-well plates. At the start of the experiment, cultures were washed and treated with 0.5 ml of DMEM containing 10% conditioned medium alone or in the presence of KICA or SSP for 3 h at 37°C. Monolayers were then washed twice in PBS and lysed in a buffer containing 50 mM HEPES, pH 7.4, 0.1 mM EDTA, 1 mM DTT, and 0.1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid (50 μl/well). The lysates were freeze fractured three times (−80°C, 10 min) and clarified by centrifugation (5 min, 10,000 × g). Supernatants were used for enzyme assays using the caspase 3 substrate (Z-Asp-Glu-Val-Asp-pNA) purchased from Biomol (Plymouth Meeting, PA). The caspase assay was carried out according to the protocol provided by the manufacturer, and absorbance was measured at 405 nm in a spectrophotometer plate reader.
Studies of Cell Respiration
Cellular oxygen consumption was measured polarographically in both intact and digitonin-permeabilized C6 cells. At the start of the experiment, C6 cells cultured on 10-cm dishes (3 × 106 cells per dish) were washed and treated with DMEM containing 0.5% FCS alone or in the presence of defined MSUD metabolites or SSP (1 μM). After 4 h, cell suspensions were prepared from treated monolayers, and measurements of respiration were performed on intact cells as described previously (Rustin et al., 1994). In addition, the succinate oxidation rate was assayed in parallel cultures permeabilized with digitonin (0.002%, wt/vol), with successive additions of rotenone (3 μM), succinate (10 mM), ATP (0.2 mM), and cytochrome c (20 μM) as described (Rustin et al., 1994). Protein content was estimated according to the method of Bradford (1976), and the results were normalized accordingly.
Cytochrome c Release
The release of cytochrome c from the mitochondria to the cytosol was investigated by Western blotting of fractionated cells. C6 cells were cultured on 10-cm dishes, washed, and incubated for defined times at 37°C in DMEM containing 0.5% FCS alone or in the presence of KICA or SSP. At the end of the experiment, mitochondrial and cytosolic fractions were prepared as described previously (Rickwood et al., 1987) and assayed (25 μg/lane) by Western blotting as described above. The primary antibodies used were 1) a mouse monoclonal antibody raised against cytochrome c (PharMingen, San Diego, CA) used at a dilution of 1:500 and 2) a mouse monoclonal antibody raised against subunit IV of cytochrome oxidase (Molecular Probes) used at a dilution of 1:500.
Determination of Mitochondrial Membrane Potential
Changes in mitochondrial membrane potential (Δψm) were measured at both the population and single-cell levels.
Determination of Δψm in Cultures of C6 Cells.
To measure Δψm changes in whole cultures of C6 cells treated with KICA, the carbocyanine dye JC-1 was used as a mitochondrial membrane potential indicator probe. When excited at 490 nm, JC-1 is able to selectively enter the mitochondria and form aggregates that emit at 585 nm (orange–red). If the Δψm is reduced, JC-1 disaggregates to monomers that emit fluorescence at 527 nm (green). Thus, the color of the dye changes reversibly from orange to green as the membrane depolarizes (Reers et al., 1991; Smiley et al., 1991; Salvioli et al., 1997).
The potassium ionophore valinomycin (1 μM) and the protontranslocator carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP, 50 μM) were used as positive controls for disrupting the electrochemical gradient of mitochondria. SSP served as a positive control for apoptosis. Aliquots of C6 cells (50 μl, equivalent to 104 cells per well) were placed in 96-well plates and treated with defined concentrations of aqueous solutions of KICA, valinomycin, FCCP, or SSP. To measure Δψm, drugs were removed at the times stated by gently washing the cells in DMEM. Next, 50 μl of treatment solution containing 6 μM JC-1 (made from a frozen stock solution of 10 mM JC-1 in dimethylfluoride) in serum-free DMEM were added to each well, and cultures were incubated for a further 20 min at 37°C. Subsequently the cell monolayers were washed in PBS, and the fluorescence was measured using a Cytofluor 2300 plate reader (Millipore, Watford, United Kingdom; excitation λ, 485 nm; emission λ, 530 and 590 nm). The results are expressed as the ratio of JC-1 monomers against aggregates to reflect changes in Δψm.
Single-Cell Fluorescence Imaging.
C6 cells or primary cerebellar granule neurons were cultured as described above and plated in 24-well plates on circular glass coverslips at a density of 105 cells/ml (0.5 ml/well). Cells were preloaded with single dyes by incubation in 6 μM JC-1 (20 min), 200 nM TMRE (90 min), or 1.3 μM Rh-123 (15 min) at 5% CO2/air and 37°C. The cells were briefly washed, and 250 μl of phenol red-free MEM, supplemented with HEPES and glucose (Sigma) were added to each well. Individual coverslips were placed in the thermostated holder (set to 37°C) of an Olympus IX70 inverted fluorescence microscope. At defined time points KICA (50 mM) or FCCP (50 μM) was gently added directly to the chamber. Images were captured using a diachroic mirror with excitation λ and emission λ, respectively, at 490 and 590 nm (JC-1), 520 and 550 nm (TMRE), and 485 and 520 nm (Rh-123) using a SpectraMASTER monochromator (Life Science Resources, Cambridge, United Kingdom). For JC-1 experiments, an Omega Optical XF32 590-nm emission filter (OF35; Molecular Probes) was fitted to visualize only the red signal from JC-1 aggregates and to prevent contamination of the emission signal with green fluorescence from disaggregated JC-1 monomers. Images were acquired using an AstroCam 12-bit digital camera, and the output was visualized with a Merlin imaging system, version 1.85 (both from Life Science Resources).
In Vivo Studies of KICA Neurotoxicity
Animal Preparation and Intracerebral Injection.
All animal procedures used were in accordance with the United Kingdom Home Office guidelines and specifically licensed under the Animals (Scientific Procedures) Act, 1986. Anesthesia in 14-d Wistar rats was induced and maintained with halothane (5 and 1–2%, respectively) in oxygen:air (1:1). The skull was exposed, and the interaural line was visualized. A 23-gauge needle with syringe was stereotactically inserted through a small burr hole into the right forebrain (2.6 mm anterior to the interaural line, 1.5 mm laterally, 3.0 mm deep). Animals received a single 2-μl bolus of 0.9% saline alone or containing KICA (50, 100, and 200 mM; all adjusted for isotonicity and pH 7.4) injected over 2 min. Two animals were used at each time point and for each KICA concentration studied, with two animals receiving saline. After wound closure, animals were returned to their dam.
Histology and ISEL.
At 24 h and 5 d after injection, animals were killed (by intraperitoneal injection of pentobarbitone, 30 mg/kg) and transcardially perfused with 25–30 ml of 0.9% NaCl followed by 25–30 ml of PFA (4%, wt/vol, in 0.9% NaCl). Brains were then removed, fixed overnight in 4% PFA at 4°C, rinsed in PBS, and then transferred to 15% (wt/vol) sucrose in PBS (4°C). Specimens were routinely dehydrated in serial alcohols and paraffin embedded before sectioning. Coronal sections (5 μm) were cut at a point corresponding to between 2.4 and 2.8 mm anterior to the interaural line (Sherwood and Timiras, 1970).
ISEL was performed as described previously (Ansari et al., 1993) with two modifications. First, before the addition of hydrogen peroxide, sections were dewaxed and digested with proteinase K (20 μg/ml) for 15 min at room temperature. Second, after incubation in diaminobenzidine reagent for 30 min, reactions were quenched in tap water followed by PBS, and slides were counterstained with Cole's hematoxylin. Slides were serially dehydrated, cleared in xylene, and mounted with DPX (BDH-Merck, Poole, United Kingdom). ISEL-positive nuclei were visualized with light microscopy.
RESULTS
MSUD Metabolites Are Toxic to Cultured Glial and Neuronal Cells
Because MSUD results in an accumulation of BCAAs and their keto acid derivatives, we first investigated the effect of the MSUD metabolite KICA on cell viability in selected glial and neuronal lines by MTT assay. KICA induced cell death in all these lines in a dose-dependent manner (Figure 1A). At the highest dose tested (50 mM KICA), viability in C6, B104, and N1E-115 cells was reduced to 34, 44, and 46%, respectively. Because C6 (astroglial) cells were the most sensitive, and because MSUD is primarily a white matter disease, this line was selected for further investigation.
Figure 1.

Effect of MSUD metabolites on the viability of defined glial and neuronal cell lines. (A) C6 (black bars), N1E-115 (gray bars), or B104 (white bars) cultures were incubated for 20 h with DMEM/0.5% FCS containing 10, 25, or 50 mM KICA as indicated or 1 μM cycloheximide (CHX). Cell death was then assessed by MTT assay. (B) Comparison of the effects of BCAAs or their corresponding keto derivatives on the viability of C6 cells. Cultures were incubated for 20 h with DMEM/0.5% FCS containing leucine, valine, isoleucine, or their keto acids: KICA, α-keto isovaleric acid (KIVA), and KILE, respectively, each at a concentration of 25 mM. Cell viability was then assessed by MTT assay. (C) MSUD metabolites act synergistically to induce apoptosis. The combined effect of increased concentrations of leucine and its keto acid on the viability of rat C6 glioma cells is shown. Cultures were incubated for 20 h with increased concentrations of leucine (white triangles), KICA (white circles), or both at equimolar concentrations (black squares). Cell viability was then assessed using the MTT assay. In all the above experiments, data are expressed as percent cell death and represent the mean ± SEM of three independent experiments.
We next compared the effect of BCAAs with their keto acid derivatives (Figure 1B). In every case the α-keto acid was more toxic than its parent BCAA. For example, in C6 cultures treated with 25 mM isoleucine or its analogue KILE alone, the maximum amount of cell death observed was 7 and 55%, respectively. KICA is the most abundant BCKA in MSUD patients and was therefore selected for further investigation.
Leucine and KICA Act Synergistically to Induce Cell Death
In MSUD, both BCAAs and their respective α-keto acids accumulate. Thus, we next investigated the effects of combined treatment of C6 cells with leucine and its keto derivative KICA. As shown in Figure 1C, leucine was not significantly toxic up to a concentration of 10 mM, whereas at the same concentration, KICA resulted in 27% cell death. The combination of 10 mM leucine and 10 mM KICA significantly reduced cell viability to 41%.
The Mechanism of Cell Death Induced by MSUD Metabolites Is Apoptosis
Morphological analysis at the subcellular level remains the most conclusive method for distinguishing apoptosis from necrosis. Hematoxylin and eosin staining revealed that cultures underwent a marked change in morphology after treatment with BCKAs. Although control cultures of C6 cells had distinct processes and large, rounded nuclei, KICA-treated cells were smaller, displaying reduced cytoplasmic volume and marked nuclear pyknosis; moreover, these shrunken (and sometimes fragmented) nuclei were significantly more basophilic than their healthy counterparts (our unpublished results).
Induction of apoptosis by KICA was confirmed by electron microscopy (Figure 2). Healthy cultures of C6 cells were spindle shaped with large, oval, euchromatic nuclei. Cytoplasm contained dark, somewhat elongated mitochondria and considerable quantities of intermediate filaments (possibly glial fibrillary acidic protein), characteristically arranged in bundles parallel to the plasma membrane. In contrast, KICA-treated cells demonstrated typical apoptotic morphology; the cells were rounded, containing condensed nuclei with chromatin crescents. Cytoplasm was shrunken and electron dense with prominently dilated endoplasmic reticulum, frequently seen to open onto the cell surface, a feature consistent with cell shrinkage caused by the expulsion of water. Organelles, including mitochondria, were tightly packed into the remaining cytosol (Figure 2A). In some cells characteristic cytoplasmic blebbing could be seen, leading to the formation of numerous apoptotic bodies from a single cell (Figure 2B). Changes in mitochondrial morphology, in particular reduction in mitochondrial volume, close intermitochondrial juxtaposition, and retention of seemingly functional intact double membrane structure, were also consistent with cell death by apoptosis.
Figure 2.

Effect of KICA on ultrastructural morphology of C6 cells in culture. Cultures were incubated for 20 h with DMEM/0.5%FCS containing 25 mM KICA, and the morphology was examined by electron microscopy. (A) This cell has a rounded, condensed appearance. The nucleus has typical chromatin crescents (arrowheads), and shrunken cytoplasm that is electron dense. Organelles (white circles) are crammed together, and the endoplasmic reticulum is highly dilated, with frequent connections to the exterior (thin arrows). (B) This cell has undergone cytoplasmic blebbing (B) before total condensation of the nucleus. Perinuclear heterochromatin is visible (thin arrow), and cytoplasmic blebs contain autophagic vacuoles (arrowheads). Magnification, 9900×.
Molecular Evidence for Apoptosis: Caspase Activation
Biochemical markers for caspase activation and endonuclease activation after treatment with BCKA provided further evidence that the mode of cell death was apoptosis. Caspase activation was first investigated by measuring cleavage of PARP (a known substrate) to an Mr 85,000 fragment. Immunoblotting analysis revealed that healthy C6 cells predominantly expressed the intact Mr 116,000 PARP protein, with only a minor band (14% of the total PARP protein) at Mr 85,000, representing the caspase-3-cleaved product. After treatment with KICA or the protein kinase inhibitor SSP for 1 h, PARP cleavage increased significantly. This effect was time dependent, with maximum cleavage (45%) occurring at 3 h after treatment with KICA (Figure 3A). In addition to intact PARP and the Mr 85,000 cleavage product, immunoblotting with the PARP antibody revealed a third protein with an apparent molecular mass of 104,000 that was detected in both control and treated cultures and did not increase significantly after treatment with KICA.
Figure 3.

Effect of KICA on PARP cleavage and caspase activity. (A) PARP cleavage in C6 cells assessed by Western blot analysis after treatment with DMEM/0.5% FCS containing 25 mM KICA for 0 (control) 1, 2, or 3 h as indicated. The amount of cleaved PARP was defined by measuring the signal intensity of the Mr 85,000 (85K) cleavage product expressed as a percentage of the total combined signal for the 85K and Mr 116,000 (116K) bands. (B) Caspase activation in C6 cells after treatment for 3 h with DMEM/0.5% FCS alone (con) or in the presence of 50 mM KICA (black bar) or 1 μM SSP (gray bar) as indicated. Caspase activation was assessed by colorimetric assay using DEVD-pNA as a substrate. Results were normalized for differences in protein content between lysates, and the results are expressed as fold increase ± SEM in caspase activity relative to untreated cultures (n = 6). (C) Effect of caspase inhibitors on the toxic effects of KICA. C6 cells were incubated for 20 h with 10% conditioned medium (10% CM), 50 mM KICA, or 1 μM SSP either alone (white bars) or in the presence of 100 μg/ml BAF (black bars), DEVD-FMK (gray bars), or IETD-FMK (hatched bars). Cell death was then assessed by MTT assay.
Caspase activity was also measured directly, using the caspase-3 substrate DEVD-pNA. After 3 h of KICA treatment, caspase activity increased in C6 cells by 220%. Although this was a significant increase over control levels, it represented only 30% of DEVD-specific caspase activation in response to SSP, which was almost eightfold higher than untreated cultures (Figure 3B). We next investigated the role of caspase activation in the toxic effects of KICA. Cells exposed to KICA or SSP were simultaneously treated with cell-permeable caspase inhibitors. Three separate inhibitors were used: the ubiquitous caspase inhibitor BAF, the preferred caspase-3 substrate DEVD-FMK, and the caspase-8 substrate IETD-FMK. As shown in Figure 3C, in cells treated with KICA or SSP alone, MTT metabolism was reduced by 55 and 67%, respectively. This reduction in MTT metabolism was significantly blocked by the presence of BAF but not by DEVD-FMK or IETD-FMK (Figure 3C).
Molecular Evidence for Apoptosis: DNA Fragmentation
Further molecular evidence of apoptosis after treatment with BCKAs was obtained from ISEL studies to detect DNA fragmentation indicative of apoptosis. In control cultures incubated in 0.5% FCS alone, <5% of the cells showed nuclear labeling, indicative of DNA fragmentation. In contrast, in KICA-treated cultures the majority of nuclei were stained positive by ISEL (Figure 4A). Similar morphological changes were observed in the B104 and N1E-115 cell lines and with other BCKAs (our unpublished results). In a separate series of experiments, we were also able to detect the endonuclease-mediated degradation of chromatin, giving rise to characteristic DNA laddering. C6 cells were incubated for 0–48 h with KICA (50 mM), and total DNA was visualized at defined time points by agarose gel electrophoresis. KICA induced DNA laddering in a time-dependent manner: cells at the beginning of the experiment contained only high-molecular-weight DNA, whereas at time points from 12 h onward, evidence of DNA fragmentation was present. From 18 h onward, low-molecular-weight DNA species could be detected that migrated as a ladder, with fragments differing by ∼200 bp (Figure 4B). In a separate series of experiments, KICA was found to induce DNA laddering in a dose-dependent manner, with maximal endonuclease activation occurring at the 50 mM (our unpublished results). Taken together, these data provide molecular evidence that apoptotic proteases and endonucleases are activated in C6 cells after KICA treatment.
Figure 4.

Effect of KICA on DNA fragmentation of in rat glioma cells in culture. (A) Cultures were incubated for 20 h with DMEM/0.5% FCS containing 25 mM KICA, and the morphology was investigated by light microscopy after ISEL. Healthy and apoptotic cells are indicated by arrowheads and arrows, respectively. Magnification, 175×. (B) Analysis of nucleosome laddering in C6 rat glioma cells at defined time points after treatment with DMEM/0.5% FCS containing 50 mM KICA. DNA molecular weight markers are indicated (in base pairs) by the arrows.
KICA Triggers a Reduction in Cell Respiration
Because MSUD metabolites have been shown to inhibit mitochondrial enzymes (Land et al., 1976; Jackson and Singer, 1983), we investigated the effect of KICA on cell respiration by polarography. As shown in Table 1, after a 4-h exposure to KICA or SSP, oxygen consumption by intact cells was impaired, although mitochondrial succinate oxidation in permeabilized cells was comparable with control values. Significantly, these parameters were not altered after the addition of exogenous cytochrome c (Figure 5B). At the same time point, however, there was a 25% (KICA) and 50% (SSP) reduction in MTT metabolism (Figure 5A), indicating that respiratory chain function was normal despite the cells being in the irreversible phase of apoptosis.
Table 1.
Polarographic studies of the effects of KICA on mitochondrial function in intact C6 glioma cells
| Control (n = 3) | KICA (n = 4) | |
|---|---|---|
| Oxygen consumption (mmol O2 · min−1 · mg protein−1) | 19.9 ± 1.0 | 4.0 ± 2.5 |
| Succinate oxidation rate without exogenous cyt c (mmol O2 · min−1 · mg protein−1) | 20.4 ± 0.7 | 21.8 ± 1.3 |
| Succinate oxidation rate with exogenous cyt c (mmol O2 · min−1 · mg protein−1) | 20.4 ± 0.7 | 21.8 ± 1.3 |
Oxygen consumption in intact cells was assessed after a 4-h exposure to 50 mM KICA or medium alone (control). Succinate oxidation was measured (after the same treatment) in cells permeabilized with digitonin in the presence or absence of cytochrome c (cyt c). Results are expressed as mean ± SEM.
Figure 5.

Effect of KICA on cellular oxygen consumption and respiratory chain function in C6 glioma cells. (A) Effect of different time exposures of C6 glioma cells to 50 mM KICA (white squares) or 1 μM SSP (white circles) on cell viability assessed by MTT assay. Data are expressed as the mean ± SEM of three experiments. (B) Effect of cytochrome c addition on respiratory function. After a 4-h exposure to 50 mM KICA, C6 rat cells were permeabilized with digitonin. Succinate was then added as an electron donor, and oxygen consumption was measured kinetically by polarography before and after the addition of cytochrome c at the time indicated. (C) Effect of KICA on the subcellular localization of cytochrome c. After a 4-h exposure to 0.5% FCS alone as a control (lanes 1 and 4), to 50 mM KICA (lanes 2 and 5), or to 1 μM SSP (lanes 3 and 6), mitochondrial fractions (lanes 1–3) and cytosolic fractions (lanes 4–6) of C6 cells were prepared, and the presence of cytochrome c was assessed by Western blotting using a mouse monoclonal antibody (top panel). The mitochondrial content of each cell fraction was assessed by Western blotting with a cytochrome oxidase-specific monoclonal antibody (bottom panel).
KICA Induces Mitochondrial Death without Cytochrome c Release
A number of recent studies have established that cytochrome c release from the mitochondria into the cytosol is a frequent feature of the apoptotic program (Krippner et al., 1996; Kluck et al., 1997; Yang et al., 1997). We therefore investigated the effect of KICA on the release of cytochrome c into the cytosol of C6 cells. As shown in Figure 5C, both cytochrome oxidase and cytochrome c were absent from the cytosol of control cultures. Similarly, exposure of C6 cells to KICA for up to 4 h resulted in a slight increase in cytochrome c in the cytosolic fraction, although increased levels were also observed in the mitochondrial fraction. In contrast, SSP-treated cultures contained large amounts of cytochrome c in the cytosol, and this was accompanied by a reduction in mitochondrial cytochrome c levels. Cytochrome oxidase was only detected in mitochondrial fractions in all samples tested and was unaltered after exposure to KICA or SSP (Figure 5C). These data confirmed our polarographic studies indicating that KICA induced impaired cellular oxygen consumption without detectable cytochrome c involvement.
KICA Does Not Trigger Early Changes in Mitochondrial Membrane Potential
To investigate possible changes in Δψm, C6 cells were loaded with the cationic fluorochrome JC-1, the aggregation of which depends on the Δψm. In control cultures, the ratio of JC-1 monomers to aggregates was <0.37, reflecting the baseline for healthy mitochondria in C6 cells. After a 20-h treatment with SSP (1 μM) or valinomycin (1 μM), this figure increased to 0.74 and 0.83, respectively, indicating significant mitochondrial membrane depolarization. In contrast, a range of concentrations of KICA up to 100 mM did not induce significant changes in Δψm (Figure 6A). To exclude the possibility that early or transient changes in Δψm were missed, a time course study was carried out, measuring changes in JC-1 fluorescence between 1 and 20 h after the addition of KICA or control drugs. Valinomycin induced rapid changes in Δψm, detected as early as 10 min after addition (our unpublished results) and sustained up to 20 h (Figure 6B). SSP induced similar changes in mitochondrial membrane potential, although the kinetics were slower; 3–4 h of treatment was needed before changes in JC-1 disaggregation could be detected (our unpublished results), whereas the effect was still present at the 20-h time point. In contrast, KICA treatment did not result in mitochondrial depolarization at any of the time points tested (Figure 6B), even 20 h after KICA treatment, although at this time point cells were not viable (Figure 1). To investigate this apparent paradox further, we expressed the fluorescence data from JC-1 monomers and aggregates separately. As shown in Figure 6B, inset, fluorescence at both 530 and 590 nm was significantly reduced at 20 h. Thus although the ratio of JC-1 monomers to aggregates was unaltered, the mitochondria were clearly compromised in many cells at this time point.
Figure 6.

Effect of KICA on the mitochondrial membrane potential (Δψm) of C6 cells. (A) Dose–response study of the effects of KICA on Δψm. Cell monolayers were incubated for 20 h with DMEM/0.5% FCS either alone (C) or containing 1, 3, 10, 30, or 100 mM KICA as indicated or 1 μM SSP (S). Δψm was assessed using the fluorescent probe JC-1, added 20 min before the end of the experiment. (B) Time course study of the effects of KICA onΔψm. Cell monolayers were incubated for defined times with DMEM/0.5% FCS containing 50 mM KICA or for 20 h with DMEM/0.5% FCS alone (C) or 1 μM SSP (S). At the times indicated (20 h for controls), Δψm was assessed as described above. For both experiments, 1 μM valinomycin (V) served as a positive control for depolarization. Results are expressed as the ratio between emission at 530 and 590 nm, reflecting the ratio between JC-1 monomers and aggregates. (B, inset) Time course of changes in JC-1 fluorescence emission at 530 nm (white circles) and 590 nm (black circles). Results are expressed as the percent change in fluorescence relative to values at time 0 (100%).
The lack of effect of KICA on Δψm was confirmed at the single-cell level by fluorescence imaging. Healthy C6 cells loaded with JC-1 contained numerous intact mitochondria arranged in a perinuclear manner. When excited at 490 nm, cells were highly fluorescent at an emission wavelength of 590 nm, indicating the presence of JC-1 aggregates. Within seconds of treatment with the proton ionophore FCCP, a significant reduction in fluorescence was observed that increased with time (our unpublished observations). In contrast, no depolarization occurred at early time points after KICA addition. At later time points only viable C6 cells contained polarized mitochondria. These data indicate that KICA impaired mitochondrial metabolism in C6 cells without affecting membrane potential at early time points.
KICA Induces Apoptosis in Primary Neurons and Glia without Early Changes in Δψm
It was important to determine whether the toxic effects of KICA could be reproduced in primary neurons. We therefore investigated the effects of KICA on rat cerebellar granule cells (CGCs). Although control cultures contained predominantly healthy nuclei (Figure 7A), KICA-treated cells (Figure 7B) underwent marked nuclear pyknosis, comparable with those treated with SSP (Figure 7C). Quantitative analysis indicated that KICA induced apoptosis in CGCs in a dose-dependent manner, with 50% death observed at KICA concentrations between 1 and 10 mM (Figure 8A). Because MSUD is primarily a white matter disease, we also investigated the toxic effects of KICA on primary rat oligodendrocytes and astrocytes. As in the case of CGCs, KICA induced cell death in oligodendrocytes and astrocytes in a dose-dependent manner. At lower concentrations (1 mM) both glial cell types were more sensitive to KICA than neurons, with oligodendrocytes (Figure 8B) slightly more sensitive than astrocytes (Figure 8C). The dead cells displayed classic morphological features of apoptosis, including cytoplasmic shrinkage and nuclear pyknosis, and were indistinguishable from those exposed to SSP (our unpublished results).
Figure 7.

Effect of KICA on the nuclear morphology and mitochondrial membrane potential (Δψm) of primary cerebellar granule neurons at the single-cell level. Cells were cultured on circular glass coverslips and treated with DMEM/0.5% FCS alone (A and D) or in the presence of 50 mM KICA (B and E) or 1 μM SSP (C and F) for 24 h. At the end of the experiment, cultures were stained either with Hoechst 33342 for nuclear morphology (A-C) or with JC-1 for Δψm (D-F). Healthy and apoptotic nuclei are indicated (in A) by arrowheads and arrows respectively. The arrows in D and E indicate cells with punctate JC-1 fluorescence, indicative of mitochondrial localization. Magnification, 400×.
Figure 8.

Effect of KICA on the viability of primary neurons and glia. Cerebellar granule neurons (A) oligodendrocytes (B), or astrocytes (C) were incubated for 24 h with MEM/5% FCS alone (con) or in the presence of KICA (black bars) at defined concentrations or 1 μM SSP. Cell death was then assessed by nuclear morphology after Hoechst 33342 staining (A) or by MTT assay with microscopic quantitation of surviving cells (B and C). Data are expressed either as percent apoptosis (A) or percent cell survival (B and C) and represent the mean ± SEM of at least two independent experiments (n = 10 [A] and 6 [B and C]).
We next investigated the effect of KICA and SSP on Δψm using three separate mitochondrial potential indicator dyes, JC-1, TMRE, and Rh-123. At early time points in SSP-treated CGC cultures, the fluorescence of JC-1 aggregates (Figure 7F) was diminished, and the dye had dispersed into the cytoplasm, indicating marked depolarization. In contrast, at comparable times in KICA-treated cells (Figure 7E), JC-I fluorescence was comparable with controls (Figure 7D): bright, punctate staining, which was colocalized to mitochondria. At later time points, the number of viable CGCs was reduced, although those remaining retained mitochondria in the polarized state. These data were confirmed using the rhodamine-based dyes Rh-123 and TMRE (our unpublished data).
Mitochondrial membrane potential was also investigated in primary CGCs using the rhodamine-based dyes TMRE and Rh-123. Solvent addition or KICA treatment did not cause a significant reduction in either TMRE (Figure 9A) or Rh-123 (Figure 9B) fluorescence. In contrast, the addition of the uncoupler FCCP (after KICA) immediately depolarized mitochondria by up to 35% (Figure 9). In both TMRE- and Rh-123-loaded cells, KICA did cause a slight reduction in fluorescence at later time points, although these cells still responded equally well to FCCP. After prolonged periods of loading, both TMRE and Rh-123 fluorescence was significantly reduced, even in control cultures. Microscopic examination of the cultures indicated that TMRE was toxic to primary CGC' (our unpublished data). These data confirmed our findings with JC-1, indicating that early changes in Δψm did not precede caspase activation in KICA-treated cells.
Figure 9.

Effect of KICA on mitochondrial membrane potential in cerebellar granule neurons. Cells were grown on coverslips and loaded with 200 nM TMRE (90 min) (A) or 1.3 μM Rh-123 (15 min) (B) and imaged 5 s before (gray bars) and 5 s after (black bars) addition of solvent (CON), 50 mM KICA, or 200 μM FCCP. In each case FCCP was added 8 min after KICA treatment. The results are expressed as percent fluorescence (measured at the appropriate emission λ for each dye) at the start of the experiment and represent the mean ± SEM from 50 individually imaged cells.
Intracerebral Injection of KICA Induces Neuronal Apoptosis In Vivo
Taken together, these results suggested that the neural impairment observed in MSUD patients was a direct consequence of KICA neurotoxicity. To test the ability of KICA to induce apoptosis in vivo, we injected defined concentrations ranging from 0 to 200 mM (corrected for osmolarity and pH 7.40) into the hippocampus of 14-d-old rats and investigated apoptosis by ISEL at 24 h and 5 d after injection. Injection of 0.9% NaCl had no detectable effect on cell survival after 24 h or 5 d (Figure 10A). In contrast, after intracerebral injection of KICA, cell apoptosis could be detected by ISEL in the area surrounding the injection site, encompassing the CA1 region and the dentate gyrus of the hippocampus. The number of apoptotic cells in this region increased with the concentration of KICA, with a maximal effect observed 5 d after injection of 200 mM KICA (Figure 10, B and C). When viewed under higher magnification, the majority of apoptotic cells were pyramidal neurons and granule cells (Figure 10C). These results indicated that KICA also triggered cell apoptosis in vivo in a dose-dependent manner.
Figure 10.

ISEL of cells in the postnatal day 14 rat hippocampus 5 d after intracerebral injection of 0.9% NaCl alone (A) or containing 200 mM KICA (B). No ISEL staining is detectable in NaCl-treated animals. However, after injection of KICA, numerous ISEL-positive cells are seen in the CA1 region and dentate gyrus of the hippocampus. Under higher magnification the majority of ISEL-positive cells (arrowheads) display the morphological characteristics of apoptotic pyramidal neurons and granule cells (C). Magnification: A and B, 250×; C, 630×.
DISCUSSION
Toxic effects of the metabolites that accumulate in MSUD have been previously demonstrated in the rat cerebellum, where myelination was severely impaired (Silberberg, 1969), and in lymphoblastoid cell lines from MSUD patients, where significant growth inhibition was observed (Skaper et al., 1976). In earlier studies the effects on C6 cells were interpreted as an increase in cell cycle time; morphological changes indicative of apoptosis were present but not commented on (Liao et al., 1978). In the present study, we demonstrate that the toxic effects of MSUD metabolites are due to the induction of apoptotic cell death, verified by classical morphological criteria, ISEL of apoptotic nuclei, evidence of caspase activation and nucleosome laddering.
It should noted that caspase 3 activity was only moderately increased after exposure to KICA compared with SSP. Moreover, the observation that neither IETD-FMK nor DEVD-FMK prevented the decrease in MTT reduction suggests that neither activation of caspase 8 nor that of caspase 3 is a major component of the KICA apoptosis pathway. This is consistent with a growing number of separate studies (Miller et al., 1997; Ha et al., 1998; Monney et al., 1998; Drenou et al., 1999; Mateo et al., 1999; Jones et al., 2000) indicating that apoptosis can proceed through caspase-independent pathways. On the other hand, the protective effects of BAF and the characteristic cleavage of PARP indicate that other caspases may be activated in KICA-induced apoptosis.
All three BCKAs tested induced significant reductions in glial and neuronal cell viability. These results are consistent with those of Bissel et al. (1974), who observed that the replication of mouse fibroblasts was inhibited by all three BCKAs (KICA, α-keto valeric acid, and KILE), Zielke et al. (1997), who recently reported that KICA reduced energy metabolism in rat brain, and Patel (1974), who found that all three BCKAs inhibited the mitochondrial BCKA dehydrogenase complex in the developing rat brain. In contrast, Silberberg (1969) did not observe any toxic effects of α-keto valeric acid on myelinating cultures of rat cerebellum. One explanation might be that these cells are known to metabolize BCAAs quickly. Alternatively, cerebellar cultures may be resistant to the effects of BCAAs but succumb to combinations of the BCAA with the corresponding keto acid. The data presented here emphasize the importance of this synergy: leucine at high concentrations is only slightly toxic but at lower concentrations acts synergistically with BCKA to trigger apoptosis. This effect may be particularly important in the brain where high aminotransferase activity rapidly metabolizes leucine to KICA (Brand et al., 1984). We found that the two other BCKAs that accumulate in MSUD are also toxic to C6 cells and may therefore contribute to neurological damage in MSUD patients, although their concentration in plasma is relatively low (Snyderman et al., 1984).
Previous studies suggested that KICA disrupts energy metabolism by inhibiting the mitochondrial pyruvate and α-ketoglutarate dehydrogenases (Dreyfus and Prensky, 1967; Walajtys-Rode and Williamson, 1980; Jackson and Singer, 1983) and the pyruvate and β-hydroxybutyrate translocases (Land et al., 1976). Moreover, it has become apparent that apoptotic execution can involve the release of mitochondrial cytochrome c into the cytosol (Kluck et al., 1997; Yang et al., 1997). The resulting impairment of mitochondrial function can be largely corrected by the addition of exogenous cytochrome c (Krippner et al., 1996).
Because mitochondrial changes at the ultrastructural level in KICA-treated cells are consistent with apoptosis, it is not clear whether these changes reflect the cause or are a consequence of apoptosis. We therefore investigated whether KICA triggers apoptosis through a mechanism impinging on cell oxidative metabolism and the release of cytochrome c. In particular we measured intact cell respiration and the rate of mitochondrial succinate oxidation in permeabilized cells (reflecting the function of respiratory chain complexes II, III, and IV of the ubiquinone pool and of cytochrome c). Our results indicate that cell respiration is significantly decreased by KICA, and this parallels an irreversible commitment to apoptotic death in C6 cells. In contrast, KICA does not affect the succinate oxidation rate, suggesting that the respiratory chain is essentially unaffected after exposure to MSUD metabolites; in particular, mitochondrial cytochrome c levels do not become limiting. This was confirmed by immunoblotting studies and by the inability of exogenous cytochrome c to correct the succinate oxidation rate. Similar observations have been made in murine T lymphocytes (Hockenbery et al., 1993). These data suggest that apoptotic execution can proceed without significant loss of cytochrome c and without changes in Δψm and agree with recent studies in lymphoid cell lines (Tang et al., 1998), Hela cells (Bossy-Wetzel et al., 1998), myeloid cells (Finucane et al., 1999), and primary cerebellar granule neurons (Paterson et al., 1998).
Our immunoblotting data are in accord with a number of recent reports that mitochondrial release of cytochrome c is not an obligatory event for apoptotic cell death but is dependent on the apoptotic trigger in a range of cell types (Adachi et al., 1997; Chauhan et al., 1997; Li et al., 1997; Tang et al., 1998). Indeed, we and others (Tang et al., 1998) have observed an increase in mitochondrial cytochrome c levels without any increase in the cytosol. So, although cell respiration is impaired after exposure to MSUD metabolites, it is not clear whether this is the trigger or a consequence of apoptotic cell death. The absence of significant early changes in mitochondrial cytochrome c or Δψm, are consistent with the latter possibility.
Our data using three separate dyes to measure Δψm conclusively show that KICA does not induce early mitochondrial depolarization. This observation is in accord with recent studies challenging the assertion that a reduction in Δψm is a ubiquitous event in the apoptotic program (Garland et al., 1997; Finucane et al., 1999). It is worthy of note that in our hands all three dyes used to measure Δψm showed inadequacies at prolonged time points: JC-1 fluorescence ratios remained constant after a 20-h exposure to KICA, although the mitochondria were metabolically dead. On closer examination, fluorescence of both JC-1 monomers and J-aggregates, probably because of severely damaged mitochondria, was equally unable to retain the dye. Using Rh-123 or TMRE, potential-dependent staining of mitochondria can also be obtained, with membrane potential measurements largely following the Nernst equation. In this study, Rh-123-loaded CGCs showed reduced fluorescence at late time points, probably because of self-quenching (Bindokas et al., 1998), and TMRE, which has been shown to inhibit cell respiration (Scaduto and Grotyohann, 1999), was toxic after prolonged treatment of CGCs.
Our results provide useful information for patient management; plasma leucine levels are routinely used to monitor the treatment of MSUD, and circulating concentrations of BCAAs are a good indicator of the risk of neurological injury (Riviello et al., 1991). On the other hand, our results indicate that keto acids are more toxic than their parent amino acids and suggest that it would be more beneficial to patients if circulating BCKAs were reduced rather than the parent BCAAs.
The present study also showed that direct intracerebral injection of KICA leads to neuronal apoptosis in the hippocampus of neonatal rats in a dose-dependent manner. To our knowledge, this is the first demonstration of the neurotoxic effect of BCKAs and, if confirmed in brain specimens from MSUD patients, will have important implications for the design of therapeutic strategies to prevent or limit cerebral injury that occurs during the acute-onset MSUD. Our observations in vivo also suggest, rather provocatively, that other conditions associated with keto acid accumulation (e.g., diabetic acidosis) may also have an apoptotic component in the neurological deficit.
In summary, we have demonstrated that the branched chain amino and keto acids that accumulate in MSUD trigger apoptosis in glial and neuronal cells in vitro and in vivo in a dose- and time-dependent manner. These observations may explain, at least in part, the neurological sequelae associated with high plasma concentrations of MSUD metabolites.
ACKNOWLEDGMENTS
We thank the Weston Foundation for continued financial support (to M.K., A.D.E., and H.M.). D.L.T. is funded by Wellcome Trust grant 046343/z/95; U.J. is an Action Research clinical research fellow; and K.G. is supported by Sir Jules Thorn Charitable Trust grant 96/76A.
REFERENCES
- Adachi S, Cross AR, Babior BM, Gottlieb RA. Bcl-2 and the outer mitochondrial membrane in the inactivation of cytochrome c during Fas-mediated apoptosis. J Biol Chem. 1997;272:21878–21882. doi: 10.1074/jbc.272.35.21878. [DOI] [PubMed] [Google Scholar]
- Ansari B, Coates P, Greenstein B, Hall P. In situ end-labeling detects DNA strand breaks in apoptosis and other physiological and pathological states. J Pathol. 1993;170:1–8. doi: 10.1002/path.1711700102. [DOI] [PubMed] [Google Scholar]
- Barres BA, Hart IK, Coles HSR, Burne JF, Voyvodic JT, Richardson WD, Raff MC. Cell death and control of cell survival in the oligodendrocyte lineage. Cell. 1992;70:31–46. doi: 10.1016/0092-8674(92)90531-g. [DOI] [PubMed] [Google Scholar]
- Bindokas VP, Lee CC, Colmers WF, Miller RJ. Changes in mitochondrial function resulting from synaptic activity in the rat hippocampal slice. J Neurosci. 1998;18:4570–4587. doi: 10.1523/JNEUROSCI.18-12-04570.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissel M, Bensch K, Herman M. Effects of maple syrup urine disease metabolites on mouse L-fibroblasts in vitro: a fine structural and biochemical study. J Neurochem. 1974;22:957–964. doi: 10.1111/j.1471-4159.1974.tb04322.x. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Newmeyer D, Green D. Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO J. 1998;17:37–49. doi: 10.1093/emboj/17.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Brand K, Hauschildt S, Lüthje J. Effect of diets on the activity of enzymes involved in branched chain α-keto acid metabolism. In: Adibi SA, Fekl W, Langenbeck U, Schauder P, editors. Branched Chain Amino and Keto Acids in Health and Disease. Vol. 1. Basel: Karger; 1984. pp. 100–111. [Google Scholar]
- Brismar J, Aqeel A, Brismar G, Coates R, Gascon G, Ozand P. Maple syrup urine disease: findings on CT and MR scans of the brain in 10 infants. AJNR Am J Neuroradiol. 1990;11:1219–1228. [PMC free article] [PubMed] [Google Scholar]
- Chauhan D, Pandey P, Ogata A, Teoh G, Krett N, Halgren R, Rosen S, Kufe D, Kharbanda S, Anderson K. Cytochrome c-dependent and -independent induction of apoptosis in multiple myeloma cells. J Biol Chem. 1997;272:29995–29997. doi: 10.1074/jbc.272.48.29995. [DOI] [PubMed] [Google Scholar]
- Collarini EJ, Kuhn R, Marshall CJ, Monuki ES, Lemke G, Richardson WD. Down-regulation of the POU transcription factor SCIP is an early event in oligodendrocyte differentiation in vitro. Development. 1992;116:193–200. doi: 10.1242/dev.116.1.193. [DOI] [PubMed] [Google Scholar]
- Drenou B, Blancheteau V, Burgess DH, Fauchet R, Charron DJ, Mooney NA. A caspase-independent pathway of MHC class II antigen-mediated apoptosis of human B lymphocytes. J Immunol. 1999;163:4115–4124. [PubMed] [Google Scholar]
- Dreyfus P, Prensky A. Further observations on the biochemical lesion in maple syrup urine disease. Nature. 1967;214:276. doi: 10.1038/214276a0. [DOI] [PubMed] [Google Scholar]
- Finucane DM, Waterhouse NJ, Amarante-Mendes GP, Cotter TG, Green DR. Collapse of the inner mitochondrial transmembrane potential is not required for apoptosis of HL60 cells. Exp Cell Res. 1999;25:166–174. doi: 10.1006/excr.1999.4527. [DOI] [PubMed] [Google Scholar]
- Garland JM, Sondergaard KL, Jolly J. Redox regulation of apoptosis in interleukin-3-dependent hemopoietic cells: absence of alteration in both mitochondrial membrane potential (delta psi (m)) and free radical production during apoptosis induced by IL3 withdrawal. Br J Haematol. 1997;99:756–765. doi: 10.1046/j.1365-2141.1997.4733277.x. [DOI] [PubMed] [Google Scholar]
- Ha HC, Woster PM, Casero RA., Jr Unsymmetrically substituted polyamine analogue induces caspase-independent programmed cell death in Bcl-2-overexpressing cells. Cancer Res. 1998;58:2711–2714. [PubMed] [Google Scholar]
- Hansen M, Nielsen S, Berg K. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J Immunol Methods. 1989;119:203–210. doi: 10.1016/0022-1759(89)90397-9. [DOI] [PubMed] [Google Scholar]
- Hockenbery D, Oltvai Z, Yin X, Milliman C, Korsmeyer S. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 1993;75:241–251. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- Jackson R, Singer T. Inhibition of the 2-ketoglutarate and pyruvate dehydrogenase complexes of beef heart by branched chain keto acids. J Biol Chem. 1983;258:1857–1865. [PubMed] [Google Scholar]
- Joashi UC, Greenwood K, Taylor D, Kozma M, Mazarakis ND, Edwards AD, Mehmet H. Poly (ADP ribose) polymerase cleavage precedes neuronal death in the hippocampus and cerebellum following injury to the developing rat forebrain. Eur J Neurosci. 1999;11:91–100. doi: 10.1046/j.1460-9568.1999.00409.x. [DOI] [PubMed] [Google Scholar]
- Jones BE, Lo CR, Liu H, Srinivasan A, Streetz K, Valentino KL, Czaja MJ. Hepatocytes sensitized to tumor necrosis factor-alpha cytotoxicity undergo apoptosis through caspase-dependent and caspase-independent pathways. J Biol Chem. 2000;275:705–712. doi: 10.1074/jbc.275.1.705. [DOI] [PubMed] [Google Scholar]
- Khan S, Kayahara M, Joashi U, Mazarakis ND, Sarraf C, Edwards AD, Hughes MN, Mehmet H. Differential induction of apoptosis in Swiss 3T3 cells by nitric oxide and the nitrosonium cation. J Cell Sci. 1997;110:2315–2322. doi: 10.1242/jcs.110.18.2315. [DOI] [PubMed] [Google Scholar]
- Kluck R, Bossy-Wetzel E, Green D, Newmeyer D. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- Krippner A, Matsuno-Yagi A, Gottlieb R, Babior B. Loss of function of cytochrome c in jurkat cells undergoing Fas-mediated apoptosis. J Biol Chem. 1996;271:21629–21636. doi: 10.1074/jbc.271.35.21629. [DOI] [PubMed] [Google Scholar]
- Laird P, Zijderveld A, Linders K, Rudnicki M, Jaenisch M, Berns A. Simplified mammalian DNA isolation procedure. Nucleic Acids Res. 1991;19:4293. doi: 10.1093/nar/19.15.4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land J, Mobray J, Clark J. Control of pyruvate and β-hydroxybutyrate utilization in rat brain mitochondria and its relevance to phenylketonuria and maple syrup urine disease. JNeurochem. 1976;26:823–830. doi: 10.1111/j.1471-4159.1976.tb04458.x. [DOI] [PubMed] [Google Scholar]
- Langenbeck U. Pathobiochemical and pathophysiologic analysis of the MSUD phenotype. In: Adibi SA, Fekl W, Langenbeck U, Schauder P, editors. Branched Chain Amino and Keto Acids in Health and Disease. Vol. 1. Basel: Karger; 1984. pp. 315–334. [Google Scholar]
- Levin M, Scheinmann A, Lewis R, Beaudet A. Cerebral edema in maple syrup urine disease. J Pediatr. 1993;122:167–168. doi: 10.1016/s0022-3476(05)83521-8. [DOI] [PubMed] [Google Scholar]
- Li F, Srinivasan A, Wang Y, Armstrong R, Tomaselli K, Fritz L. Cell-specific induction of apoptosis by microinjection of cytochrome c. Bcl-xL has activity independent of cytochrome c release. J Biol Chem. 1997;272:30299–30305. doi: 10.1074/jbc.272.48.30299. [DOI] [PubMed] [Google Scholar]
- Liao C, Herman M, Bensch K. Prolongation of G1 and S phase in C-6 glioma cells treated with maple syrup urine disease metabolites. Morphologic and cell cycle studies. Lab Invest. 1978;38:122–133. [PubMed] [Google Scholar]
- Mateo V, Lagneaux L, Bron D, Biron G, Armant M, Delespesse G, Sarfati M. CD47 ligation induces caspase-independent cell death in chronic lymphocytic leukemia. Nat Med. 1999;5:1277–1284. doi: 10.1038/15233. [DOI] [PubMed] [Google Scholar]
- Miller TM, Molder KL, Knudson CM, Creedon DJ, Deshmukh M, Korsmeyer SJ, Johnson EM., Jr Bax deletion further orders the cell death pathway in cerebellar granule cells and suggests a caspase-independent pathway to cell death. J Cell Biol. 1997;139:205–217. doi: 10.1083/jcb.139.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monney L, Otter I, Olivier R, Ozer HL, Haas AL, Omura S, Borner C. Defects in the ubiquitin pathway induce caspase-independent apoptosis blocked by Bcl-2. J Biol Chem. 1998;273:6121–6131. doi: 10.1074/jbc.273.11.6121. [DOI] [PubMed] [Google Scholar]
- Patel M. Inhibition by the branched chain 2-oxoacids of the 2-oxoglutarate dehydrogenase complex in developing rat and human brain. Biochem J. 1974;144:91–97. doi: 10.1042/bj1440091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson I, Zhang D, Warrington R, Boulton A. R-deprenyl and R-2-heptyl-N-methylpropargylamine prevent apoptosis in cerebellar granule neurons induced by cytosine arabinoside but not low extracellular potassium. J Neurochem. 1998;70:515–523. doi: 10.1046/j.1471-4159.1998.70020515.x. [DOI] [PubMed] [Google Scholar]
- Pocock JM, Cousin MA, Nicholls DG. The Ca2+ channel coupled to the exocytosis of l-glutamate from cerebellar granule cells is inhibited by the spider toxin Aga-GI. Neuropharmacology. 1993;32:1185–1194. doi: 10.1016/0028-3908(93)90012-r. [DOI] [PubMed] [Google Scholar]
- Reers M, Smith T, Chen L. J-aggregate formation of a carbocyanine as a quantitative fluorescent indicator of membrane potential. Biochemistry. 1991;30:4480–4486. doi: 10.1021/bi00232a015. [DOI] [PubMed] [Google Scholar]
- Rickwood D, Wilson M, Darky-Usmar V. Mitochondria: A Practical Approach, ed V. Darky-Usmar, D. Rickwood, and M. Wilson. Oxford: IRL Press; 1987. Isolation and characteristics of intact mitochondria; pp. 1–16. [Google Scholar]
- Riviello J, Rezvani I, DiGeorge A, Foley C. Cerebral edema causing death in children with maple syrup urine disease. J Pediatr. 1991;119:42–45. doi: 10.1016/s0022-3476(05)81036-4. [DOI] [PubMed] [Google Scholar]
- Rustin P, Chretien D, Bourgeron T, Gérard B, Rštig A, Saudubray JM, Munnich A. Biochemical and molecular investigations in respiratory chain deficiencies. Clin Chim Acta. 1994;228:35–51. doi: 10.1016/0009-8981(94)90055-8. [DOI] [PubMed] [Google Scholar]
- Salvioli S, Ardizzoni A, Franceschi C, Cossarizza A. JC-1, but not DiOC6 (3) or rhodamine 123, is a reliable fluorescent probe to assess Δψ changes in intact cells: implications for studies on mitochondrial functionality during apoptosis. FEBS Lett. 1997;411:77–82. doi: 10.1016/s0014-5793(97)00669-8. [DOI] [PubMed] [Google Scholar]
- Scaduto RC, Jr, Grotyohann LW. Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys J. 1999;76:469–477. doi: 10.1016/S0006-3495(99)77214-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood N, Timiras P. A Stereotaxic Atlas of the Developing Rat Brain. London: University of California Press; 1970. pp. 78–141. [Google Scholar]
- Silberberg D. Maple syrup urine disease metabolites studied in cerebellum cultures. J Neurochem. 1969;16:1141–1146. doi: 10.1111/j.1471-4159.1969.tb05959.x. [DOI] [PubMed] [Google Scholar]
- Skaper S, Molden D, Seegmiller J. Maple syrup urine disease: branched-chain amino acid concentrations and metabolism in cultured human lymphoblasts. Biochem Genet. 1976;14:527. doi: 10.1007/BF00485832. [DOI] [PubMed] [Google Scholar]
- Smiley S, Reers M, Mottola-Hartshorn C. Intracellular heterogeneity in mitochondrial membrane potential revealed by a J-aggregate-forming lipophilic cation JC-1. Proc Natl Acad Sci USA. 1991;88:3671–3675. doi: 10.1073/pnas.88.9.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyderman S. Treatment outcome of maple syrup urine disease. Acta Paediatr Jpn. 1988;30:417–424. doi: 10.1111/j.1442-200x.1988.tb02531.x. [DOI] [PubMed] [Google Scholar]
- Snyderman S, Goldstein F, Sansaricq C, Norton P. The relationship between the branched chain amino acids and their α-keto acids in maple syrup urine disease. Pediatr Res. 1984;18:851–853. doi: 10.1203/00006450-198409000-00009. [DOI] [PubMed] [Google Scholar]
- Steinlin M, Blaser S, Boltshauser E. Cerebellar involvement in metabolic disorders: a pattern-recognition approach. Neuroradiology. 1998;40:347–354. doi: 10.1007/s002340050597. [DOI] [PubMed] [Google Scholar]
- Tang D, Li L, Zhu Z, Joshi B. Apoptosis in the absence of cytochrome c accumulation in the cytosol. Biochem Biophys Res Commun. 1998;242:380–384. doi: 10.1006/bbrc.1997.7969. [DOI] [PubMed] [Google Scholar]
- Walajtys-Rode E, Williamson J. Effects of branched chain α-ketoacids on the metabolism of isolated rat liver cells. J Biol Chem. 1980;255:413–418. [PubMed] [Google Scholar]
- Wyllie A, Arends M, Morris R, Walker S, Evan G. The apoptosis endonuclease and its regulation. Semin Immunol. 1992;4:389–397. [PubMed] [Google Scholar]
- Wyllie A, Duvall E. Cell injury and death. In: Jo'D McGee PI, Wright NA, editors. Oxford Textbook of Pathology. Oxford: Oxford University Press; 1992. pp. 141–193. [Google Scholar]
- Yan GM, Ni B, Weller M, Wood KA, Paul SM. Depolarisation of glutamate receptor activations blocks apoptotic cell death of cultured cerebellar granule neurons. Brain Res. 1994;656:43–51. doi: 10.1016/0006-8993(94)91364-1. [DOI] [PubMed] [Google Scholar]
- Yang J, Liu X, Bhalla K, Kim C, Ibrado A, Cai J, Peng T, Jones D, Wang W. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- Zielke HR, Huang Y, Baab PJ, Collins RM, Zielke CL, Tildon JT. Effect of alpha-ketoisocaproate and leucine on the in vivo oxidation of glutamate and glutamine in the rat brain. Neurochem Res. 1997;22:1159–1164. doi: 10.1023/a:1027325620983. [DOI] [PubMed] [Google Scholar]