

Abstract
One hallmark of AIDS progression is a decline in CD4+ T lymphocytes, though the mechanism is poorly defined. There is ample evidence that increased apoptosis is responsible for some, if not all, of the decline. Prior studies have shown that binding of cellular calmodulin to the envelope glycoprotein (Env) of HIV-1 increases sensitivity to fas-mediated apoptosis and that calmodulin antagonists can block this effect. We show that individual mutation of five residues in the C-terminal calmodulin-binding domain of Env is sufficient to significantly reduce fas-mediated apoptosis in transfected cells. The A835W mutation in the cytoplasmic domain of gp41 eliminated co-immunoprecipitation of Env with calmodulin in studies with stably transfected cells. Four point mutations (A835W, A838W, A838I, and I842R) and the corresponding region of HIV-1 HXB2 were cloned into the HIV-1 proviral vector pNL4-3 with no significant effect on viral production or envelope expression, although co-immunoprecipitation of calmodulin and Env was decreased in three of these mutant viruses. Only wild-type envelope-containing virus induced significantly elevated levels of spontaneous apoptosis by day 5 post-infection. Fas-mediated apoptosis levels positively correlated with the degree of calmodulin co-immunoprecipitation, with the lowest apoptosis levels occurring in cells infected with the A835W envelope mutation. While spontaneous apoptosis appears to be at least partially calmodulin-independent, the effects of HIV-1 Env on fas-mediated apoptosis are directly related to calmodulin binding.
Keywords: HIV-1, Calmodulin, Apoptosis
Introduction
While increased apoptosis during HIV infection is a well-documented phenomenon both in vitro (Katsikis et al., 1995) and in vivo (Badley et al., 1998), its role in the pathogenesis of AIDS remains controversial (Bell and Dockrell, 2003). Although lymphocyte turnover during the asymptomatic stage of HIV infection is thought to be as high as 109 cells/day (Ho et al., 1995), the fate of these cells remains undefined. Hypotheses for this event include apoptosis (Alimonti et al., 2003) direct cell killing by the virus (Cao et al., 1996) and altered homing of lymphocytes to different immune compartments (Alimonti et al., 2003; Cloyd et al., 2000) It is likely that these all occur in vivo, and they may turn out to be related events. Peripheral blood mononuclear cells (PBMCs), which include CD4+ lymphocytes, from HIV-infected patients undergo more spontaneous apoptosis than PBMCs from uninfected donors (Pan et al., 1998). This increase in apoptosis correlates with stage of disease (Cotton et al., 1997) and with fas (Silvestris et al., 1996) and fas ligand (Badley et al., 1998) expression, two proteins responsible for regulating physiological apoptosis in lymphocytes (Mountz et al., 1994).
Fas is a transmembrane receptor in the tumor necrosis factor receptor (TNFR) superfamily that transduces an apoptotic signal following binding to fas ligand, (for review, see Peter et al., 1999). This binding induces recruitment of proteins to the death inducing signaling complex (DISC) (Scaffidi et al., 1999). Included in the DISC are upstream initiator caspases, including caspases 8 (Boatright et al., 2003) and 10 (Cohen, 1997), which are activated upon recruitment. This causes activation of downstream effector caspases, including caspases 3, 6, and 7, release of cytochrome C from the mitochondria (Green and Reed, 1998), cleavage of survival-promoting proteins (Geng et al., 1998; Janicke et al., 1998), externalization of phosphatidyl serine on the cell membrane (Denecker et al., 2000), cell and nuclear shrinkage, DNA cleavage, and cell death (Cidlowski et al., 1996). This simplified view of apoptosis includes events that do not occur in every type of apoptosis. Additionally, there is an opposing survival pathway in operation at the same time (Gomez-Angelats et al., 2000), and the relative strength of the two signals determines the ultimate fate of the cell.
Within this expanding concept of apoptotic signaling is a growing body of evidence that calcium (Krebs, 1998) and calmodulin (Ahn et al., 2003) play a role in mediating apoptosis. A slow, sustained increase in intracellular calcium concentration has been observed in many types of apoptosis, with calcium coming from both extracellular and intracellular sources. Calcium channel blockers inhibit apoptosis (Ares et al., 1997), and calcium release by thapsigargin can induce apoptosis in certain cell types (Tombal et al., 2000). Calpain, a calcium-dependent protease, has been shown to be active during apoptosis (Ghibelli et al., 2003), and the endonuclease responsible for the chromatin cleavage that occurs in most forms of apoptosis is calcium-dependent (Yakovlev et al., 2000). Death-associated protein (DAP) kinase is both calcium- and calmodulin-dependent and functions in fas-and TNFα-induced apoptosis downstream of receptor ligation but upstream of effector caspases (Cohen et al., 1997). Other calmodulin-dependent enzymes have proposed roles in apoptosis, including calmodulin-dependent protein kinases (CaM kinase) II (Wright et al., 1997) and IV (Means et al., 1997) and the calmodulin-dependent phosphatase, calcineurin (Ankarcrona et al., 1996). Calcineurin has been shown to induce apoptosis by dephosphorylating the pro-apoptotic bcl-2 homologue, bad (Wang et al., 1999), which subsequently translocates to the mitochondria. Overexpression of calcineurin alone can induce apoptosis (Shibasaki and McKeon, 1995), and cyclosporine and FK506, which inhibit calcineurin (Wiederrecht et al., 1993), can prevent many types of apoptosis (Higashigawa et al., 1997). Recently, Ahn et al. demonstrated that calmodulin binds to the cytoplasmic domain of Fas and that this binding is altered during fas-mediated apoptosis (Ahn et al., 2004).
HIV attachment and entry into cells occur through the action of the viral envelope glycoproteins, gp120 and gp41. The surface subunit of this Env complex is gp120, and the transmembrane subunit (TM) is gp41. The binding and entry process is multi-step and involves first the binding of gp120 to the host CD4 molecule which allows gp120 to then bind the chemokine receptor, CXCR4, on T cells (Choe et al., 1998). The binding of the external subunit of envelope to the host cell causes a conformational change that exposes the external fusion domain of gp41 and allows viral entry (Hunter, 1997). The cytoplasmic region of gp41 appears to play an important role in viral assembly and infectivity, by directing the glycoprotein to the correct subcellular location (Nguyen and Hildreth, 2000) and by mediating interactions between Env and the assembling capsid (Murakami and Freed, 2000). The cytoplasmic tail of gp41 is approximately 150 amino acids long and is fairly well-conserved in different strains of HIV-1 (Modrow et al., 1987). Especially well-conserved are two calmodulin-binding domains near the C-terminus of gp41 (Tencza et al., 1997). These regions are predicted to form amphipathic helices, and peptides corresponding to these regions bind to calmodulin (Srinivas et al., 1993) and have been called lentiviral lytic peptides 1 and 2 (LLP1 and 2) based on their ability to lyse cells (Miller et al., 1991). Expression of Env increases sensitivity to fas-mediated apoptosis (Micoli et al., 2000) and has also been reported to induce apoptosis in the absence of external stimuli (Ishikawa et al., 1998).
Cells expressing full-length HIV Env, in contrast to those expressing gp120 alone, display increased intracellular levels of calcium that can be prevented by treatment with calmodulin antagonists (Sasaki et al., 1996). The two highly conserved calmodulin-binding regions of gp160/41 (Tencza et al., 1997) were identified by homology to the calmodulin-binding domain of myosin light chain kinase and confirmed by peptide binding studies (Micoli et al., 2000; Srinivas et al., 1993). Calmodulin has also been shown to associate with gp160/gp41, but not with gp120 or various truncation mutants of gp160/gp41 (Ishikawa et al., 1998; Radding et al., 1996). Calmodulin expression increases in cells expressing Env with intact calmodulin-binding domains, but not in cells expressing truncated forms of Env or a point mutant that disrupts calmodulin binding (Micoli et al., 2000; Radding et al., 1996).
Expression of Env alone has been shown to increase apoptosis induced by fas (Ishikawa et al., 1998), and this could be blocked by calmodulin antagonists or by point mutation of the C-terminal calmodulin-binding domain of Env (Micoli et al., 2000; Pan et al., 1996). Additionally, calmodulin antagonists reduce spontaneous apoptosis of infected PBMCs cultured from AIDS patients (Pan et al., 1998). Therefore, apoptosis may play a key role in the progression of AIDS and one of the apoptotic triggers may be the interaction between Env and calmodulin. However, the precise molecular mechanism underlying the apoptotic events, including upregulation of calmodulin by Env, remains to be determined and represents a potentially useful area for the development of new therapies for HIV-infected patients. Furthermore, it is not known whether calmodulin binding to Env plays a role in viral infectivity (Dubay et al., 1992) and/or replication (Srinivas et al., 1994), processes clearly critical in the pathogenesis of AIDS.
Results
Effect of point mutation of gp160 on fas-mediated apoptosis
Our laboratory previously reported that expression of Env enhanced fas-mediated apoptosis and that a single point mutation in the C-terminal calmodulin-binding domain of gp160/gp41, A835W, eliminated enhanced apoptosis (Micoli et al., 2000). This mutation was chosen based on comparison of the C-terminal calmodulin-binding domain of gp160 to the calmodulin-binding domain of myosin light chain kinase (MLCK), a model for calmodulin-binding proteins (see Fig. 1 for diagram of gp41 and selected point mutations). Four other residues of gp160/gp41, alanine 838 and isoleucines 829, 839, and 842, corresponded to residues of MLCK that make contact with calmodulin and were mutated to isoleucine or tryptophan (A838) or arginine (I829, I839, and I842). These mutants of gp160/gp41, as well as an internal deletion mutant, ΔSD1, spanning residues 819–838, were made in the envelope expression vector pSRHS and transiently transfected into Jurkat cells to determine their effect on fas-mediated apoptosis (Fig. 2). The transfected cells were treated with 500 ng/ml anti-fas (clone CH11) for 3 h followed by TUNEL assay for apoptosis. Cells transfected with wild-type Env underwent significantly more apoptosis when treated with anti-fas (45 ± 3%) than empty vector-transfected cells (27 ± 2%, n = 6, P < 0.001) or EnvA835W-transfected cells treated with anti-fas (23 ± 3%, n = 6, P < 0.001). All of the mutants described above showed significant reductions in fas-mediated apoptosis compared to wild-type gp160: I829R (35 ± 1%, n = 5, P < 0.03), A835I (26 ± 1.7%, n = 3, P < 0.002), A838I (30 ± 1.5%, n = 5, P < 0.003) A838W (33 ± 1.4%, n = 4, P < 0.02), I839R (28 ± 3%, n = 5, P < 0.003), I842R (34 ± 2.5%, n = 5, P < 0.02), and ΔSD1 (28 ± 1.7%, n = 6, P < 0.001). Spontaneous apoptosis was unaffected by the expression of wild-type envelope or any of the envelope point mutations compared to mock-transfected Jurkat cells (10 ± 3%, n = 5). Statistical analyses were performed using an unpaired t test with Welch’s correction.
Fig. 1.
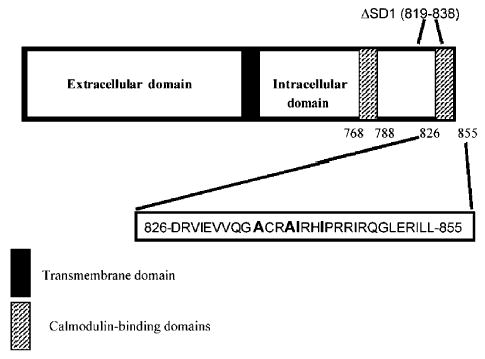
Diagram of gp41 and point mutants in the C-terminal calmodulin-binding domain. Bar diagram of gp41, the transmembrane and cytoplasmic subunit of gp160. The transmembrane region (black) and the N- and C-terminal calmodulin-binding domains (cross-hatched boxes) are shown. The 30 amino acids of the C-terminal binding domain are shown, with residues selected for mutation in bold. The internal deletion ΔSD1 is also indicated.
Fig. 2.
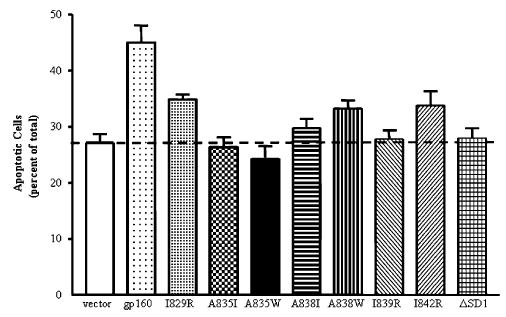
Effect of gp41 point mutations on fas-mediated apoptosis in transiently transfected Jurkat cells. The mutations shown in Fig. 1 were placed in the vector pSRHS containing the cDNA for HIV-1 HXB2 gp160. Jurkat cells were transiently transfected then treated with 500 ng/ml anti-fas (CH11) for 3 h. Apoptosis was quantified by TUNEL staining and manually counting >300 cells/sample. The graph represents the mean percentage of apoptotic cells, ± SEM, of 3–6 independent experiments. Cells transfected with an Env-expressing plasmid were significantly more fas-sensitive than empty vector-transfected cells (P < 0.002), and all mutant-transfected cells had significantly reduced fas sensitivity compared to Env-transfected cells (P < 0.05).
Effect of A835W mutation on calmodulin co-immunoprecipitation with gp160/gp41
To determine the importance of gp160/gp41 binding calmodulin on the enhancement of apoptosis, Jurkat Tet-off cells stably transfected with wild-type Env or Env A835W were incubated for 48 h with or without 2 μg/ml tetracycline to inhibit or induce, respectively, gp160 expression. We previously demonstrated that these two cell lines expressed Env only when tetracycline was removed from the culture medium and that apoptosis is unaffected by Env expression in the absence of fas-stimulation in this time frame (Micoli et al., 2000). Following induction of Env, cells were labeled with 35S-cysteine/methionine, lysed, and incubated with HIV+ patient serum to immunoprecipitate Env. Following immunoprecipitation, proteins were separated by SDS-PAGE, after which the gel was dried and autoradiography performed to determine Env expression (Fig. 3A, top). Alternatively, proteins were transferred following SDS-PAGE and Western blotted for calmodulin (Fig. 3A, bottom). Expression of wild-type and A835W mutant Env was equivalent and was seen only in cells incubated without tetracycline. Calmodulin strongly co-immunoprecipitated with wild-type Env, but this interaction was eliminated or greatly reduced by the A835W mutation. Additionally, neither calmodulin nor envelope was co-immunoprecipitated from cells incubated with tetracycline to block expression of Env.
Fig. 3.
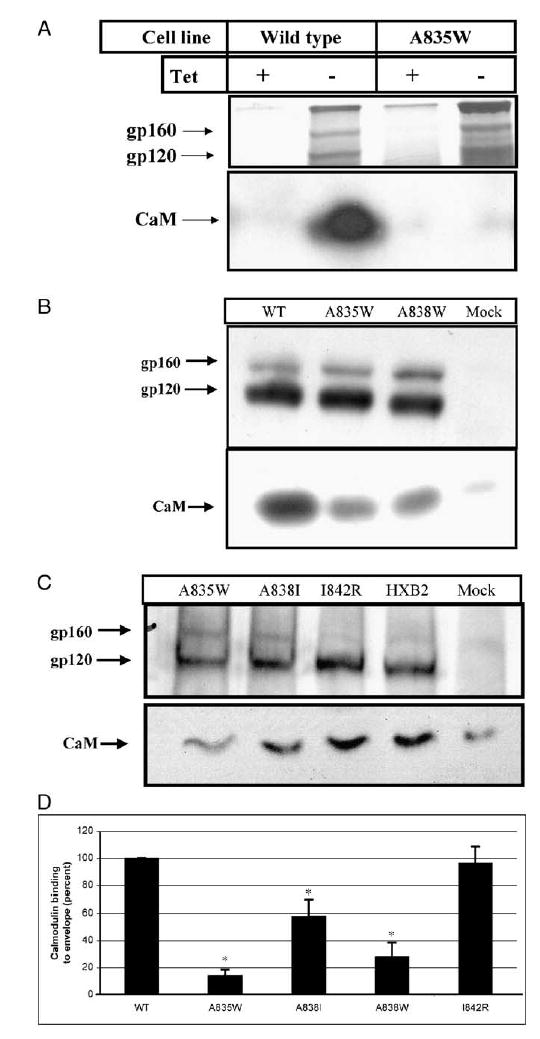
Effect of Env mutations on calmodulin co-immunoprecipitation. (A) Jurkat Tet-off cells stably transfected with wild-type env (wild-type) or the A835W env gene (A835W) were incubated in the presence of 2 μg/ml tetracycline (+Tet) to inhibit Env expression or in the absence of tetracycline (−Tet) for 48 h to induce expression of Env. Cells were metabolically labeled with 35S-cysteine/methionine followed by immunoprecipitation with HIV+ patient serum. Immunoprecipitated proteins were separated by SDS-PAGE and visualized by autoradiography for gp160/120 (upper panel) or by Western blot for calmodulin (CaM) (lower panel). (B) H9 cells were infected as described in panel A, and, on day 11 post-infection, approximately 1 × 106 cells were collected and lysed. Viral proteins were immunoprecipitated with HIV+ patient serum, separated by SDS-PAGE, and Western blotted for gp120 (top) and calmodulin (bottom). Shown is a representative Western blot of two independent experiments and is identical to results using cell lysates made on day 7 and 14 post-infection (data not shown). (C) H9 cells were infected with HIV-1 NL4-3 containing the env BamHI/XhoI fragments from HXB2. This region was either unchanged (HXB2) or changed as indicated (A835W, A838I, I842R). On day 8 post-infection, cells were lysed and viral proteins immunoprecipitated as described in Materials and methods. Shown is a representative Western blot (n = 3) and is similar to results obtained on days 5 and 12 post-infection (not shown). (D) Co-immunoprecipitation of calmodulin with envelope was quantified by densitometry. For each mutation of envelope tested, the band density corresponding to calmodulin was determined, and this value was expressed as a percentage of the band density from HXB2 Env-infected cells. Data shown are the means ± SEM, n = 6 for HXB2 and A835W and n = 3 for A838I, A838W and I842R. Asterisks indicate a significant decrease in calmodulin co-immunoprecipitation compared to HXB2 (P < 0.05).
Effect of point mutations of Env on calmodulin co-immunoprecipitation
To determine what effect, if any, mutations of Env have on calmodulin binding in the context of the entire virus, proviral vectors were designed that inserted various Env point mutations (illustrated in Fig. 1) into the pNL4.3 viral genome. These were transfected into 293T cells to produce infectious virus, which was collected and assayed for reverse transcriptase (RT) activity. Approximately 5 × 104 RT units were then used to infect 3 × 106 H9 cells, which were cultured for 28 days. At 3-to 4-day intervals, cells were split to 3 × 106 cells/ml, and aliquots of culture medium were assayed for RT activity. All of the different viral constructs peaked between day 7 and day 11 post-infection (Fig. 4A).
Fig. 4.
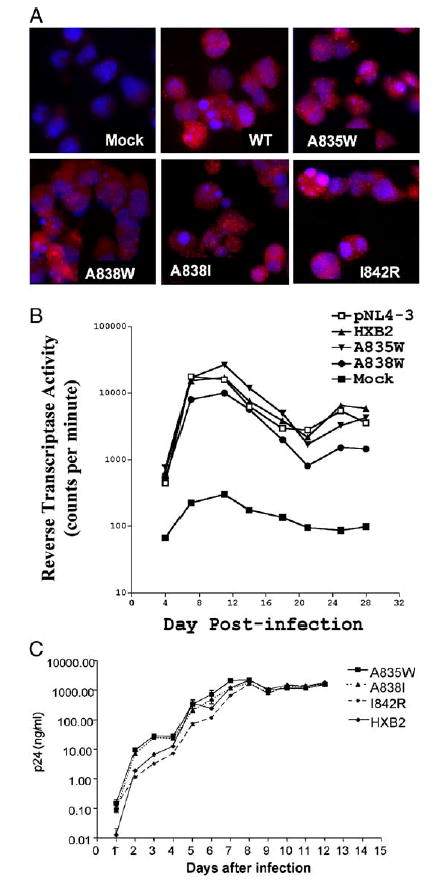
Effect of gp41 point mutations on HIV production in H9 cells infected with HIV-1. (A) H9 cells were infected with HIV-1 NL4.3, NL4.3 with the BamHI/XhoI fragment of wild type HXB2 env (HXB2) inserted, or NL4.3 containing the gp41 mutations A835W, A838W, A838I, or I842R. On day 10 post-infection, cells were collected and stained for gp120 by immunohistochemistry (red), and with Hoechst dye to identify nuclei (blue). Shown is a representative photomicrograph of three independent experiments. Identical results were obtained in separate infections on day 7 post-infection (data not shown). (B) H9 cells were infected as in panel A and at three day intervals beginning on day 4 following infection, culture supernatants were collected and assayed for reverse transcriptase (RT) activity. The mean RT activities of triplicate samples are shown. (C) H9 cells were infected as in panel A with env point mutants A835W, A838I, and I842R. Cell culture supernatants were collected at daily intervals following infection and viral p24 levels were determined in triplicate by ELISA. Points represent averages of three independent infections, ± SEM.
Cells infected as described above with HXB2 or the point mutants A835W and A838W were lysed on day 7, 11, or 14 post-infection. HIV proteins were immunoprecipitated from the cell lysates by HIV+ patient serum and separated by SDS-PAGE. Following SDS-PAGE, the gel was cut at 35 kDa and transferred for Western blot. The lower portion was probed for calmodulin and the upper portion for Env. As shown in Fig. 3B, Env could be detected in equal amounts in samples from all three viral constructs, but not from mock-infected samples. In contrast, calmodulin strongly co-immunoprecipitated with wild-type Env samples, but weakly with the Env mutants A835W and A838W. Shown is a representative Western blot from cells lysed on day 11 post-infection (n = 3). Similar results were obtained using samples taken on days 7 and 14 post-infection (not shown).
In further experiments, cells were infected as described with wild-type HXB2, A835W, A838I, and I842R, and cells were collected and lysed on day 8 post-infection. A representative Western blot shown in Fig. 3C indicates that gp120 production was equivalent for all viruses and that, as in Fig. 3B, wild-type HXB2 strongly co-immunoprecipitated with calmodulin while A835W co-immunoprecipitation was greatly reduced. A838I was also reduced in calmodulin co-immunoprecipitation, while I842R co-immunoprecipitated calmodulin as strongly as HXB2. The Western blots shown are representative of three separate experiments and are similar to results obtained from samples obtained on day 5 and day 12 post-infection (data not shown).
The amount of calmodulin co-immunoprecipitated with envelope was quantitated by densitometry (Fig. 3D) and shows that the A835W mutation results in an 85% reduction in calmodulin binding compared to HXB2 (P < 0.0001) and that the A838W mutation results in a 70% reduction (P < 0.001). The A838I mutation reduced binding to calmodulin by 40% and was significantly reduced compared to HXB2 (P < 0.05). The I842R mutation retained the same ability to bind calmodulin as HXB2.
The apparent differences in the amount of uncleaved gp160 immunoprecipitated (Figs. 3A–C) in HXB2-env-infected samples are due to different exposure times for each Western blot. Longer exposures showed that uncleaved gp160 is immunoprecipitated from all infected samples, and densito-metric analysis of the gp160 band was not significantly different between HXB2 and any of the mutants (data not shown).
Effect of Env point mutations on HIV production
To verify that Env point mutants did not alter expression or subcellular distribution of Env, cells were stained for gp120 by immunofluorescence (Fig. 4A) after infection. Shown is a representative photomicrograph of H9 cells infected with HIV encoding wild-type Env or the listed Env point mutants on day 7 post-infection following staining for gp120 (red). Nuclei were stained with Hoechst to demonstrate total cell number. As shown, on day 7 post-infection, all cells express approximately equal amounts of gp120 expression (red staining). Staining performed on days 5 and 10 post-infection also had equal gp120 staining in all constructs tested, and no envelope staining was detected on days 1 or 3 post-infection (data not shown).
To determine what effect, if any, mutations of Env have on viral production, proviral vectors were designed that inserted various Env point mutations (illustrated in Fig. 1) into the pNL4.3 viral genome. These were transfected into 293T cells to produce infectious virus, which was collected and assayed for reverse transcriptase (RT) activity. Approximately 5 × 104 RT units were then used to infect 3 × 106 H9 cells, which were cultured for 28 days. At 3- to 4-day intervals, cells were split to 3 × 106 cells/ml, and aliquots of culture medium were assayed for RT activity. All of the different viral constructs peaked between day 7 and day 11 post-infection (Fig. 4B).
In later experiments, verification that the point mutations did not cause defects in viral production was shown by p24 assay (Fig. 4C). Production of p24 was monitored by ELISA on the culture supernatants of virally infected cells on each day for 12 days post-infection. In all cases, p24 production was substantial by day 2 post-infection, increasing to peak values between days 7 and 8 post-infection. These data indicate that HIV production was not significantly impaired by any of the point mutants. In fact, p24 values were higher for the A835W and A838I mutants than for the I842R mutation or wild-type envelope-containing virus prior to reaching peak values. After day 8, levels of p24 dropped slightly and remained at the same level for the duration of the experiment, with no difference among the mutants and wild-type viruses.
Effect of Env point mutations on spontaneous apoptosis following HIV infection
H9 cells were infected with HIV as described above, and apoptosis was determined on day 5 post-infection. Spontaneous apoptosis was determined in the absence of activating fas antibody treatment and on day 5 post-infection. Significantly elevated levels of apoptosis were seen only in cells infected with WTHXB2, 13% versus 9% in mock-infected cells, P < 0.01 (39.6% increased apoptosis) (Fig. 5A).
Fig. 5.
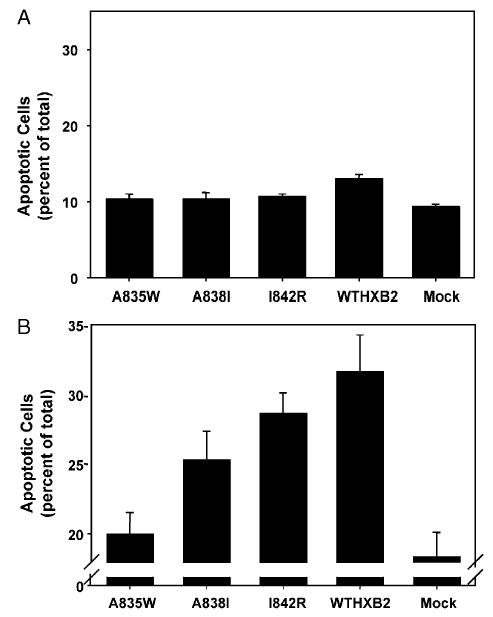
(A) Spontaneous apoptosis in H9 cells infected with HIV-1. H9 cells were infected with HIV-1 NL4.3 with the BamHI/XhoI fragment of the wild-type HXB2 env gene (WTHXB2) inserted or the same viral construct containing the indicated point mutations in gp41. Spontaneous apoptosis was determined on day 5 post-infection by Hoechst staining and morphological analysis of nuclei. Shown are the results of three independent infections, indicating mean percentage of apoptotic cells ±SEM. (B) Fas-mediated apoptosis in H9 cells infected with HIV-1. H9 cells were infected with HIV-1 NL4.3 with the BamHI/XhoI fragment of the wild-type HXB2 env gene (WTHXB2) inserted or the same viral construct containing the indicated point mutations in gp41. Cells were treated with 500 ng/ml anti-fas on day 5 post-infection for 1 h followed by fixation and Hoechst staining and morphological analysis of nuclei. Fas-mediated apoptosis was calculated by subtracting the value for spontaneous apoptosis from the value for apoptosis following fas antibody treatment. Data represent the mean percentage of apoptotic cells from three independent experiments, ±SEM.
Effect of Env point mutations on fas-mediated apoptosis following HIV infection
Fig. 5B demonstrates the effect of HIV-1 infection of H9 cells on fas-mediated apoptosis 5 days post-infection. On day 5 post-infection, 1 × 106 cells were treated for 1 h with 500 ng/ml anti-fas to induce apoptosis followed by fixation and Hoechst staining for apoptosis. Fas-mediated apoptosis was calculated by subtracting spontaneous apoptosis values from apoptosis values measured after treatment with activating fas antibody for each sample. The axis break in Fig. 5B is to better illustrate the difference in apoptosis levels and does not reflect subtraction of spontaneous apoptosis. Cells infected with HIV encoding wild-type Env had significantly higher levels of apoptosis than mock-infected cells (74% increase, 31% apoptotic cells versus 18% in mock-infected cells, P < 0.02). The Env mutant I842R enhanced fas-mediated apoptosis 60% above mock-infected cells, P < 0.02. In contrast, infection with the A835W mutant resulted in levels of fas-mediated apoptosis that were not significantly elevated above mock-infected levels (10% increase). The levels of fas-mediated apoptosis in the A835W-infected cells (20 ± 1.5%) were significantly lower than in wild-type-infected cells (31.7 ± 2.6%), P < 0.02. The effect of the A838I mutation on fas-mediated apoptosis was intermediate, with lower apoptosis levels compared to wild-type (25% vs. 31% P > 0.05, N.S.), and although this level of apoptosis was higher than the levels in mock-infected cells (increased by 40%), the difference was not statistically significant (P > 0.05). Apoptosis levels after fas antibody treatment did not change on day 12 post-infection in any of the infected cells, despite increases in spontaneous apoptosis in all of the infected cells (data not shown).
Discussion
While it has been known for some time that HIV infection leads to a depletion of the CD4+ lymphocytes and that this depletion is a major component of the eventual failure of the immune system, the mechanism of this loss remains controversial (Alimonti et al., 2003). Apoptosis of infected and uninfected cells in the immune system is one possible mechanism, and there is abundant evidence indicating that this occurs both in vitro and in vivo (Cotton et al., 1997). The mechanism of HIV-induced apoptosis is also controversial as multiple HIV proteins possess the ability to induce or enhance apoptosis, including envelope (gp160) (Ishikawa et al., 1998; Micoli et al., 2000), Tat (Bartz and Emerman, 1999), Vpr (Jian and Zhao, 2003; Shostak et al., 1999), Nef (Zauli et al., 1999), Vpu (Casella et al., 1999), and Rev (Trillo-Pazos et al., 2000). Two of these proteins, Tat (McCloskey et al., 1997) and Vpr (Fukumori et al., 1998), have been shown to possess anti-apoptotic properties as well.
Calmodulin, the major cellular calcium sensing and binding protein, has been reported in numerous systems to play a role in apoptotic signaling (Ahn et al., 2004), and two HIV proteins besides envelope, Gag (Radding et al., 2000) and Nef (Hayashi et al., 2002), have been shown to bind calmodulin, adding further complexity to the issue. Additionally, the cytoplasmic tail of HIV-1 gp41 contains two well-conserved domains that bind calmodulin (Tencza et al., 1997). These domains have been identified by peptide studies and by comparison to the calmodulin-binding domain of skeletal muscle myosin light chain kinase (Micoli et al., 2000). Truncation of the cytoplasmic tail of gp41 reduced the sensitivity of T cells to fas-mediated apoptosis (Pan et al., 1996), and one point mutant in the C-terminal calmodulin-binding domain eliminated both enhanced apoptosis and calmodulin binding to a synthetic peptide with that mutation (Micoli et al., 2000). We therefore created a panel of point mutants predicted to prevent calmodulin binding and tested their effect on apoptosis (Fig. 2). All of these Env mutants led to a significant decrease in fas-mediated apoptosis compared to wild-type Env when expressed in transiently transfected Jurkat cells, strengthening the hypothesis that this region of Env is critical for this effect. Expression of Env in the absence of fas stimulation did not result in increased apoptosis or altered membrane permeability, tested by TUNEL assay, Trypan blue exclusion, and annexin V-FITC staining (data not shown), indicating that the levels of Env expression in this model are not sufficient to grossly alter membrane permeability. The data presented in this manuscript indicate that the more C-terminal calmodulin-binding domain plays a critical role in binding calmodulin since mutating either A835 or A838 to W eliminates co-immunoprecipitation of Env and calmodulin (Fig. 3).
Although the C-terminal domain appears to be the more important for binding calmodulin, these experiments cannot rule out the possibility that both calmodulin-binding domains are necessary for binding. However, the N-terminal calmodulin-binding domain may be too constrained in the context of the entire gp41 protein to form a helix in vivo (Srinivas et al., 1993) and thus may not be able to bind calmodulin. This hypothesis is strengthened by work with the SIV calmodulin-binding domain using NMR and circular dichroism that indicated the helical nature of the region increased upon addition of calmodulin and that this interaction appeared to be in a 1:1 ratio (Yuan et al., 1995). Nevertheless, higher order multimers were also detected, and the possibility that more than one calmodulin molecule was binding to each gp41 molecule could not be ruled out.
It is interesting that disruption of the calmodulin-binding domain did not diminish viral infectivity or Env production (Fig. 3). However, as HIV-1 is able to replicate efficiently in vitro with many other mutations, including deletion of entire genes, the in vitro assay system utilized here may not be sufficiently sensitive to detect subtle changes in viral fitness. It will be of interest to determine whether the point mutations tested in these studies have a detrimental effect on viral production in vivo in the SHIV animal model.
Despite the fact that none of the point mutations in gp41 led to a significant impairment of viral production in vitro, several of these mutations did significantly reduce HIV-enhanced fas-mediated apoptosis (Fig. 5). Interestingly, although spontaneous apoptosis significantly increased over time with all of the infectious virus constructs, fas-mediated apoptosis was virtually unchanged from day 5 compared to day 12 post-infection (Fig. 5 and data not shown). This indicates that the molecular events necessary to fully enhance fas-mediated apoptosis occur early in infection and are maintained at the same level over time despite large increases in viral production (Fig. 3). In fact, the point mutant A835W reduced fas-mediated apoptosis to the level of mock-infected cells on day 5 post-infection. The data implicate gp160/gp41 as the major factor enhancing fas-mediated apoptosis in HIV-infected cells. Importantly, the results suggest that there is a direct link between the ability of gp160/gp41 to bind calmodulin and enhance fas-mediated apoptosis as A835W which co-immunoprecipitates little or no calmodulin (Fig. 4) has an apoptotic profile identical to mock-infected cells (Fig. 5), and I842R, which co-immunoprecipitates calmodulin at levels equal to wild-type HXB2, has an apoptotic profile identical to that virus. The A838I mutant, which shows an intermediate apoptotic phenotype, was also intermediate in its ability to co-immunoprecipitate calmodulin.
The issue of whether infected or uninfected (bystander) cells undergo apoptosis during natural infection is very important but could not be addressed by the studies reported here. Staining for envelope and observing nuclei for apoptotic morphology were inconclusive as envelope expression was observed in all cells at all time points where apoptosis levels increased (Figs. 4A and 5).
A significant amount of uncleaved gp160 was immunoprecipitated from infected cells, and it is not known whether this form of Env plays a role in apoptosis since uncleavable Env mutants do not produce infectious virus.
It is not currently known, and beyond the scope of these investigations, whether the pro-apoptotic effects of gp160/gp41 are mediated by increasing or decreasing calmodulin signaling. We have shown previously that expression of full-length Env, but not A835W or Δ147 Env, leads to increases in calmodulin expression (Micoli et al., 2000; Radding et al., 1996). This raises the possibility that pro-apoptotic calmodulin-binding proteins are more active in this situation and that the increase in calmodulin expression may by the key event in enhancing apoptosis by HIV. Calmodulin-dependent enzymes, most notably calcineurin, have been shown to play important roles in apoptotic signaling (Ankarcrona et al., 1996). Increased calmodulin expression could lead to increased activity of calcineurin, which has been shown to cause apoptosis in certain cell types (Shirane and Nakayama, 2003; Tombal et al., 2000). Calcineurin can activate the pro-apoptotic protein bad by dephosphorylation (Wang et al., 1999), inducing bad to translocate from the cytosol to the mitochondrial membrane where it presumably interferes with bcl-2 function. Calcineurin also functions in activating NF-AT (nuclear factor of activated T cells) (Rao et al., 1997), a transcription factor that can increase expression of fas ligand (Latinis et al., 1997). Upregulation of fas ligand (Badley et al., 1996) and its receptor, fas (McCloskey et al., 1998), have been observed in HIV-infected individuals, and this upregulation correlates with increased apoptosis and disease progression.
It has also been speculated that the binding of gp41 to calmodulin may disrupt anti-apoptotic calmodulin signaling in the cell by either reducing the amount of free calmodulin in the cytoplasm or altering its subcellular localization (Srinivas et al., 1993). The recent report that calmodulin binds directly to fas in the absence of stimulation raises the possibility that calmodulin functions to negatively regulate fas signaling (Ahn et al., 2004). This scenario is strengthened by data showing that calmodulin binding to fas is increased immediately following activation of fas but that with further incubation with activating antibody, binding is diminished. The fact that calmodulin binds both gp160/gp41 and fas and that gp160/gp41 enhances fas-mediated apoptosis strengthens the hypothesis that enhanced apoptosis may be due to altered localization of calmodulin.
Changes in calmodulin expression and localization can also impact calcium regulation in cells as calmodulin binds to and modulates the activity of the plasma membrane calcium pump (Carafoli et al., 1986), the ryanodine receptor, RYR1 (Rodney et al., 2000), and the IP3 receptor (Hirota et al., 1999), both at the endoplasmic reticulum (ER) membrane. The interaction between calmodulin and these proteins is calcium-dependent, and the effect of calmodulin on the ryanodine receptor changes from activation to inhibition with increases in calcium. Both RYR1 and the IP3 receptor have been shown to have roles in mediating apoptosis (Diaz and Bourguignon, 2000; Hajnoczky et al., 2000). Increased cytosolic calcium is seen in multiple cell types undergoing apoptosis (Tombal et al., 2000), and there is good evidence that the calcium-dependent protease calpain plays a role in apoptosis as well (Johnson, 2000).
Overall, the data presented here indicate that calmodulin binding to gp160/gp41 is necessary for enhanced fas-mediated apoptosis in two transfection models and in cells infected with HIV-1. The data show that point mutations in the C-terminal calmodulin-binding domain of Env diminish calmodulin binding and apoptosis, indicating a possibility that interruption of the interaction of gp160/gp41 with calmodulin by pharmaceutical means could lead to a disruption of the sequence of events leading to the CD4+ lymphocyte depletion and immune system failure characteristic of AIDS.
Materials and methods
Cells and cell culture
Jurkat and H9 cells were purchased from the ATCC (Rockville, MD) and grown at 37 °C in 5% CO2 in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM l-glutamine.
Plasmids
Plasmid pSRHS containing the HIV-1 envelope gene encoding gp160, and the truncated forms of gp160, Δ147, which lacks the C-terminal 147 amino acids of gp41, and Δ67, which lacks the C-terminal 67 amino acids of gp41, the plasmid pKS8, containing the cDNA for β-galactosidase under control of the human β-actin promoter, and the HIV-1 proviral construct pNL4.3 containing the BamHI/Xho fragment of HXB2 envelope have been previously described (Piller et al., 2000) and were used in all transfection and infection experiments.
Transfections
All transient transfections were performed using the cationic lipid, DMRIE-C (Gibco-BRL, Rockville, MD). Briefly, lipid/DNA complexes were allowed to form for 45 min at room temperature in serum-free medium. Cells were added to the complex in serum-free medium and incubated for 5 h at 37 °C in 5% CO2. RPMI 1640 medium (Gibco-BRL) was added, and cells were cultured an additional 48 h. Transfection efficiency was monitored by simultaneous transfection of the β-galactosidase expression vector, pKS8. Efficiencies ranged from 50 to 90% in Jurkat cells.
Antibodies and reagents
Monoclonal antibody to calmodulin was developed as previously described (Sacks et al., 1991; now available from Upstate Biotechnology Inc. Lake Placid, NY). Mouse anti-human fas monoclonal antibody CH11 (Upstate Bio-technology Inc.) was used to induce apoptosis. Mouse anti-gp120 Mab (ID6) was used for immunohistochemistry, and sheep antiserum to HIV-1 IIIB was used for gp160/120 Western blot, both obtained from NIH AIDS Research and Reference Reagent Program (Rockville, MD). HIV immunoprecipitation was performed with pooled sera from HIV+ patients.
Site-directed mutagenesis
Stratagene’s (La Jolla, CA) Quikchange Site-directed Mutagenesis Kit was used to make point mutations of gp160 according to manufacturer’s instructions. Primers for the mutagenesis were purchased from Gibco-BRL and correspond to the desired mutations of I829 to R, A835 to W or I, A838 to W or I, I839 to R, and I842 to R flanked by 10–15 base pairs of correct sequence, using the sequence of HIV-1 strain HXB2 as a template.
Creation of gp160 and gp160A835W expressing cell lines
Tet-off Jurkat cells (Clontech, Palo Alto, CA) were transfected with the pTRE expression vector containing gp160 or gp160A835W cDNA by electroporation. Selection of stable cell lines was initiated 48 h after transfection using 100 μg/ml G418 (Gibco BRL), 300 μg/ml hygromycin, and 2 μg/ml tetracycline in RPMI 1640 complete medium changed every 4 days. After 5–7 days, living cells were separated from dead cells by Ficoll–Hypaque centrifugation and plated at a lower density. After serial dilution, isolated single cell clones were cultured in 96-well plates then transferred to 75 cm2 tissue culture flasks.
TUNEL staining
In situ apoptosis staining was performed using terminal deoxynucleotide transferase (Boehringer Mannheim, Indiana-polis, IN). Briefly, cells were collected and cytospun onto microscope slides and fixed in 10% formalin in PBS. Samples were washed and treated with 20 μg/ml Proteinase K (Fisher, Pittsburgh, PA) for 15 min at room temperature followed by several washes with water and incubated at 37 °C with TdT (0.3 U/ml), digoxigenin-labeled dUTP, and buffer (30 mM Tris base, pH 7.2, 140 mM sodium cacodylate, 1 mM cobalt chloride). After a brief wash, samples were blocked in 1% BSA, 0.1% gelatin in PBS for 15 min at room temperature. Samples were incubated with alkaline-phosphatase-conjugated anti-digoxigenin antibody for 1 h at room temperature, washed in PBS, and developed with NBT/BCIP (Sigma, St. Louis, MO) at room temperature for 30 min. Following development, cells were counted, scored as apoptotic or non-apoptotic, and the apoptotic cells calculated as a percentage of the total number of cells counted.
Hoechst staining
Following HIV infection, cells were fixed in 10% buffered formalin overnight, cytospun onto slides, washed 3× in PBS, and incubated in 10 μg/ml bisbenzimide (Sigma) in PBS for 1 min. Slides were washed 3× with PBS, mounted, and coverslipped. Apoptosis was determined by microscopic evaluation of nuclei, with at least 250 cells/slide counted and scored.
Immunoprecipitation and Western blot for calmodulin and Env
Cells were treated as indicated and lysed in buffer containing 50 mM HEPES, pH 7.4, 150 mM NaCl, 0.5% NP-40, 1 mM sodium orthovanadate, 1 mM EGTA, 1 mM ammonium molybdate, 50 mM NaF, 0.5 μM okadaic acid, 5 mM benzamidine, and 50 μg/ml pepstatin. After brief centrifugation at 3000 × g to remove debris, approximately 500 μg of protein was incubated with 1 μl HIV patient serum for 1–3 h at 4 °C with rotation. In addition to protein assay to determine protein levels in the cell lysates, total protein was assessed by Coomassie stain of the post-transfer gels, and total calmodulin was determined by Western blot for calmodulin in the post-IP supernatants. Immunoprecipitated proteins were pulled down using Protein A–agarose (60 μl of 1:1 slurry) or an equivalent amount of heat-inactivated Staphylococcus aureus. After four washes with lysis buffer and one wash with TBS, immunoprecipitated proteins were separated on 12.5% SDS-PAGE and transferred to Immobilon P membrane (Millipore, Bedford, MA). Membranes were fixed in 0.2% glutaraldehyde in Tris-buffered saline (TBS) for calmodulin only, blocked in TBS containing 2% BSA and 0.2% gelatin, and then incubated with a monoclonal calmodulin antibody (1:3000) in TBS–Tween 20 (TTBS) and anti-mouse–HRP-conjugated antibody (Amersham, Piscataway, NJ), 1:5000 in TTBS followed by development with enhanced chemiluminescence (ECL; Amersham). For gp160 Western blot, membranes were incubated with sheep anti-gp120 antibody at 1:1000 in TTBS followed by anti-sheep–HRP antibody and development by ECL.
Densitometric analysis
Quantitation of co-immunoprecipitated calmodulin was performed using densitometry by the following method. Films were scanned and converted to TIFF images. Using Adobe Photoshop Elements, the image was inverted, and bands were selected and analyzed for average pixel luminescence. A sample of identical size in an adjacent region was analyzed and subtracted as background for each lane. To normalize densitometric ranges, the band density for mock-infected cells was subtracted from all samples, and the value from HXB2-infected cells was set as 100%. Band density for each Env mutation-infected sample was expressed as a percent of HXB2 band density, and the mean ± SEM was determined. Statistical analysis was performed using a one-tailed, paired t test.
Infection of H9 cells with HIV-1
Proviral constructs encoding wild-type pNL4.3 or various envelope mutations were transfected into 293T cells using Fugene 6 (Roche Applied Science, Indianapolis, IN). Forty-eight hours post-transfection, cell culture supernatants were collected and filtered (0.45 μm), and viral activity measured by reverse transcriptase (RT) or p24 assay. Approximately 5 × 104 RT units were used to infect 3 × 106 H9 cells. Briefly, H9 cells were counted, collected by centrifugation (1000 × g for 5 min), and resuspended in 5 ml media (RPMI, 10%FBS, pen/strep) containing 5 × 104 RT units. After overnight incubation, cells were collected and resuspended in 10 ml fresh media. Cells were counted and culture supernatant collected on the indicated days for RT determination.
In other infections, DNA transfections were performed on 60–70% confluent monolayer culture of 293T cells grown in six-well plates using FuGENE 6 Transfection Reagent (Roche Applied Science) in accordance to manufacturer’s protocol. Supernatants from transfected cultures were collected 72 h post-transfection, clarified by low-speed centrifugation (1000–1500 × g, 15 min), filtered through 0.45 μm low protein binding filters (Pall Corporation, East Hills, NY), aliquoted and stored at −80 °C.
HIV-1 p24 antigen concentration was determined by enzyme-linked immunosorbent assay (ELISA) (Beckman-Coulter Inc., Fullerton, CA).
Infectious titers of the viral stocks were determined by infecting JC53 BL cells with serial five-fold dilutions in the presence of DEAE-dextran (40 μg/ml) during 2 h at 37 °C. Cells were cultured in Dulbecco’s modified Eagle medium containing 10% fetal bovine serum (FBS), 100 U/ml of penicillin, and 100 μg/ml of streptomycin. After 2 days in culture, cells were fixed and stained to detect β-Gal expression as described (Piller et al., 2000).
H9 cells were infected with equal amounts of the viral stocks (MOI = 0.5 IU/cell) in RPMI 1640 medium containing 1% FBS and 8 μg/ml of Hexadimethrin Bromide (formerly Polybrene, Sigma) during 4 h at 37 °C. After that, cells were washed two times with Hank’s solution and cultured in RPMI 1640 medium supplemented with 10% FBS, 100 U/ml of penicillin, and 100 μg/ml of streptomycin. Cells were split 1:2 every 3–4 days, and fresh medium was added. Aliquots of the culture supernatants were taken every 24 h and kept at −80 °C for further analysis of HIV-1 p24 concentration.
Reverse transcriptase (RT) assay
RT activity in culture supernatants was assayed by taking 25 μl culture supernatant from infected cultures and incubating with 75 μl reaction mix composed of 67 mM Tris–HCl (pH 8.0), 6.7 mM dithiothreitol, 6.7 mM MgCl2, 200 mM KCl, 0.133% Triton X-100, 0.67 mM EGTA, 5 μCi 35S-TTP (10 mCi/ml), 1.25 μg poly(A) (dT). The reaction was allowed to proceed for 90 min at 37 °C then stopped by addition of 50 μl of 200 mM NaPPi and blotted onto NA45 paper (Schleicher and Schuell, Keene, NH). The samples were quantitated by a radioanalytical imaging system (AMBIS Systems Inc., San Diego, CA).
HIV-1 p24 antigen assay
Production of HIV was also monitored by p24 assay (Beckman Coulter) following the manufacturer’s instructions. Briefly, aliquots of cell culture conditioned medium were collected at various time points following infection with HIV-1 pNL4.3, beginning on day 1 and continuing at 3- to 4-day intervals until day 18 post-infection. Culture supernatant was diluted with RPMI 1640 (1:10–1:10,000) and added to HIV-1 p24 antibody-coated microtiter plates with lysis buffer for 1 h. Plates were washed six times with wash buffer supplied by manufacturer followed by incubation for 1 h with biotinylated anti-HIV IgG. Following incubation with streptavidin/horse-radish peroxidase, color was developed with tetramethylbenzidine (TMB) and hydrogen peroxide. The reaction was stopped with 4N H2SO4, and A450/570 was determined. Actual ng/ml p24 values were determined by plotting sample values on a standard curve of known p24 quantities.
Immunofluorescence for gp120
Expression of gp120 was verified in HIV-infected cells on various days post-infection by immunofluorescent staining. Cells were fixed and spun onto slides then incubated in 2% BSA in PBS for 30 min. Primary antibody was a monoclonal anti-gp120 (ID6) and was diluted 1:200 in PBS prior to 1-h incubation with slides. Following 6 washes (1 min each) in PBS, slides were incubated with a goat anti-mouse antibody (1:200 dilution) conjugated to Alexafluor 594 (Invitrogen, Molecular Probes, Carlsbad, CA) for 30 min. Slides were washed, mounted under coverslips with anti-fade mounting medium, and analyzed by fluorescence microscopy.
Acknowledgments
This work was supported by grants CA/72823 and AI49090 (to Jay McDonald) and AI33319 (to Eric Hunter) from the National Institutes of Health and a Veterans Administration Merit Review (to Jay McDonald).
References
- Ahn EY, Pan G, Oh JH, Tytler EM, McDonald JM. The combination of calmodulin antagonists and interferon-gamma induces apoptosis through caspase-dependent and -independent pathways in cholangiocarcinoma cells. Am J Pathol. 2003;163:2053–2063. doi: 10.1016/s0002-9440(10)63563-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn EY, Lim ST, Cook WJ, McDonald JM. Calmodulin binding to the Fas death domain. Regulation by Fas activation. J Biol Chem. 2004;279:5661–5666. doi: 10.1074/jbc.M311040200. [DOI] [PubMed] [Google Scholar]
- Alimonti JB, Ball TB, Fowke KR. Mechanisms of CD4+ T lymphocyte cell death in human immunodeficiency virus infection and AIDS. J Gen Virol. 2003;84:1649–1661. doi: 10.1099/vir.0.19110-0. [DOI] [PubMed] [Google Scholar]
- Ankarcrona M, Dypbukt JM, Orrenius S, Nicotera P. Calcineurin and mitochondrial function in glutamate-induced neuronal cell death. FEBS Lett. 1996;394:321–324. doi: 10.1016/0014-5793(96)00959-3. [DOI] [PubMed] [Google Scholar]
- Ares MP, Porn-Ares MI, Thyberg J, Juntti-Berggren L, Berggren PO, Diczfalusy U, Kallin B, Bjorkhem I, Orrenius S, Nilsson J. Ca2+ channel blockers verapamil and nifedipine inhibit apoptosis induced by 25-hydroxycholesterol in human aortic smooth muscle cells. J Lipid Res. 1997;38:2049–2061. [PubMed] [Google Scholar]
- Badley AD, McElhinny JA, Leibson PJ, Lynch DH, Alderson MR, Paya CV. Upregulation of Fas ligand expression by human immunodeficiency virus in human macrophages mediates apoptosis of uninfected T lymphocytes. J Virol. 1996;70:199–206. doi: 10.1128/jvi.70.1.199-206.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badley AD, Dockrell DH, Algeciras A, Ziesmer S, Landay A, Lederman MM, Connick E, Kessler H, Kuritzkes D, Lynch DH, Roche P, Yagita H, Paya CV. In vivo analysis of Fas/FasL interactions in HIV-infected patients. J Clin Invest. 1998;102:79–87. doi: 10.1172/JCI2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartz SR, Emerman M. Human immunodeficiency virus type 1 Tat induces apoptosis and increases sensitivity to apoptotic signals by up-regulating FLICE/caspase-8. J Virol. 1999;73:1956–1963. doi: 10.1128/jvi.73.3.1956-1963.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell DJ, Dockrell DH. Apoptosis in HIV-1 infection. J Eur Acad Dermatol Venereol. 2003;17:178–183. doi: 10.1046/j.1468-3083.2003.00681.x. [DOI] [PubMed] [Google Scholar]
- Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H, Pedersen IM, Ricci JE, Edris WA, Sutherlin DP, Green DR, Salvesen GS. A unified model for apical caspase activation. Mol Cell. 2003;11:529–541. doi: 10.1016/s1097-2765(03)00051-0. [DOI] [PubMed] [Google Scholar]
- Cao J, Park IW, Cooper A, Sodroski J. Molecular determinants of acute single-cell lysis by human immunodeficiency virus type 1. J Virol. 1996;70:1340–1354. doi: 10.1128/jvi.70.3.1340-1354.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carafoli E, Zurini M, Benaim G. The calcium pump of plasma membranes. Ciba Found Symp. 1986;122:58–72. doi: 10.1002/9780470513347.ch5. [DOI] [PubMed] [Google Scholar]
- Casella CR, Rapaport EL, Finkel TH. Vpu increases susceptibility of human immunodeficiency virus type 1-infected cells to fas killing. J Virol. 1999;73:92–100. doi: 10.1128/jvi.73.1.92-100.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe H, Martin KA, Farzan M, Sodroski J, Gerard NP, Gerard C. Structural interactions between chemokine receptors, gp120 Env and CD4. Semin Immunol. 1998;10:249–257. doi: 10.1006/smim.1998.0127. [DOI] [PubMed] [Google Scholar]
- Cidlowski JA, King KL, Evans-Storms RB, Montague JW, Bortner CD, Hughes FM., Jr The biochemistry and molecular biology of glucocorticoid-induced apoptosis in the immune system. Recent Prog Horm Res. 1996;51:457–490. (discussion 490–491) [PubMed] [Google Scholar]
- Cloyd MW, Chen JJ, Wang I. How does HIV cause AIDS? The homing theory. Mol Med Today. 2000;6:108–111. doi: 10.1016/s1357-4310(99)01663-9. [DOI] [PubMed] [Google Scholar]
- Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997;326 (Pt 1):1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen O, Feinstein E, Kimchi A. DAP-kinase is a Ca2+/calmodulin-dependent, cytoskeletal-associated protein kinase, with cell death-inducing functions that depend on its catalytic activity. EMBO J. 1997;16:998–1008. doi: 10.1093/emboj/16.5.998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotton MF, Ikle DN, Rapaport EL, Marschner S, Tseng PO, Kurrle R, Finkel TH. Apoptosis of CD4+ and CD8+ T cells isolated immediately ex vivo correlates with disease severity in human immunodeficiency virus type 1 infection. Pediatr Res. 1997;42:656–664. doi: 10.1203/00006450-199711000-00018. [DOI] [PubMed] [Google Scholar]
- Denecker G, Dooms H, Van Loo G, Vercammen D, Grooten J, Fiers W, Declercq W, Vandenabeele P. Phosphatidyl serine exposure during apoptosis precedes release of cytochrome c and decrease in mitochondrial transmembrane potential. FEBS Lett. 2000;465:47–52. doi: 10.1016/s0014-5793(99)01702-0. [DOI] [PubMed] [Google Scholar]
- Diaz F, Bourguignon LY. Selective down-regulation of IP(3)receptor subtypes by caspases and calpain during TNF alpha-induced apoptosis of human T-lymphoma cells. Cell Calcium. 2000;27:315–328. doi: 10.1054/ceca.2000.0126. [DOI] [PubMed] [Google Scholar]
- Dubay JW, Roberts SJ, Hahn BH, Hunter E. Truncation of the human immunodeficiency virus type 1 transmembrane glycoprotein cytoplasmic domain blocks virus infectivity. J Virol. 1992;66:6616–6625. doi: 10.1128/jvi.66.11.6616-6625.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumori T, Akari H, Iida S, Hata S, Kagawa S, Aida Y, Koyama AH, Adachi A. The HIV-1 Vpr displays strong anti-apoptotic activity. FEBS Lett. 1998;432:17–20. doi: 10.1016/s0014-5793(98)00824-2. [DOI] [PubMed] [Google Scholar]
- Geng YJ, Azuma T, Tang JX, Hartwig JH, Muszynski M, Wu Q, Libby P, Kwiatkowski DJ. Caspase-3-induced gelsolin fragmentation contributes to actin cytoskeletal collapse, nucleolysis, and apoptosis of vascular smooth muscle cells exposed to proinflammatory cytokines. Eur J Cell Biol. 1998;77:294–302. doi: 10.1016/S0171-9335(98)80088-5. [DOI] [PubMed] [Google Scholar]
- Ghibelli L, Mengoni F, Lichtner M, Coppola S, De Nicola M, Bergamaschi A, Mastroianni C, Vullo V. Anti-apoptotic effect of HIV protease inhibitors via direct inhibition of calpain. Biochem Pharmacol. 2003;66:1505–1512. doi: 10.1016/s0006-2952(03)00505-7. [DOI] [PubMed] [Google Scholar]
- Gomez-Angelats M, Bortner CD, Cidlowski JA. Protein kinase C (PKC) inhibits fas receptor-induced apoptosis through modulation of the loss of K+ and cell shrinkage. A role for PKC upstream of caspases. J Biol Chem. 2000;275:19609–19619. doi: 10.1074/jbc.M909563199. [DOI] [PubMed] [Google Scholar]
- Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Hajnoczky G, Csordas G, Madesh M, Pacher P. Control of apoptosis by IP(3) and ryanodine receptor driven calcium signals. Cell Calcium. 2000;28:349–363. doi: 10.1054/ceca.2000.0169. [DOI] [PubMed] [Google Scholar]
- Hayashi N, Matsubara M, Jinbo Y, Titani K, Izumi Y, Matsushima N. Nef of HIV-1 interacts directly with calcium-bound calmodulin. Protein Sci. 2002;11:529–537. doi: 10.1110/ps.23702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashigawa M, Komada Y, Shimono Y, Nagata T, Inamochi H, Mao XY, M’Soka T, Hori H, Kawasaki H, Sakurai M. FK506 inhibits anti-IgM antibody-induced apoptosis and 17 kD endonuclease activity in the human B-cell line, MBC-1, established from Burkitt’s lymphoma. Br J Haematol. 1997;99:908–913. doi: 10.1046/j.1365-2141.1997.4783287.x. [DOI] [PubMed] [Google Scholar]
- Hirota J, Michikawa T, Natsume T, Furuichi T, Mikoshiba K. Calmodulin inhibits inositol 1,4,5-trisphosphate-induced calcium release through the purified and reconstituted inositol 1,4,5-trisphosphate receptor type 1. FEBS Lett. 1999;456:322–326. doi: 10.1016/s0014-5793(99)00973-4. [DOI] [PubMed] [Google Scholar]
- Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- Hunter, E., 1997. Viral entry and receptors. In: Coffin, S.H.H. John M., Varmus, Harold E. (Eds.), Retroviruses. Cold Spring Harbor Laboratory Press, Plainview, pp. 71–121. [PubMed]
- Ishikawa H, Sasaki M, Noda S, Koga Y. Apoptosis induction by the binding of the carboxyl terminus of human immunodeficiency virus type 1 gp160 to calmodulin. J Virol. 1998;72:6574–6580. doi: 10.1128/jvi.72.8.6574-6580.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janicke RU, Ng P, Sprengart ML, Porter AG. Caspase-3 is required for alpha-fodrin cleavage but dispensable for cleavage of other death substrates in apoptosis. J Biol Chem. 1998;273:15540–15545. doi: 10.1074/jbc.273.25.15540. [DOI] [PubMed] [Google Scholar]
- Jian H, Zhao LJ. Pro-apoptotic activity of HIV-1 auxiliary regulatory protein Vpr is subtype-dependent and potently enhanced by nonconservative changes of the leucine residue at position 64. J Biol Chem. 2003;278:44326–44330. doi: 10.1074/jbc.C300378200. [DOI] [PubMed] [Google Scholar]
- Johnson DE. Noncaspase proteases in apoptosis. Leukemia. 2000;14:1695–1703. doi: 10.1038/sj.leu.2401879. [DOI] [PubMed] [Google Scholar]
- Katsikis PD, Wunderlich ES, Smith CA, Herzenberg LA. Fas antigen stimulation induces marked apoptosis of T lymphocytes in human immunodeficiency virus-infected individuals. J Exp Med. 1995;181:2029–2036. doi: 10.1084/jem.181.6.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs J. The role of calcium in apoptosis. Biometals. 1998;11:375–382. doi: 10.1023/a:1009226316146. [DOI] [PubMed] [Google Scholar]
- Latinis KM, Norian LA, Eliason SL, Koretzky GA. Two NFAT transcription factor binding sites participate in the regulation of CD95 (Fas) ligand expression in activated human T cells. J Biol Chem. 1997;272:31427–31434. doi: 10.1074/jbc.272.50.31427. [DOI] [PubMed] [Google Scholar]
- McCloskey TW, Ott M, Tribble E, Khan SA, Teichberg S, Paul MO, Pahwa S, Verdin E, Chirmule N. Dual role of HIV Tat in regulation of apoptosis in T cells. J Immunol. 1997;158:1014–1019. [PubMed] [Google Scholar]
- McCloskey TW, Oyaizu N, Bakshi S, Kowalski R, Kohn N, Pahwa S. CD95 expression and apoptosis during pediatric HIV infection: early upregulation of CD95 expression. Clin Immunol Immunopathol. 1998;87:33–41. doi: 10.1006/clin.1997.4496. [DOI] [PubMed] [Google Scholar]
- Means AR, Ribar TJ, Kane CD, Hook SS, Anderson KA. Regulation and properties of the rat Ca2+/calmodulin-dependent protein kinase IV gene and its protein products. Recent Prog Horm Res. 1997;52:389–406. (discussion 406–407) [PubMed] [Google Scholar]
- Micoli KJ, Pan G, Wu Y, Williams JP, Cook WJ, McDonald JM. Requirement of calmodulin binding by HIV-1 gp160 for enhanced FAS-mediated apoptosis. J Biol Chem. 2000;275:1233–1240. doi: 10.1074/jbc.275.2.1233. [DOI] [PubMed] [Google Scholar]
- Miller MA, Garry RF, Jaynes JM, Montelaro RC. A structural correlation between lentivirus transmembrane proteins and natural cytolytic peptides. AIDS Res Hum Retroviruses. 1991;7:511–519. doi: 10.1089/aid.1991.7.511. [DOI] [PubMed] [Google Scholar]
- Modrow S, Hahn BH, Shaw GM, Gallo RC, Wong-Staal F, Wolf H. Computer-assisted analysis of envelope protein sequences of seven human immunodeficiency virus isolates: prediction of antigenic epitopes in conserved and variable regions. J Virol. 1987;61:570–578. doi: 10.1128/jvi.61.2.570-578.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mountz JD, Bluethmann H, Zhou T, Wu J. Defective clonal deletion and anergy induction in TCR transgenic lpr/lpr mice. Semin Immunol. 1994;6:27–37. doi: 10.1006/smim.1994.1005. [DOI] [PubMed] [Google Scholar]
- Murakami T, Freed EO. The long cytoplasmic tail of gp41 is required in a cell type-dependent manner for HIV-1 envelope glycoprotein incorporation into virions. Proc Natl Acad Sci USA. 2000;97:343–348. doi: 10.1073/pnas.97.1.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DH, Hildreth JE. Evidence for budding of human immunodeficiency virus type 1 selectively from glycolipid-enriched membrane lipid rafts. J Virol. 2000;74:3264–3272. doi: 10.1128/jvi.74.7.3264-3272.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Z, Radding W, Zhou T, Hunter E, Mountz J, McDonald JM. Role of calmodulin in HIV-potentiated Fas-mediated apoptosis. Am J Pathol. 1996;149:903–910. [PMC free article] [PubMed] [Google Scholar]
- Pan G, Zhou T, Radding W, Saag MS, Mountz JD, McDonald JM. Calmodulin antagonists inhibit apoptosis of CD4+ T-cells from patients with AIDS. Immunopharmacology. 1998;40:91–103. doi: 10.1016/s0162-3109(98)00018-6. [DOI] [PubMed] [Google Scholar]
- Peter ME, Scaffidi C, Medema JP, Kischkel F, Krammer PH. The death receptors. Results Probl Cell Differ. 1999;23:25–63. doi: 10.1007/978-3-540-69184-6_3. [DOI] [PubMed] [Google Scholar]
- Piller SC, Dubay JW, Derdeyn CA, Hunter E. Mutational analysis of conserved domains within the cytoplasmic tail of gp41 from human immunodeficiency virus type 1: effects on glycoprotein incorporation and infectivity. J Virol. 2000;74:11717–11723. doi: 10.1128/jvi.74.24.11717-11723.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radding W, Pan ZQ, Hunter E, Johnston P, Williams JP, McDonald JM. Expression of HIV-1 envelope glycoprotein alters cellular calmodulin. Biochem Biophys Res Commun. 1996;218:192–197. doi: 10.1006/bbrc.1996.0034. [DOI] [PubMed] [Google Scholar]
- Radding W, Williams JP, McKenna MA, Tummala R, Hunter E, Tytler EM, McDonald JM. Calmodulin and HIV type 1: interactions with Gag and Gag products. AIDS Res Hum Retroviruses. 2000;16:1519–1525. doi: 10.1089/088922200750006047. [DOI] [PubMed] [Google Scholar]
- Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- Rodney GG, Williams BY, Strasburg GM, Beckingham K, Hamilton SL. Regulation of RYR1 activity by Ca(2+) and calmodulin. Biochemistry. 2000;39:7807–7812. doi: 10.1021/bi0005660. [DOI] [PubMed] [Google Scholar]
- Sacks DB, Porter SE, Ladenson JH, McDonald JM. Monoclonal antibody to calmodulin: development, characterization, and comparison with polyclonal anti-calmodulin antibodies. Anal Biochem. 1991;194:369–377. doi: 10.1016/0003-2697(91)90243-m. [DOI] [PubMed] [Google Scholar]
- Sasaki M, Uchiyama J, Ishikawa H, Matsushita S, Kimura G, Nomoto K, Koga Y. Induction of apoptosis by calmodulin-dependent intracellular Ca2+ elevation in CD4+ cells expressing gp 160 of HIV. Virology. 1996;224:18–24. doi: 10.1006/viro.1996.0502. [DOI] [PubMed] [Google Scholar]
- Scaffidi C, Krammer PH, Peter ME. Isolation and analysis of components of CD95 (APO-1/Fas) death-inducing signaling complex. Methods. 1999;17:287–291. doi: 10.1006/meth.1999.0742. [DOI] [PubMed] [Google Scholar]
- Shibasaki F, McKeon F. Calcineurin functions in Ca(2+)-activated cell death in mammalian cells. J Cell Biol. 1995;131:735–743. doi: 10.1083/jcb.131.3.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirane M, Nakayama KI. Inherent calcineurin inhibitor FKBP38 targets Bcl-2 to mitochondria and inhibits apoptosis. Nat Cell Biol. 2003;5:28–37. doi: 10.1038/ncb894. [DOI] [PubMed] [Google Scholar]
- Shostak LD, Ludlow J, Fisk J, Pursell S, Rimel BJ, Nguyen D, Rosenblatt JD, Planelles V. Roles of p53 and caspases in the induction of cell cycle arrest and apoptosis by HIV-1 vpr. Exp Cell Res. 1999;251:156–165. doi: 10.1006/excr.1999.4568. [DOI] [PubMed] [Google Scholar]
- Silvestris F, Cafforio P, Frassanito MA, Tucci M, Romito A, Nagata S, Dammacco F. Overexpression of Fas antigen on T cells in advanced HIV-1 infection: differential ligation constantly induces apoptosis. AIDS. 1996;10:131–141. doi: 10.1097/00002030-199602000-00002. [DOI] [PubMed] [Google Scholar]
- Srinivas SK, Srinivas RV, Anantharamaiah GM, Compans RW, Segrest JP. Cytosolic domain of the human immunodeficiency virus envelope glycoproteins binds to calmodulin and inhibits calmodulin-regulated proteins. J Biol Chem. 1993;268:22895–22899. [PubMed] [Google Scholar]
- Srinivas RV, Bernstein H, Oliver C, Compans RW. Calmodulin antagonists inhibit human immunodeficiency virus-induced cell fusion but not virus replication. AIDS Res Hum Retroviruses. 1994;10:1489–1496. doi: 10.1089/aid.1994.10.1489. [DOI] [PubMed] [Google Scholar]
- Tencza SB, Mietzner TA, Montelaro RC. Calmodulin-binding function of LLP segments from the HIV type 1 transmembrane protein is conserved among natural sequence variants. AIDS Res Hum Retroviruses. 1997;13:263–269. doi: 10.1089/aid.1997.13.263. [DOI] [PubMed] [Google Scholar]
- Tombal B, Weeraratna AT, Denmeade SR, Isaacs JT. Thapsigargin induces a calmodulin/calcineurin-dependent apoptotic cascade responsible for the death of prostatic cancer cells. Prostate. 2000;43:303–317. doi: 10.1002/1097-0045(20000601)43:4<303::aid-pros10>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Trillo-Pazos G, McFarlane-Abdulla E, Campbell IC, Pilkington GJ, Everall IP. Recombinant nef HIV-IIIB protein is toxic to human neurons in culture. Brain Res. 2000;864:315–326. doi: 10.1016/s0006-8993(00)02213-7. [DOI] [PubMed] [Google Scholar]
- Wang HG, Pathan N, Ethell IM, Krajewski S, Yamaguchi Y, Shibasaki F, McKeon F, Bobo T, Franke TF, Reed JC. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science. 1999;284:339–343. doi: 10.1126/science.284.5412.339. [DOI] [PubMed] [Google Scholar]
- Wiederrecht G, Lam E, Hung S, Martin M, Sigal N. The mechanism of action of FK-506 and cyclosporin A. Ann. N Y Acad Sci. 1993;696:9–19. doi: 10.1111/j.1749-6632.1993.tb17137.x. [DOI] [PubMed] [Google Scholar]
- Wright SC, Schellenberger U, Ji L, Wang H, Larrick JW. Calmodulin-dependent protein kinase II mediates signal transduction in apoptosis. FASEB J. 1997;11:843–849. doi: 10.1096/fasebj.11.11.9285482. [DOI] [PubMed] [Google Scholar]
- Yakovlev AG, Wang G, Stoica BA, Boulares HA, Spoonde AY, Yoshihara K, Smulson ME. A role of the Ca2+/Mg2+-dependent endonuclease in apoptosis and its inhibition by Poly(ADP-ribose) polymerase. J Biol Chem. 2000;275:21302–21308. doi: 10.1074/jbc.M001087200. [DOI] [PubMed] [Google Scholar]
- Yuan T, Mietzner TA, Montelaro RC, Vogel HJ. Characterization of the calmodulin binding domain of SIV transmembrane glycoprotein by NMR and CD spectroscopy. Biochemistry. 1995;34:10690–10696. doi: 10.1021/bi00033a045. [DOI] [PubMed] [Google Scholar]
- Zauli G, Gibellini D, Secchiero P, Dutartre H, Olive D, Capitani S, Collette Y. Human immunodeficiency virus type 1 Nef protein sensitizes CD4(+) T lymphoid cells to apoptosis via functional upregulation of the CD95/CD95 ligand pathway. Blood. 1999;93:1000–1010. [PubMed] [Google Scholar]