

Abstract
Numerous factors have been theorized to affect the development of antimicrobial resistance, including those specific to the host, the organism, the environment, the drug, and the drug prescriber. One variable under the control of the prescriber is the drug dosing regimen. Dosing regimens can vary in dose level, dosing interval, and treatment duration. The current studies examined the relationships between antimicrobial dosing regimens and resistance development by use of an in vivo model. A murine model of systemic Candida albicans infection was used to examine resistance emergence during exposure to the triazole antifungal fluconazole. Data from this experimental model demonstrated that the more frequently administered dosing prevented selection of the isogenic resistant cell populations. Conversely, dosing regimens producing prolonged sub-MIC effects appeared to contribute to the outgrowth of isogenic resistant strains. The association between dosing and resistance emergence observed in the current investigation is disparate from that described for antimicrobial compounds with cidal killing characteristics. The inhibitory or static antimicrobial activity of the triazole compounds may explain these differences.
The rapid development and spread of antimicrobial resistance has become an increasingly serious public health problem in a wide range of infectious diseases (3, 28, 29, 37, 38, 44, 45, 53, 58). New drugs for the treatment of these resistant infections are unlikely to appear soon enough and in sufficient numbers to solve many of the resistance problems. Thus, it is imperative to understand the factors that lead to the evolution of resistance and to design strategies to prevent or delay the emergence of antimicrobial-resistant pathogens.
The need to stem the growing problem of antimicrobial resistance has prompted multiple calls for change in the use of antimicrobial agents to maximize the life spans of these drugs (6, 8, 10, 16, 21, 22, 30, 31, 38, 42, 53, 55). However, the relationship in the context of use pattern and resistance development is complex and remains, for the most part, undefined. One dosing regimen approach that has been shown to reduce the amplification of resistant strains involves the use of large, infrequent doses of antimicrobials to eliminate not only the susceptible populations but also any resistant mutants (7, 13, 15, 18, 25, 26, 28). The success of this dosing strategy has been demonstrated with a few antimicrobial drug class-organism combinations in which antimicrobial drugs exhibit extensive organism killing or “cidal” activity. For example, Jumbe et al. used an in vivo model system to define the fluoroquinolone concentration likely to select for resistant cell populations (25). However, studies have not considered the large group of antimicrobials that exhibit only inhibitory or “static” effects. These drugs are unable to effectively eliminate pathogen populations. Drug effectiveness is primarily the result of limiting additional organism generations. It is possible that alternative dosing strategies will be necessary to affect the emergence of resistance with these groups of antimicrobials. The goal of the current studies was to define the relationship between dose level and dosing frequency with a single “static” antimicrobial and modulation of clinically isogenic susceptible and resistant cell populations in an in vivo test system. For these investigations, a triazole antifungal, fluconazole, was chosen as the antimicrobial and the fungus Candida albicans was used as the infecting pathogen (14, 17, 20, 32, 33, 46, 49, 50, 51, 57). Animals were infected with an inoculum mixture of each strain pair in which the isogenic resistant strain represented a defined, small percentage of the total inoculum. Subsequent analysis characterized the pharmacokinetic (PK)/pharmacodynamic (PD) exposure associated with the outgrowth of the resistant cell populations.
MATERIALS AND METHODS
Microorganisms, antifungal drug, and in vitro susceptibility testing.
The six clinical strain pairs of Candida albicans used in this study are listed in Table 1. The strain pairs were chosen to include variations in MIC differences, stable drug resistance, and multiple resistance mechanisms. These strains were kindly provided by both T. White and J. Lopez-Ribot (32, 33, 57, 58, 59). The strains were isolated sequentially from human immunodeficiency virus-infected patients with oropharyngeal candidiasis. The first isolate listed for each pair represents the strain recovered prior to antifungal drug therapy. In each case the isolate was susceptible to fluconazole. The second listed isolate represents a (stably) fluconazole-resistant strain isolated from the same patient after drug therapy (and who, at the time of isolate recovery, was clinically failing antifungal therapy). The genetic relatedness of the strain pairs has previously been confirmed by restriction length polymorphism and DNA fingerprinting with the moderately repetitive probe Ca3 (47, 52). The mechanistic explanation for fluconazole resistance was also previously identified by either T. White or J. Lopez-Ribot and is listed in Table 1 (32, 33, 57, 58, 59). Pharmaceutical-grade fluconazole was obtained from Pfizer. Stock solutions were prepared in sterile water. Susceptibility testing was performed in duplicate on two occasions by using CLSI (formerly NCCLS) M27-A methods (39). The final results are expressed as the geometric means of these results.
TABLE 1.
C. albicans strain pairs, fluconazole MICs, resistance mechanism, and in vivo fitness
| Strain | MIC (μg/ml) | Resistance mechanism | Strain growth in untreated control animal (log10 CFU/kidney) |
|---|---|---|---|
| 1649 | 0.25 | NAa | 3.20 ± 0.48 |
| 2183 | 4.0 | CDR | 2.20 ± 0.19 |
| 1002 | 1.0 | NA | 3.09 ± 0.12 |
| 2823 | 32 | MDR, ERG11 | 3.06 ± 0.30 |
| 2-76 | 1.0 | NA | 3.44 ± 0.24 |
| 12-99 | 64 | CDR, MDR, ERG11 | 5.28 ± 0.14 |
| 412 | 0.25 | NA | 3.43 ± 0.21 |
| 2307 | 128 | CDR, ERG11 | 0.60 ± 0.11 |
| 580 | 0.50 | NA | 3.65 ± 0.32 |
| 2438 | 32 | Unknown | 3.38 ± 0.16 |
| FH1 | 1.0 | NA | 2.94 ± 0.20 |
| FH5 | 4.0 | CDR | 2.40 ± 0.12 |
NA, not applicable.
Inoculum preparation.
An inoculum consisting of both the isogenic susceptible and resistant strains was prepared for each of the strain pairs. In each case a 99% susceptible isolate and 1% resistant isolate mixture was used. For a subset of experiments, inocula containing the resistant strain at 0.1 and 10% were prepared. Inoculum cell counts were confirmed both with a hemocytometer and by plating.
Animal infection model.
The disseminated candidiasis model was chosen for these studies to allow consistent comparison of the results to those of prior fluconazole pharmacodynamic studies. Briefly, 6-week-old, specific-pathogen-free female CD1 mice weighing 23 to 27 g were rendered neutropenic prior to infection by intraperitoneal injection of cyclophosphamide at 4 days (150 mg/kg) and 1 day (100 mg/kg). The cyclophosphamide protocol results in a profound reduction in neutrophil levels (<100/mm3) throughout the period of study, allowing adequate growth of the Candida albicans strains in the mice (Harlan Sprague-Dawley, Indianapolis, IN) (1, 2). The inoculum of C. albicans K1 was injected via the lateral tail vein of the neutropenic mice. Drug treatment was initiated 2 h after infection. Groups of two animals were used for each treatment regimen. We chose the kidney as the initial end organ of study, as this is the most extensively characterized in vivo infection model for this pathogen (1, 2). The kidneys were processed for CFU enumeration in duplicate. The lower limit of detection for both susceptible and resistant cells was 100 CFU/kidney. The results were expressed as the mean CFU/two kidneys from two mice.
Tracking susceptible and resistant cell populations.
After infection and fluconazole therapy, kidney homogenates were quantitatively cultured. To quantify all viable organisms the homogenate was grown on Sabouraud dextrose agar plates. To quantify the fluconazole-resistant strain the homogenate was grown on CHROMagar plates containing fluconazole (4 μg/ml), as described previously (43). The quantity of the susceptible isolate was then calculated as the log-transformed difference in growth between the two plates. For each of the strain pairs, we confirmed the differentiating capability of this method by plating mixtures (0.1% resistant strain/99.9% susceptible strain, 1% resistant strain/99% susceptible strain, and 10% resistant strain/90% susceptible strain) of known concentrations of each strain (Table 2).
TABLE 2.
Validation of drug-containing plates used for tracking strain populations in vivo
| Strain pair | Plate | Log-transformed difference (%) in growth between the two plates, confirmed with mixtures containing the following concn (% susceptible/% resistant)
|
||
|---|---|---|---|---|
| 90/10 | 99/1 | 99.9/0.1 | ||
| 1649, 2183 | CHROMagar | 5.56 (15) | 3.98 (0.9) | 3.07 (0.11) |
| SDA | 6.40 (85) | 6.11 (99.1) | 6.10 (99.99) | |
| 1002, 2823 | CHROMagar | 5.18 (21) | 4.28 (1.7) | 3.11 (0.13) |
| SDA | 5.85 (79) | 5.99 (98.3) | 5.98 (99.87) | |
| 2-76, 12-99 | CHROMagar | 5.42 (14) | 4.00 (1.0) | 3.04 (0.16) |
| SDA | 5.85 (86) | 5.71 (99.0) | 5.82 (99.84) | |
| 412, 2307 | CHROMagar | 5.11 (8.0) | 4.12 (1.1) | 3.42 (0.11) |
| SDA | 6.01 (92) | 6.08 (98.9) | 6.38 (99.11) | |
| 580, 2438 | CHROMagar | 4.96 (13) | 4.36 (2.4) | 2.96 (0.09) |
| SDA | 5.84 (87) | 5.97 (97.6) | 6.02 (99.91) | |
| FH1, FH5 | CHROMagar | 4.99 (8.7) | 4.41 (2.3) | 3.00 (0.11) |
| SDA | 6.05 (91.3) | 6.04 (97.7) | 5.93 (99.89) | |
To confirm the capability of these culture methods to accurately track the quantity of the resistant and susceptible strains, we used a dominant selectable marker in one strain pair (5, 27). P. Magee kindly provided a vector containing a mycophenolic acid (MPA) resistance cassette [IMH3r flanked by ARG4 (p3394)] (24). C. albicans 12-99 was transformed with the linearized plasmid p3394 (19, 48). The strain was easily selected with MPA-containing plates (2.5 μg/ml) after 48 h of incubation. For the MPA-resistant strain 12-99 and susceptible strain 2-76, we confirmed the differentiating capability of MPA-containing plates by plating mixtures (0.1% resistant strains/99.9% susceptible strains, 1% resistant strains/99% susceptible strains, and 10% resistant strains/90% susceptible strains) of known concentrations of each strain.
C. albicans strain growth in vivo.
Because we wished to primarily test the impact of drug exposure on the emergence of each strain, we first determined the relative impact of drug resistance on fitness in the animal model. Animals were infected with each strain alone and for each strain pair in a 50/50 mixture (to estimate competitive growth). Growth in untreated control animals was determined over 24 h as a measure of in vivo fitness (Table 1). Strains which grew to a similar burden (within 1 log10 CFU/kidney) were considered “normally fit.” Strains which grew less (less than 1 log10 CFU/kidney than normal) or more were considered less fit or superfit (more than 1 CFU log10/kidney than normal), respectively.
Efficacy of fluconazole in single-isolate infection for each strain.
Groups of two animals were treated for 24 h with fourfold increasing total doses of fluconazole administered every 6 h. Total doses ranged from 0.8 to 800 mg/kg of body weight/24 h. The mice were euthanized after 24 h of therapy, and the kidneys were removed for CFU determination. Saline-treated control mice were euthanized just before treatment and at the end of the experiment. The relationship between fluconazole treatment and efficacy against each strain was examined on the basis of total dose per 24 h and the 24-h area under the concentration-time curve (AUC)/MIC for each dosing regimen.
Impact of fluconazole exposure on emergence of resistant isolates.
Mice had a total Candida burden of 4.36 ± 0.11 CFU/kidneys at the start of therapy. For all strain pairs a mixture of 99% susceptible strains and 1% resistant strains was used. For one strain pair we also studied ratios of 99.1% susceptible strains/0.1% resistant strains and 90% susceptible strains/10% resistant strains. Eighteen fluconazole dosing regimens (six total dose levels and three dosing intervals) were used in the treatment studies to vary PD magnitudes. Twenty-four-hour total doses similarly ranged from 0.8 to 800 mg/kg/day. The total doses were fractionated into one dose (every 24 h), two doses (every 12 h), and four doses (every 6 h) per day. We examined the outcomes after 72-h (all six pairs) and 24-h (one pair) treatment durations.
Fluconazole pharmacokinetics.
Serum kinetics were determined after the administration of single fluconazole doses of 6.25, 25, and 100 mg/kg in neutropenic infected mice. For each dose examined, groups of three mice were sampled one time by intracardiac puncture at 1- to 12-h intervals. Following collection, the blood was allowed to clot at 4°C. After the blood clotted, the samples were centrifuged (model MB centrifuge; International Equipment Co.) at 10,000 × g for 5 min, and the serum was removed and stored at −80°C until it was processed. Serum drug concentrations were determined by gas chromatography. Pharmacokinetic indices, including the elimination half-life, the area under the serum concentration-time curve from time zero to infinity (AUC0-∞), and peak concentration (Cmax), were calculated by using a noncompartmental model.
Pharmacodynamic analyses.
The pharmacokinetic/pharmacodynamic index values (percentage of time that the serum levels remain above the MIC [T > MIC], the ratio of Cmax relative to the MIC [Cmax/MIC], and AUC relative to the MIC [AUC/MIC]) were calculated for each of the 18 dosing regimens. Calculations were performed relative to the MIC of both the susceptible parent and the resistant cell populations. A sigmoid Emax model was used to characterize the relationship among each of the PK/PD indices for the treatment regimens and quantitation of the viable burden of both the susceptible and resistant populations for each of the six strain pairs (1, 2). The model is derived from the Hill equation, E = (Emax × DN)/(ED50N × DN), where E is the observed effect (change in log10 CFU/kidney compared with that for the untreated controls at the end of the treatment period), D is the total dose, Emax is the maximum effect, ED50 is the dose required to achieve 50% of the Emax, and N is the slope of the dose-effect relationship. The correlation between efficacy and each of the three indices studied was determined by nonlinear least-squares multivariate regression analysis. The coefficient of determination (R2) was used to estimate the percent variance in the change in log10 CFU/kidney over the treatment period for the different dosing regimens that could be attributed to each of the pharmacodynamic indices. The various relationships were examined to define the PK/PD exposure(s) that led to the emergence of resistant cell populations and that prevented the emergence of the resistant cell populations.
RESULTS
In vitro susceptibility testing.
The MICs for the initial (susceptible) strains ranged from 0.25 to 1.0 μg/ml (Table 1). The MICs for the later isolate ranged from 4.0 to 128 μg/ml. The rises in the MICs between the strain pairs ranged from 16- to 512-fold.
C. albicans strain growth in vivo.
The burden of viable organisms recovered from animals with single-pathogen infections was similar for four of the six strain pairs. The competitive growth of the strains from the 50/50 mixture was also similar for these four strain pairs, suggesting equivalent fitness in this infection model. The in vivo growth of fluconazole-resistant strain 2307 was less than 1 log over the 24-h experiment (the strain was categorized as less fit) and was nearly sixfold less than that of the drug-susceptible parent strain (strain 412). The growth of drug-resistant strain 12-99 and MPA-transformed strain 12-99 (DRA) showed that it appeared to be more fit in vivo than the drug-susceptible counterpart (and was categorized as superfit). The increase in kidney burden over the study period was more than 1.5-fold greater for this drug-resistant strain. Variations in fitness for the two strain pairs did not appear to be explained by a drug resistance mechanism(s).
Differential growth on drug-containing plates.
The fluconazole-susceptible strains did not grow after 48 h of incubation on the CHROMagar plates containing fluconazole. Similarly, the agar plates containing 2.5 μg/ml of MPA supported the growth of only pinpoint colonies for the MPA-susceptible cell population. Conversely, the entire drug-resistant population counted with a hemocytometer was recovered on each of the drug-containing plates. The MPA-resistant strain (strain 12-99) grew as a large colony at between 24 and 48 h. In the pilot experiment with different degrees of organism doping, the amounts of susceptible and resistant populations estimated by the use of hemocytometer counts were appropriately recovered on the respective tracking plates (Table 2).
Fluconazole pharmacokinetics.
The time course of the serum fluconazole concentrations following administration of subcutaneous doses of 6.25, 25, and 100 mg/kg is shown in Fig. 1. Total drug levels were used in the calculations due to the very low degree of protein binding (<10%). The inter- and intraday coefficients of determination ranged from 6.4 to 9.8% and 1.4 and 11.4%, respectively. The pharmacokinetic profile was linear, with the peak level and AUC0-∞ increasing proportionally for each dose escalation. Peak levels occurred within 1 h of administration and ranged from 5.2 to 100 μg/ml. The elimination half-life ranged from 2.8 to 3.5 h. The peak levels ranged from 5 to 100 μg/ml, and the 24-h AUC0-∞ values ranged from 24 to 460 mg · h/liter.
FIG. 1.

Single-dose pharmacokinetics of fluconazole in neutropenic infected mice. Each symbol represents the mean and standard deviation of the serum concentration values from three mice. t1/2, elimination half-life.
Efficacy of fluconazole against each strain in single-pathogen infection.
The dose-response relationship in the fluconazole treatment studies with each isolate alone is shown in Fig. 2. The dose-response curves for the isogenic resistant strains are shifted to the right, demonstrating reducing in vivo efficacy. However, when the dose-response relationship is expressed by use of the 24-h AUC/MIC in place of the total dose, the dose-response curves are similar. When the dosing regimens were expressed as the 24-h AUC/MIC ratio, the outcomes appeared to be independent of the MIC or the resistance mechanism. The relationship between the 24-h AUC/MIC and efficacy among the strains was quite strong (R2 = 84%).
FIG. 2.

(A) Relationship between the total fluconazole dose level (mg/kg) and change in C. albicans viable burden in the kidneys of neutropenic mice. Each symbol shape represents data for a different isogenic strain pair. Solid symbols, data for the susceptible strain; hollow symbols, data for the fluconazole-resistant strain. (B) The same data set used for panel A; however, the fluconazole drug exposure is expressed as the 24-h AUC/MIC ratio. Each symbol represents the mean values from two mice.
Impacts of fluconazole dose and dose fractionation on emergence of resistant strains.
We observed both the emergence and the prevention of emergence of the isogenic resistant strain with four of six strain pairs (Fig. 3). The data for the fluconazole-containing CHROMagar and MPA plates were identical (data not shown). Among the treatment regimens used, the more frequently administered dosing interval best suppressed the emergence of the resistant strain. For each total dose level administered, the growth of the resistant strain was less when the total dose was administered at smaller more frequent dose levels than at larger infrequent dose levels. A similar relationship was observed for each of the strain pairs. This relationship suggests that maintenance of drug concentrations above the MIC of the infecting pathogen best prevented replication of the resistant subpopulation. Conversely, a dosing strategy that resulted in drug concentrations many fold greater than the MIC followed by concentrations that fell below the MIC over time did not suppress the emergence of resistant subpopulations. For strain pair 412 and 2307, the isogenic resistant strain was less fit in this animal model. In each of the treatment groups and in the untreated controls, strain 2307 (the resistant isolate) did not grow. For the more fit, drug-resistant strain, strain 12-99, the organism grew as well as the susceptible strain, despite the drug exposure. Similar results were obtained when the inoculum contained a lower percentage of the resistant strain (0.1%). However, when the resistant strain was present as 10% of the starting inoculum, no treatment regimen prevented the additional emergence of the resistant strain (Fig. 4). The single experiment that examined the impact of treatment duration demonstrated the relevance of this factor. The longer treatment duration provided additional drug pressure and more time for the resistant strain to emerge (data not shown).
FIG. 3.

Impact of dosing frequency on the modulation of a strain of susceptible and resistant C. albicans in mice for six isogenic strain pairs. The fluconazole exposure is expressed as the total dose level, and the outcome is reported as the growth of susceptible (hollow symbols) and resistant (solid symbols) strains in the kidneys of neutropenic mice. Symbols represent the mean values from two mice. SAB, Sabouraud dextrose agar plates; CHROM, CHROMagar plates; q24h, q12h, q6h, dosing every 24, 12, and 6 h, respectively.
FIG. 4.

Impact of the starting percentage of the resistant isolate on the emergence of the resistant strain in the pair following in vivo fluconazole exposure. The fluconazole exposure is expressed as the total dose level, and the outcome is reported as the growth of susceptible (hollow symbols) and resistant (solid symbols) strains in the kidneys of neutropenic mice. The symbols represent the mean values from two mice. (Top three panels) Data in which the resistant isolate made up 0.1% of the total inoculum; (middle three panels) data in which the resistant isolate made up 1% of the total inoculum; (bottom three panels) data in which the resistant isolate made up 10% of the total inoculum. SAB, Sabouraud dextrose agar plates; CHROM, CHROMagar plates; q24h, q12h, q6h, dosing every 24, 12, and 6 h, respectively.
Relationship between drug exposure PK/PD and resistance emergence.
The pharmacokinetic/pharmacodynamic exposures resulting from the 18 fluconazole treatment regimens included T > MIC values that ranged from 0 to 100% and more than 1,000-fold ranges in the 24-h AUC/MIC and the Cmax/MIC values. The pharmacodynamic relationships (T > MIC, 24-h AUC/MIC, and Cmax/MIC) were expressed relative to the MICs of both the susceptible and the resistant strains in the strain pair. The relationships between the range of T > MIC and 24-h AUC/MIC values and the modulation of the emergence of the resistant cell populations were relatively strong (Fig. 5). However, there were datum points which fit less well. These data were for both the “less” and the “more” fit strains. The association between the fluconazole Cmax/MIC exposure and the emergence of a resistant cell phenotype was less evident (data not shown). The achievement of serum levels above the MIC of the resistant strain for nearly 50% of the dosing interval prevented the emergence of the resistant strain.
FIG. 5.

(Left panel) Relationship between the fluconazole 24-h AUC/MIC and modulation of the susceptible (hollow symbols) and resistant (solid symbols) C. albicans strains (six strain pairs); (right panel) relationship between the fluconazole T > MIC and modulation of the susceptible (hollow symbols) and resistant (solid symbols) C. albicans strains (six strain pairs). Each symbol represents a mean value from two mice.
Pharmacodynamic model development.
Nonlinear regression with a sigmoid Emax model reasonably described the data sets. To improve the model, two additional variables were considered and were built into the final mathematical model. Both factors improved the fit of the data (Fig. 6). The first factor included a descriptor of the in vivo fitness of the resistant strains. Three fitness groups (superfit, normal, and less fit) were considered, as defined above. The second factor built into the final model included representation of the drug exposure to include both AUC and dose fractionation. Because the data regressed reasonably well with both the AUC/MIC and the T > MIC indices, we attempted to develop a model to better distinguish between these interrelated indices. To accomplish this, the AUC/MIC index was used as the primary exposure variable. However, for each AUC/MIC value, a coefficient was included that assigned the dose fractionation schedule (dosing every 24, 12, and 6 h) used for each AUC/MIC exposure value. These additional variables were included in the final formula for the NONLIN model, as follows:
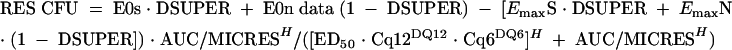 |
where RES CFU is the burden of organism CFU/kidney for the resistant strains, E0s is the effect for the more fit or the superfit strain, E0n is the effect for the normally fit strains, DSUPER is a dummy variable to engage the fitness data for each group, EmaxS is the maximal effect (CFU/kidney) for the more fit or the superfit strain, EmaxN is the maximal effect (CFU/kidney) for the normally fit strains, AUC/MICRES is the 24-h AUC/MIC or exposure, H is the Hill constant, ED50 is the 240h AUC/MIC associated with 50% of the maximal effect for the regimen administered every 24 h, Cq12 is the coefficient associated with dose fractionation every 12 h, DQ12 is a dummy variable to engage the dose fractionation every 12 h (when appropriate), Cq6 is the coefficient associated with dose fractionation every 6 h, and DQ6 is a dummy variable to engage the dose fractionation every 6 h (when appropriate).
FIG. 6.

(Left panel) Relationship between the fluconazole 24-h AUC/MIC and viable burden of six fluconazole-susceptible C. albicans strains in mice from mixed infection studies; (right panel) relationship between the fluconazole 24-h AUC/MIC and the viable burden of six fluconazole-resistant C. albicans strains in mice from mixed infection studies. Triangles, data for a “less” fit organism; circles, data for “normally” fit strains (same fitness as the susceptible parent); squares, data for “superfit” strains.
As noted above the model described the data well, with an even higher R2 value of 0.869. The parameter estimates were as follows: E0s = 7.04, E0n = 4.44, EmaxS = 3.77, EmaxN = 2.21, H = 1.37, ED50 (administration every 24 h) = 18.05, Cq12 = 0.69, and Cq6 = 0.013. These dose fractionation coefficients demonstrate the importance of T > MIC in these dose-response relationships. The 24-h AUC/MIC associated with achieving 50% of the maximal effect against these resistant cell populations was 87% less for the most fractionated regimen (every 6 h versus every 24 h) and 31% less for the intermediate fractionated regimen (every 12 h versus every 24 h).
DISCUSSION
The emergence of resistance to antimicrobial agents threatens the successful treatment of infections caused by numerous important bacterial, viral, and fungal pathogens (25, 29, 31, 37, 38). It is generally accepted that anti-infective drug exposure is an important factor in the development and emergence of drug resistance. However, the relationship between anti-infective dosing (drug level, dosing interval, duration of therapy) and drug resistance remains poorly understood. Few studies have attempted to identify the dose level, and fewer still have attempted to identify the dosing frequency as a strategy to ameliorate this problem (13, 18, 25). The goal of the majority of these investigations has been to identify an antimicrobial drug concentration or dose level that prevents the emergence or amplification of resistant cell populations (15, 25). For example, Jumbe et al. (25) used an in vivo infection model to identify the drug concentration associated with mutant selection. Fluoroquinolone dose levels relative to the MIC or the 24-h AUC/MIC that limited amplification of drug-resistant populations of Pseudomonas aeruginosa were identified. Similar in vivo and in vitro studies with aminoglycoside antimicrobials have demonstrated that infrequent dosing of large dose levels that maximize peak concentrations best prevents the emergence of resistant cell populations (7, 18). The studies undertaken thus far have considered antibacterial compounds that exhibit cidal killing characteristics. The current investigations were based upon the premise that the relationship between dosing and resistance emergence observed with static antimicrobials may be different from that observed with cidal antimicrobials.
This study considers the impact of the dosing regimen for a static antifungal compound, fluconazole, on resistance emergence in the fungal pathogen Candida albicans. Azole-resistant organisms have emerged in patients with human immunodeficiency virus infection and less commonly in patients with other systemic immunodeficiencies (14, 17, 23, 32, 33, 35, 36, 40, 41, 46, 49, 50, 51, 54, 57, 58). Study of resistance development with this drug-organism combination include in vitro studies in which drug concentration exposures have ranged from 4- to 128-fold greater than the parent MIC and exposure durations that have ranged from 24 h to nearly 20 days (4, 11, 34, 35). However, these studies have not specifically attempted to identify a drug concentration or dose level that drove, delayed, or prevented the emergence of resistant cells. In addition, there have been attempts to examine retrospectively the association between the emergence of azole resistance and the dosing regimens prescribed for patients (14, 20, 56). Despite numerous small analyses, no clear correlation between azole drug dose level, dosing interval, or duration was identified.
The current experimental analyses examined pathogen population dynamics relative to a wide variety of fluconazole pharmacodynamic exposures by use of an in vivo model. The goal of the studies was to create a paradigm for the selection of drug dosing regimens that will suppress the outgrowth of resistant clones. The experimental design mimics a heterogeneous cell population (mixture) consisting of a predominately susceptible cell type and a very small percentage of a resistant cell type. The dosing regimens used in the studies resulted in both the emergence (outgrowth) and the prevention of resistant cell populations. Each of the well-described triazole resistance mechanisms was represented among the study organisms. The resistance mechanism did not affect the relationship between the drug exposure and resistance emergence. Additional study variables considered in these experiments with strain mixtures included the duration of drug exposure, the percentage of the starting subpopulation that was resistant, and the relative fitness of the susceptible and the resistant cell populations. More prolonged in vivo drug exposure resulted in the emergence of a larger population of resistant cells compared to the size of resistant cell population that emerged after the shorter treatment period for each dose level examined. The impact of the treatment duration observed in the current investigation is similar to that observed in studies with other antimicrobials. The percentage of the total population assumed by the resistant cell type did not affect the outcome over the 1,000-fold range examined until that percentage reached 1/10 of the entire population. When the infecting inoculum prior to the start of drug exposure contained 10% resistant cells, the population appeared to behave phenotypically as an entirely resistant population in response to treatment.
The fitness cost of resistance in numerous resistant pathogens has been well documented (9, 12, 38). Despite resistance to the effects of the antimicrobial, these pathogens often either cannot amplify in the host or do not exhibit many of the fitness traits associated with disease production. This appeared to be the case for one of the six strain pairs examined in the experiments with mixtures of strains. Regardless of the treatment exposure, the less fit, resistant cell did not emerge. However, the relative fitness of the remaining resistant strains was either similar to or exceeded that of the susceptible parent. The relationship between dose level and dosing frequency was similar for each of these strain pair replicates. Current and prior studies with single pathogens have clearly shown that the relationship between triazole exposure and effect is best described by the 24-h AUC/MIC indices. However, the current study examining the modulation of a mixed population of susceptible and resistant cells demonstrated the importance of the T > MIC index. For each dose level examined, the emergence of a resistant population occurred with the more widely spaced dosing intervals. The more frequent administration of fluconazole suppressed the emergence of the resistant cell populations. These data suggest that sub-MIC exposures drove the emergence of these resistant populations. These relationships are distinct from those observed with the cidal antibacterial compounds, where sub-MICs appeared to have little effect on either of the cell populations (7, 15, 18, 25). For these cidal compounds there appears to be a range of supra-MICs that favor the emergence of resistant cells and a yet higher dose level or peak concentration that eliminates the entire cell population. It is perhaps not surprising that an inhibitory drug is not capable of eliminating either population. These findings have a remarkable impact on the development of mathematical models examining resistance development. Prior models created to investigate the relationship between drug exposure and the emergence of resistance often relied heavily on the rate or extent of organism killing (25). Successful development of the current model relied upon consideration of both concentration and time in the description of the drug exposures. These data also point to the importance of accounting for the fitness cost that can be associated with the evolution of these resistant strains.
In sum, the experimental model system used in the current studies was able to monitor resistance development over time in relation to in vivo antimicrobial exposures. The model provides insight into the relationship between drug dosing and treatment failure due to drug resistance. Furthermore, examination of the relationships among drug exposures and resistance modulation demonstrated that pharmacodynamic variables have a remarkable impact on resistance development. The emergence of resistance with the triazole antifungal was most closely related to the time-above-the-MIC index. These data demonstrate that the static killing characteristics and the presence of sub-MIC effects contribute to the selection of resistant mutants. We hypothesize that the dosing regimen relationships identified as important for affecting drug resistance in the current studies may be similar for other static antimicrobials. Predictions from these studies may be valuable for the redesign of dosing regimens for current triazole antifungals.
Acknowledgments
D.A. is supported by NIH grant AI01767-01A1.
REFERENCES
- 1.Andes, D., and M. Van Ogtrop. 1999. Characterization and quantitation of the pharmacodynamics of fluconazole in a neutropenic murine disseminated candidiasis model. Antimicrob. Agents Chemother. 43:2116-2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andes, D. 2003. In-vivo pharmacodynamics of antifungal drugs in treatment of candidiasis. Antimicrob. Agents Chemother. 47:1179-1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baddley, J. W., and S. A. Moser. 2004. Emerging fungal resistance. Clin. Lab. Med. 24:721-735. [DOI] [PubMed] [Google Scholar]
- 4.Barchiesi, F., D. Calabrese, D. Sanglard, L. DiFancesco, F. Caselli, D. Giannini, A. Giacometti, S. Gavaudan, and G. Scalise. 2000. Experimental induction of fluconazole resistance in Candida tropicalis ATCC 750. Antimicrob. Agents Chemother. 44:1578-1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beckerman, J., H. Chibana, J. Turner, and P. T. Magee. 2001. Single-copy IMH3 allele is sufficient to confer resistance to mycophenolic acid in Candida albicans and to mediate transformation of clinical Candida species. Infect. Immun. 69:108-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bergstrom, C. T., L. Monique, and M. Lipsitch. 2005. Ecological theory suggests that antimicrobial cycling will not reduce antimicrobial resistance in hospitals. Proc. Natl. Acad. Sci. USA 101:13285-13290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blaser, J., B. B. Stone, M. C. Groner, and S. H. Zinner. 1987. Comparative study with enoxacin and netilmicin in a pharmacodynamic model to determine importance of ratio of antibiotic peak concentration to MIC for bactericidal activity and emergence of resistance. Antimicrob. Agents Chemother. 31:1054-1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonhoeffer, S., M. Lipsitch, and B. R. Levin. 1997. Evaluating treatment protocols to prevent antibiotic resistance. Proc. Natl. Acad. Sci. USA 94:12106-12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bouma, J. E., and R. E. Lenski. 1988. Evolution of a bacteria/plasmid association. Nature 335:351-352. [DOI] [PubMed] [Google Scholar]
- 10.Brown, E. M., and D. Nathwani. 2005. Antibiotic cycling or rotation: a systematic review of the evidence of efficacy. J. Antimicrob. Chemother. 55:6-9. [DOI] [PubMed] [Google Scholar]
- 11.Calvet, H. M., M. R. Yeaman, and S. G. Filler. 1997. Reversible fluconazole resistance in Candida albicans: a potential in vitro model. Antimicrob. Agents Chemother. 41:535-539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohen, T., B. Sommers, and M. Murray. 2003. The effect of drug resistance on the fitness of Mycobacterium tuberculosis. Lancet Infect. Dis. 3:13-21. [DOI] [PubMed] [Google Scholar]
- 13.Drusano, G. L., D. E. Johnson, M. Rosen, and H. C. Standiford. 1993. Pharmacodynamics of a fluoroquinolone antimicrobial agent in a neutropenic rat model of Pseudomonas sepsis. Antimicrob. Agents Chemother. 37:483-490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fan-Harvard, P., D. Capano, S. M. Smith, A. Mangia, and R. H. K. Eng. 1991. Development of resistance in Candida isolates from patients receiving prolonged antifungal therapy. Antimicrob. Agents Chemother. 35:2302-2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Firsov, A. A., S. N. Vostrov, I. Y. Lubenko, K. Drlica, Y. A. Portnoy, and S. H. Zinner. 2003. In vitro pharmacodynamic evaluation of the mutant selection window hypothesis using four fluoroquinolones against Staphylococcus aureus. Antimicrob. Agents Chemother. 47:1604-1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fish, D. N., S. C. Piscitelli, and L. H. Danziger. 1995. Development of resistance during antimicrobial therapy: a review of antibiotic classes and patient characteristics in 173 studies. Pharmacotherapy 15:279-291. [PubMed] [Google Scholar]
- 17.Franz, R., S. L. Kelly, D. C. Lamb, D. E. Kelly, M. Ruhnke, and J. Morschhauser. 1998. Multiple molecular mechanisms contribute to a stepwise development of fluconazole resistance in clinical Candida albicans strains. Antimicrob. Agents Chemother. 42:3065-3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gerber, A. U., A. P. Vastola, J. Brandel, and W. A. Craig. 1982. Selection of aminoglycoside-resistant variants of Pseudomonas aeruginosa in an in vivo model. J. Infect. Dis. 146:691-697. [DOI] [PubMed] [Google Scholar]
- 19.Gietz, R. D., and R. A. Woods. 2002. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 350:87-96. [DOI] [PubMed] [Google Scholar]
- 20.Goetz, M. D. 1996. Relationship between fluconazole dosing regimens and the emergence of fluconazole-resistant Candida albicans. AIDS 10:335-336. [DOI] [PubMed] [Google Scholar]
- 21.Gould, I. M. 2002. Antibiotic policies and control of resistance. Curr. Opin. Infect. Dis. 15:395-400. [DOI] [PubMed] [Google Scholar]
- 22.Hall, B. G. 2004. Predicting the evolution of antibiotic resistance genes. Nat. Rev. Microbiol. 2:430-435. [DOI] [PubMed] [Google Scholar]
- 23.Hernaez, M. L., C. Gil, J. Pla, and C. Nombela. 1998. Induced expression of the Candida albicans multidrug resistance gene CDR1 in response to fluconazole and other antifungals. Yeast 14:517-526. [DOI] [PubMed] [Google Scholar]
- 24.Hoyer, L. L., B. B. Magee, E. H. Rikkerink, and S. Scherer. 1994. The ARG4 gene of Candida albicans. Gene 142:213-218. [DOI] [PubMed] [Google Scholar]
- 25.Jumbe, N., A. Louie, R. Leary, W. Liu, M. R. Deziel, V. H. Tam, R. Bachhawat, C. Freeman, J. B. Kahn, K. Bush, M. N. Dudley, M. H. Miller, and G. L. Drusano. 2003. Application of a mathematical model to prevent in vivo amplification of antibiotic-resistant bacterial populations during therapy. J. Clin. Investig. 112:275-285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaatz, G. W., S. M. Seo, S. L. Barriere, L. M. Albrecht, and M. J. Rybak. 1991. Development of resistance to fleroxacin during therapy of experimental methicillin-susceptible Staphylococcus aureus endocarditis. Antimicrob. Agents Chemother. 35:1547-1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kohler, G. A., T. C. White, and N. Agabian. 1997. Overexpression of a cloned IMP dehydrogenase gene of Candida albicans confers resistance to the specific inhibitor mycophenolic acid. J. Bacteriol. 179:2331-2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Korting, H. C., H. Behrendt, K. H. Roth, and U. Neubert. 1984. Repeated exposition to subinhibitory concentrations of antibiotics in vitro readily decreases susceptibility of Neisseria gonorrhoeae to rifampicin, but not to new cephalosporins. Chemotherapy 30:366-372. [DOI] [PubMed] [Google Scholar]
- 29.Levy, S. B., and B. Marshall. 2004. Antibacterial resistance worldwide: causes, challenges and responses. Nat. Med. 10(12 Suppl.):S122-S129. [DOI] [PubMed]
- 30.Lipsitch, M., and B. R. Levin. 1997. The population dynamics of antimicrobial chemotherapy. Antimicrob. Agents Chemother. 41:363-373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lipsitch, M., C. T. Bergstrom, and B. R. Levin. 2000. The epidemiology of antibiotic resistance in hospitals: paradoxes and prescriptions. Proc. Natl. Acad. Sci. USA 97:1938-1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lopez-Ribot, J. L., R. K. McAtee, L. N. Lee, W. R. Kirkpatrick, T. C. White, D. Sanglard, and T. F. Patterson. 1998. Distinct patterns of gene expression associated with development of fluconazole resistance in serial Candida albicans isolates from human immunodeficiency virus-infected patients with oropharyngeal candidiasis. Antimicrob. Agents Chemother. 42:2932-2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lopez-Ribot, J. L., R. K. McAtee, S. Perea, W. R. Kirkpatrick, M. G. Rinaldi, and T. F. Patterson. 1999. Multiple resistant phenotypes of Candida albicans coexist during episodes of oropharyngeal candidiasis in human immunodeficiency virus-infected patients. Antimicrob. Agents Chemother. 43:1621-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marr, K. A., C. N. Lyons, K. Ha, T. R. Rustad, and T. C. White. 2001. Inducible azole resistance associated with a heterogeneous phenotype in Candida albicans. Antimicrob. Agents Chemother. 45:52-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marr, K. A., C. N. Lyons, T. R. Rustad, R. A. Bowden, and T. C. White. 1998. Rapid, transient fluconazole resistance in Candida albicans is associated with increased mRNA levels of CDR. Antimicrob. Agents Chemother. 42:2584-2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marr, K. A., T. C. White, J. A. H. van Burik, and R. A. Bowden. 1997. Development of fluconazole resistance in Candida albicans causing disseminated infection in a patient undergoing marrow transplantation. Clin. Infect. Dis. 25:908-910. [DOI] [PubMed] [Google Scholar]
- 37.Martinez, J. L., and F. Baquero. 2000. Mutation frequencies and antibiotic resistance. Antimicrob. Agents Chemother. 44:1771-1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martinez, J. L., and F. Baquero. 2002. Interactions among strategies associated with bacterial infection: pathogenicity, epidemicity, and antibiotic resistance. Clin. Microbiol. Rev. 15:647-679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.National Committee for Clinical Laboratory Standards. 1997. Reference method for broth dilution antifungal susceptibility testing of yeasts. Approved standard M27-A. National Committee for Clinical Laboratory Standards, Wayne, Pa.
- 40.Nolte, F. S., T. Parkinson, D. J. Falconer, S. Dix, J. Williams, C. Gilmore, R. Geller, and J. R. Wingard. 1997. Isolation and characterization of fluconazole and amphotericin B resistant Candida albicans from blood of two patients with leukemia. Antimicrob. Agents Chemother. 41:196-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pappas, P. G., J. H. Rex, J. D. Sobel, S. G. Filler, W. E. Dismukes, T. J. Walsh, J. E. Edwards, and Infectious Diseases Society of America. 2004. Guidelines for treatment of candidiasis. Clin. Infect. Dis. 38:161-189. [DOI] [PubMed] [Google Scholar]
- 42.Paterson, D. L. 2003. Restrictive antibiotic policies are appropriate in intensive care units. Crit. Care Med. 31(1 Suppl.):S25-S28. [DOI] [PubMed] [Google Scholar]
- 43.Patterson, T. F., S. G. Revankar, W. R. Kirkpatrick, O. Dib, A. W. Fothergill, S. W. Redding, D. A. Sutton, and M. G. Rinaldi. 1996. A simple method for detecting fluconazole-resistant yeasts with chromogenic agar. J. Clin. Microbiol. 34:1794-1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perea, S., and T. F. Patterson. 2002. Antifungal resistance in pathogenic fungi. Clin. Infect. Dis. 35:1073-1080. [DOI] [PubMed] [Google Scholar]
- 45.Pfaller, M. A., and D. J. Diekema. 2004. Rare and emerging opportunistic fungal pathogens: concern for resistance beyond Candida albicans and Aspergillus fumigatus. J. Clin. Microbiol. 42:4419-4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prasad, R., W. P. De, A. Goffeau, and E. Balzi. 1995. Molecular cloning and characterization of a novel gene of Candida albicans, CDR1, conferring multiple resistance to drugs and antifungals. Curr. Genet. 27:320-329. [DOI] [PubMed] [Google Scholar]
- 47.Pujol, C., M. Pfaller, and D. R. Soll. 2002. Ca3 fingerprinting of Candida albicans bloodstream isolates from the United States, Canada, South America, and Europe reveals a European clade. J. Clin. Microbiol. 40:2729-2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 49.Sanglard, D., R. Ischer, M. Monod, and J. Bille. 1997. Cloning of Candida albicans genes conferring resistance to azole antifungal agents: characterization of CDR2, a new multidrug ABC transporter. Microbiology 143(Pt 2):405-416. [DOI] [PubMed] [Google Scholar]
- 50.Sanglard, D., K. Kuchler, F. Ischer, J. L. Pagani, M. Monod, and J. Bille. 1995. Mechanisms of resistance to azole antifungal agents in Candida albicans isolates from AIDS patients involve specific multidrug transporters. Antimicrob. Agents Chemother. 39:2378-2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sanglard, D., F. Ischer, L. Koymans, and J. Bille. 1998. Amino acid substitutions in the cytochrome P-450 lanosterol 14α -demethylase (CYP51A1) from azole-resistant Candida albicans clinical isolates contribute to resistance to azole antifungal agents. Antimicrob. Agents Chemother. 42:241-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schmid, J., E. Voss, and D. R. Soll. 1990. Computer-assisted methods for assessing strain relatedness in Candida albicans by fingerprinting with the moderately repetitive sequence Ca3. J. Clin. Microbiol. 28:1236-1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Selkon, J. B., S. Devadatta, K. G. Kulkarni, D. A. Mitchison, A. S. L. Narayana, C. Narayanan Nair, and K. Ramachandran. 1964. The emergence of isoniazid-resistant cultures in patients with pulmonary tuberculosis during treatment with isoniazid alone or isoniazid plus PAS. Bull. W. H. O. 31:273-294. [PMC free article] [PubMed] [Google Scholar]
- 54.Sheehan, D. J., C. A. Hitchcock, and C. M. Sibley. 1999. Current and emerging azole antifungal agents. Clin. Microbiol. Rev. 12:40-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith, S. V., and I. M. Gould. 2004. Optimization of antibiotic dosing schedules in the light of increasing antibiotic resistance. Expert Rev. Antiinfect. Ther. 2:227-234. [DOI] [PubMed] [Google Scholar]
- 56.Tumbarello, M., G. Caldarola, E. Tacconelli, G Morace, B. Posteraro, R. Cauda, and L. Ortona. 1996. Analysis of risk factors associated with the emergence of azole resistant oral candidosis in the course of HIV infection. J. Antimicrob. Chemother. 38:691-699. [DOI] [PubMed] [Google Scholar]
- 57.White, T. C. 1997. Increased mRNA levels of ERG16, CDR, and MDR1 correlate with increases in azole resistance in Candida albicans isolates from a patient infected with human immunodeficiency virus. Antimicrob. Agents Chemother. 41:1482-1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.White, T. C., K. A. Marr, and R. A. Bowden. 1998. Clinical, cellular, and molecular factors that contribute to antifungal drug resistance. Clin. Microbiol. Rev. 11:382-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.White, T. C., M. A. Pfaller, R. G. Rinaldi, J. Smith, and S. W. Redding. 1997. Stable azole drug resistance associated with a substrain of Candida albicans from an HIV-infected patient. Oral Dis. 3(Suppl. 1):102-109. [DOI] [PubMed] [Google Scholar]