

Abstract
During the SOS response of Escherichia coli to DNA damage, the umuDC operon is induced, producing the trimeric protein complexes UmuD2C, a DNA damage checkpoint effector, and UmuD′2C (DNA polymerase V), which carries out translesion synthesis, the basis of ‘SOS mutagenesis’. UmuD′2, the homodimeric component of DNA pol V, is produced from UmuD by RecA-facilitated self-cleavage, which removes the 24 N-terminal residues of UmuD. We report the solution structure of UmuD′2 (PDB ID 1I4V) and interactions within UmuD′–UmuD, a heterodimer inactive in translesion synthesis. The overall shape of UmuD′2 in solution differs substantially from the previously reported crystal structure, even though the topologies of the two structures are quite similar. Most significantly, the active site residues S60 and K97 do not point directly at one another in solution as they do in the crystal, suggesting that self-cleavage of UmuD might require RecA to assemble the active site. Structural differences between UmuD′2 and UmuD′– UmuD suggest that UmuD′2C and UmuD2C might achieve their different biological activities through distinct interactions with RecA and DNA pol III.
Keywords: SOS response/structure/translesion synthesis/UmuD
Introduction
During the SOS response of Escherichia coli to damage by UV radiation or mutagenic chemicals, >40 proteins are induced that enable the cell to survive (Sutton et al., 2000; Courcelle et al., 2001). Among these are DNA repair proteins and proteins that maintain genomic continuity even at the cost of mutating the DNA. The latter include UmuD, UmuD′ (a truncated version of UmuD) and UmuC, products of the umuDC operon. UmuD′ and UmuC constitute DNA polymerase V (UmuD′2C), an error-prone polymerase that bypasses DNA lesions that cannot be replicated by DNA pol III (Reuven et al., 1999; Tang et al., 1999, 2000). Because DNA pol V introduces mutations into DNA, its level is regulated carefully by both post-translational processing of UmuD to UmuD′ and regulated degradation of UmuD′ and UmuD by the ClpXP and Lon proteases (Frank et al., 1996; Gonzalez et al., 1998, 2000).
Induction of the SOS response requires a cascade of events. DNA damage results in a variety of lesions that block replication by DNA pol III. When this polymerase stalls, regions of single-stranded DNA form that are bound by RecA, generating RecA::ssDNA nucleoprotein filaments. These filaments facilitate site-specific autodigestion of LexA, the repressor of the SOS regulon, at its Ala84–Gly85 bond (Little, 1993). This cleavage in turn relieves repression of the regulon, allowing expression of the SOS operons including umuDC (Figure 1).

Fig. 1. Regulation of the expression and activities of UmuD2 and UmuD′2. Upon induction of the SOS response, LexA is cleaved, allowing expression of UmuD2 and UmuC. The UmuD2C complex effects a DNA damage checkpoint. Further processing of UmuD2 in the presence of RecA filaments yields UmuD′2, which inhibits homologous recombination by RecA and forms DNA polymerase V upon inter action with UmuC. UmuD and UmuD′ also form heterodimers that are not known to share any of the functions of the homodimers.
Induction of umuDC initially produces UmuD and UmuC. These proteins form UmuD2UmuC (UmuD2C), which effects a DNA damage cell cycle checkpoint (Opperman et al., 1999). However, in the continued presence of RecA::ssDNA nucleoprotein filaments, UmuD undergoes a self-cleavage reaction that removes its N-terminal 24 amino acids, generating UmuD′ (Burckhardt et al., 1988; Nohmi et al., 1988; Shinagawa et al., 1988). This reaction is analogous to the cleavage of LexA: both utilize a serine–lysine dyad (Slilaty and Little, 1987; Nohmi et al., 1988) and UmuD′ is homologous to the dimerization domain of LexA (Perry et al., 1985). As this homology implies, UmuD′ and UmuD are dimers, both in complex with UmuC and when isolated in vitro (Woodgate et al., 1989; Sutton and Walker, 2001). They also form UmuD′–UmuD heterodimers, which are more stable than either homodimer (Battista et al., 1990). All three dimers interact with UmuC, but only UmuD′2C can serve as DNA pol V in the presence of RecA and SSB (Nohmi et al., 1988; Reuven et al., 1999; Maor-Shoshani et al., 2000). UmuD′2C is also uniquely capable of inhibiting RecA-mediated recombination both in vivo, when UmuD′2C is constitutively expressed at elevated levels (Sommer et al., 1993; Boudsocq et al., 1997), and in vitro (Rehrauer et al., 1998). This may serve to turn off recombination in favor of translesion synthesis.
Once SOS mutagenesis is no longer required, DNA pol V activity must be switched off quickly. UmuD′– UmuD heterodimers are key to this process: once regions of DNA that contain lesions are filled in, RecA::ssDNA nucleoprotein filaments do not form, UmuD is not cleaved and UmuD′–UmuD heterodimers form that inhibit translesion synthesis (Battista et al., 1990). Heterodimer formation also targets UmuD′ for degradation by the ATP-dependent protease ClpXP, completely removing UmuD′ from the cell (Gonzalez et al., 2000). UmuD2 is then degraded by Lon protease (Gonzalez et al., 1998), shutting down the entire system.
Throughout this intricately regulated process, there are a myriad of critical protein–protein interactions. These include (i) formation of UmuD2 and UmuD′2 and their complexes with UmuC; (ii) the interaction of UmuD with RecA::ssDNA filaments required for cleavage to UmuD′; (iii) interactions of UmuD2 and UmuD′2 with DNA pol III subunits; (iv) UmuD′2C interactions with RecA, SSB and DNA polymerase III during translesion synthesis; (v) UmuD′2C interactions with RecA that inhibit homologous recombination; and (vi) the interactions of UmuD2 and UmuD′–UmuD with Lon and ClpXP. How do the structures of the two small proteins UmuD′ and UmuD (115 and 139 residues, respectively) enable them to participate in so many important interactions? Here we report the solution structure of the UmuD′2 homodimer, which differs in striking and informative ways from the crystal structure. In addition, we characterize interactions within the UmuD′–UmuD heterodimer. These studies, together with previously reported cross-linking data, suggest a model for UmuD cleavage in which the interaction of UmuD2 with a RecA::ssDNA filament creates the catalytic site in UmuD by bringing together the serine–lysine dyad. This model may be relevant to the RecA-facilitated cleavages of the LexA and λ repressors, and may provide additional insights into the structural relationship between UmuD′ and the signal peptidases. The heterodimer interface also suggests that the surfaces of UmuD′2 and UmuD2 are quite different. This could explain how two such closely related proteins function so differently while interacting with many of the same partner proteins.
Results
Determination of the UmuD′2 structure
Backbone resonance assignments for UmuD′2 were obtained using standard triple resonance experiments (Kay et al., 1990; Montelione and Wagner, 1990; Yamazaki et al., 1994), while side chains were assigned from the HCCH-TOCSY and 13C-NOESY-HSQC spectra (Fesik and Zuiderweg, 1988; Kay et al., 1993). Amides for all but the two N-terminal residues and one loop residue (S49) were observed, and complete side chain assignments were made for all but 11 amino acids. Table I summarizes the data used for structure calculations. Out of 100 starting structures, 30 had no nuclear Overhauser effect (NOE) violation or dihedral angle violation >0.5 Å or 5°, respectively. The 20 lowest energy structures are shown in Figure 2. Each monomer within the dimer is well defined and consists of two lobes separated by a cleft (Figure 2A). One lobe contains residues in the dimerization interface and the catalytic S60, while the other contains catalytic residue K97. Since the relative orientation of the two regions is flexible, r.m.s. deviations were calculated separately for the dimerization and the K97 subdomains (the dimer’s ‘wings’ in Figure 2B). The backbone r.m.s. deviations for the secondary structure elements within these regions are 0.54 and 0.49 Å, respectively. Complete structure statistics are given in Table I.
Table I. Statistics for the final 20 structures of UmuD′2.
| No. of restraints |
|
| Intra-monomer restraints | |
| NOE distances | |
| intra-residue | 540 |
| sequential | 353 |
| medium range (2 ≤|i – j| ≤4) | 137 |
| long range (|i – j| = 5) | 277 |
| hydrogen bond restraints (two per bond) | 42 |
| dihedral angles | 117 |
| total | 1466 |
| Inter-monomer restraints | |
| NOE distance restraints | 76 |
| hydrogen bond restraints (two per bond) | 12 |
| Ambiguous NOEs | 63 |
| Total restraints for dimer |
3083 |
| Restraint violations |
|
| Distance restraint violations | |
| no. of violations >0.3 Å | 0–6 |
| largest violation (Å) | 0.482 |
| mean r.m.s. violation (Å) | 0.026 |
| Dihedral angle restraint violations | |
| no. of violations >3° | 0–4 |
| largest violation | 4.16 |
| mean r.m.s violation |
0.41 |
| R.m.s. deviations from ideal geometrya |
|
| Bond lengths (Å) | 0.0031 |
| Bond angles (°) | 0.59 |
| Improper angles (°) |
0.41 |
| Ramachandran plotb |
|
| Most favorable region | 59.1 |
| Additionally allowed region | 35.2 |
| Generously allowed region | 5.1 |
| Disallowed region | 0.6 |
| Mean coordinate r.m.s. deviations from the mean structure (Å) | ||
| |
Secondary structure |
All residues |
| Core of monomer (residues 80–130) | ||
| backbone | 0.49 | 0.71 |
| all heavy atoms | 0.95 | 1.20 |
| Dimerization interface (residues 38–75 and 131–139 of both monomers) | ||
| backbone | 0.54 | 0.86 |
| all heavy atoms | 0.84 | 1.15 |
| Complete dimer (residues 38–139 of both monomers) | ||
| backbone | 0.95 | 1.20 |
| all heavy atoms | 1.25 | 1.52 |
aIdeal geometries were based on the X-PLOR parameters (parallhdg.pro).
bPROCHECK_nmr was used to assess the stereochemical quality of the structures (residues 38–139).

Fig. 2. Structure of UmuD′. (A) Stereo depiction of the 20 UmuD′ structures showing the backbone carbon and nitrogen atoms of residues 38–139 in one monomer of the dimer. Only residues in the left lobe of the protein are superimposed. Molscript rendering of the UmuD′ structure closest to the mean. The unstructured residues 25–37 are not shown. (B) Stereo view of the UmuD′2 structures with the dimerization interface superimposed.
Description of the structure and comparison with the crystal structure
UmuD′ is composed primarily of β-strands with two short helical regions in the dimerization half of the protein. One of these helices (α2) contains the catalytic S60 and is connected to the rest of the dimerization region by loops (Figure 3A). The remainder of the dimerization subdomain comprises antiparallel strands β1 and β2, the C-terminus of β7 and helix α1. The α1 helices pack against each other in the dimerization interface, while the C-termini form antiparallel β-strands, leading to a six-stranded β-sheet spanning the two monomers (Figures 2B and 3A). Each ‘wing’ of UmuD′ consists entirely of antiparallel β-strands and loops, with two highly curved strands in the middle, one of which contains K97 (β4). This structure confirms our previous identification of the dimerization interface in solution (Ferentz et al., 1997), which differs from the originally reported crystallographic dimer (Peat et al., 1996). In the following, the interface in solution will be used in discussing both the solution and crystal structures.

Fig. 3. Comparison of the solution and crystal structures. (A) Molscript renderings of the UmuD′2 solution and crystal structures. (B) Stereo view of the catalytic site in solution. (C) The catalytic site of the solution structure closest to the mean. (D) The corresponding region of the crystal structure. In (C) and (D) green lines connect protons between which NOEs are observed, while short distances in the crystal are indicated by purple lines.
Figure 3A shows the UmuD′2 solution conformation closest to the mean alongside the crystal structure in the same orientation. The overall dimensions of the dimers are strikingly different and the r.m.s. deviation between the backbones of the two dimers (residues 40–139) is 4.59 Å, yet the secondary structures and topology of the protein in solution are very similar to those of the crystal structure (Peat et al., 1996). The main difference stems from the fact that, in solution, the outer region of the protein is mobile relative to the dimerization interface. When only the dimerization subdomains are superimposed, the r.m.s. deviation between the secondary structure elements of the solution and crystal structures is 1.67 Å. This smaller difference is still significant and primarily reflects the different orientations of the α1 and α2 helices in the two structures. Significantly, in solution, residues near α2 do not contact one another across the interface as they do in the crystal (Figure 3A). Since α2 contains the catalytic S60, this difference alters the configuration of the active site of the protein (Figure 3B–D).
The NOEs from α2 across the catalytic cleft clearly distinguish the solution structure from the crystal structure: the NOEs between D63/G64 and V96/K97 are inconsistent with the crystal structure and there are no long-range NOEs from V96 to V54, A56, S57 and I66 or from K97 to S60, as would be predicted from the crystal coordinates (Figure 3C and D). These differences may well reflect the malleability of UmuD′, which is a small protein with a great deal of surface area: only nine out of 115 residues are completely buried in the monomer, with an additional five residues of each monomer being buried in the dimerization interface. UmuD′ would thus be particularly susceptible to crystal packing forces, which primarily affect protein surfaces.
Another difference between the solution and crystal structures that could be attributed to crystal packing affects the catalytic K97 in β4. When the wings of the solution and crystal structures are superimposed, the regions of secondary structure differ by 1.95 Å, largely because of the different orientations of β3, β4 and the intervening loop (Figure 3A). Residues in β4 (93–102) are directly involved in contacts across the crystallographic dimerization interface: F94 is at the center of that interface, while E93 participates in a salt bridge to K55 in the adjacent monomer (Peat et al., 1996). The resulting differences, along with the altered orientation of α2, mean that the S60 and K97 side chains do not point directly at each other in solution as they do in the crystal (Figure 3C and D). Thus the catalytic dyad is not poised for cleavage as might be inferred from the crystal structure. This point will be discussed further below.
Resonance assignments of UmuD′ in the UmuD′–UmuD heterodimer and identification of the UmuD-binding site
We were interested in comparing the structures of UmuD′2, UmuD2 and UmuD′–UmuD to understand how UmuD′2 can participate in translesion synthesis while UmuD2 acts in a DNA damage checkpoint and UmuD′–UmuD does neither. Possibly due to aggregation, the UmuD2 spectra were too poor to assign, so UmuD2 could not be studied directly. The UmuD′–UmuD spectra were of high enough quality that most of the heterodimer could be assigned, but a complete structure could not be determined. Since UmuD′–UmuD is more stable than either UmuD′2 or UmuD2, samples were prepared by mixing equimolar quantities of the two homodimers (Battista et al., 1990). Spectral analysis was simplified by labeling either UmuD′2 or UmuD2, so that each half of the heterodimer could be assigned separately.
When unlabeled UmuD2 was added to labeled UmuD′2, there were dramatic changes in the 15N-HSQC spectrum (Figure 4A). The UmuD′ side of the heterodimer was assigned using standard triple resonance experiments for the backbone and the CC(CO)NH-TOCSY and HCC(CO)NH-TOCSY experiments for the side chains (Lin and Wagner, 1999). Mapping the chemical shift differences in the amide resonances onto the structure of UmuD′2 revealed that much of the UmuD′ core is affected by UmuD binding (Figure 4B and C). Such dramatic differences could indicate a significant change in the structure of UmuD′ upon binding to UmuD. Since chemical shift index analysis indicated that the secondary structures of UmuD′ within the homodimer and heterodimer are virtually identical (Figure 5), the chemical shift changes could arise either from a slight rearrangement of the secondary structure elements or from contacts between UmuD′ and UmuD. To distinguish between these scenarios, we mapped the dimerization interface of UmuD′– UmuD by identifying NOEs from UmuD′ to UmuD in a 15N-NOESY-HSQC spectrum of [2H,15N]UmuD′ mixed with unlabeled UmuD. In addition to NOEs across the C-terminal strands, which appear to be the same as in UmuD′2, there are NOEs from UmuD′ to UmuD involving residues near the α1 helices that differ from those observed in UmuD′2 and at many residues near the catalytic cleft of UmuD′ (Figure 6). The interface between UmuD′ and UmuD thus includes regions previously identified as the UmuD′2 dimerization interface (Ferentz et al., 1997) along with a large patch of the surface adjacent to the intersecting α1 helices. The latter region contains most of the residues showing chemical shift changes, accounting for these data without altering the core of UmuD′.

Fig. 4. Mapping the dimerization interface in the UmuD′–UmuD heterodimer. (A) 1H,15N-HSQC spectrum of the UmuD′ side of the heterodimer (blue) superimposed on the spectrum of the UmuD′2 homodimer (red). (B) Differences in backbone HN and N chemical shifts {calculated as [(δHN2 + δN2/25)/2]1/2} between the UmuD′ (top) and UmuD (bottom) sides of the UmuD′–UmuD heterodimer and the UmuD′2 homodimer. In (C) and (D), chemical shift differences for the UmuD′ (C) and UmuD (D) sides of the heterodimer are mapped onto surface and worm depictions of the UmuD′2 structure closest to the mean. Residues 1–24 of UmuD have been appended to one monomer in an arbitrary orientation to clarify visualization of the heterodimer data. The color of a residue varies from white to red in proportion to the chemical shift difference. S60 of UmuD′ and C24–G25 of UmuD are in blue.

Fig. 5. Secondary structure comparison between the UmuD′2 NMR structure, the UmuD′–UmuD heterodimer and the UmuD′2 crystal structure. Secondary structure elements in the UmuD′ and UmuD sides of the heterodimer are based on Cα, Cβ and C chemical shifts. Open ellipses indicate residues for which backbone assignments could not be obtained.

Fig. 6. The interface between UmuD′ and UmuD. (A) Residues in UmuD′ that show NOEs to UmuD within the UmuD′–UmuD heterodimer, but do not show inter-monomer NOEs in UmuD′2 are indicated in green. (B) Model showing how the UmuD N-terminal arm might contact these regions of UmuD′. The model is based on the crystal structure of UmuD′, which may better reflect the conformation of the protein when in contact with other proteins.
Resonance assignments of UmuD in the UmuD′–UmuD heterodimer
When labeled UmuD2 was mixed with unlabeled UmuD′2, the spectra for the UmuD side of the heterodimer were consistently poorer than for the UmuD′ side. Nonetheless, most of the UmuD backbone could be assigned, and chemical shift index analysis indicated that its secondary structure is the same as that of UmuD′ (Figure 5). Regions that could not be assigned include the N-terminal 24 residues and several sets of residues in the core of the protein (53–55, 72–74, 96–97, 121–122 and 132–134). Of the latter, residues 53–55, 72–74 and 132–134 are next to one other in the center of UmuD′2 and residue 96 is also buried. Since the UmuD in the sample was fully deuterated, the absence of these resonances from the spectra probably reflects the failure of the buried amide protons to exchange with H2O after initial protein expression in D2O. The absence of other amides could be due to conformational exchange (Cavanagh et al., 1996), but major structural changes near 121–122 are doubtful since the chemical shifts of the surrounding residues are not perturbed. Chemical shift differences between the UmuD side of the heterodimer and UmuD′2 are primarily near α1 (Figure 4B and D), consistent with rearrangement of this region in the heterodimer.
Although the N-terminus of UmuD could not be assigned, the data that could be obtained indicate that the arm of UmuD contacts the core of UmuD′ within the heterodimer. The cores of UmuD′–UmuD and UmuD′2 are very similar according to chemical shift analysis, so UmuD′2 is a good starting point for modeling the structure of the heterodimer. The α1 helices are rearranged in the heterodimer, as indicated by chemical shift differences near α1 in both sides of UmuD′–UmuD and the altered NOE patterns across the interface: NOEs from UmuD′ to UmuD are observed at residues 34 and 36–40, in contrast to the contacts at residues 41 and 44 seen in UmuD′2. In addition, residues at the surface of UmuD′ near the catalytic cleft (Figures 4 and 6) interact with UmuD in the heterodimer. Since there is no significant chemical shift difference between the cores of UmuD and UmuD′2, the only portion of UmuD that could make such contacts is the N-terminal arm. We envisage that residues of UmuD N-terminal to α1 fold down to make extensive contacts with the adjacent region of UmuD′. Thus, the arm of UmuD is not entirely mobile, as had been suggested previously (McDonald et al., 1999).
An intriguing feature of the new contact surface within UmuD′ is that it includes residues lying in what was originally reported as the dimerization interface of UmuD′2 (Peat et al., 1996), an interface that is distinct from that used for dimerization in solution (Ferentz et al., 1997). Residues 52–55 and 94 lie in the heterodimer interface determined here and were also involved in UmuD′–UmuD′ contacts at the alternative dimerization interface in the crystal. Thus, it is possible that the crystal conformation of UmuD′ better reflects the protein’s conformation in complex with UmuD than does the solution structure, since crystal contacts are made to both regions of UmuD′ that interact with UmuD in solution.
A model for UmuD2
The extended heterodimer interface might also be used by the UmuD2 homodimer. Although the spectra of UmuD2 were too poor to assign, cross-linking experiments support such a model. The pattern of cross-linking near the α1 helices in UmuD2 (Lee et al., 1994) agrees well with the NOEs observed at the UmuD′–UmuD interface and is very similar to the cross-linking pattern for the heterodimer (M.Sutton and G.Walker, in preparation). Both differ clearly from the cross-linking pattern of UmuD′2: in both UmuD2 and UmuD′–UmuD, single-cysteine mutations at residues 37 and 38, which did not cross-link in UmuD′2, cross-link to one another, while residue 44, which cross-linked efficiently in UmuD′2, shows almost no cross-linking in UmuD2 or UmuD′– UmuD. These observations are consistent with the inter-monomer NOEs in both UmuD′2 and UmuD′–UmuD, suggesting that the cross-linking data have predictive value and that UmuD2 may closely resemble UmuD′– UmuD. In such a model, each N-terminal arm of UmuD2 would contact the core of the opposite monomer just as the N-terminus of UmuD contacts UmuD′ in UmuD′–UmuD.
Discussion
Both UmuD2 and UmuD′2 function within complexes that include UmuC, the RecA::ssDNA nucleoprotein filament and, quite probably, DNA pol III. Differences between their structures dictate how the dimers contact these other proteins, and ultimately how UmuD2 participates in a DNA damage checkpoint while the closely related UmuD′2 acts in translesion synthesis and inhibition of homologous recombination. There currently are no data describing interactions with UmuC, but information is beginning to accumulate on other interactions.
Unique interactions of UmuD′2 with RecA explain how UmuD′2C can inhibit homologous recombination and carry out translesion synthesis while UmuD2C cannot
Our finding that the N-terminal arm of UmuD is partially immobilized, while the N-terminus of UmuD′ is free in solution, offers insights into the different interactions of these proteins with the RecA::ssDNA nucleoprotein filament. Such contacts are critical for the two activities of UmuD′2C, inhibition of homologous recombination and translesion synthesis, and for self-cleavage of UmuD to UmuD′. Since the conformation of the N-termini is the major difference between UmuD2 and UmuD′2, contacts involving the N-terminal arms or the region of the core buried by the UmuD arm are expected to contribute to the distinctive interactions of UmuD′2. Indeed, most of the mutations that enhance UmuD′2C-dependent inhibition of homologous recombination lie within the N-terminal arms of UmuD′2, with E35K having the greatest effect on binding (Sommer et al., 2000). Since the corresponding residues of UmuD2 would lie on opposite sides of the dimer, our structure provides a reasonable model for how UmuD′2 could bind to the RecA filament using its free arms, thereby inhibiting homologous recombination, while UmuD2 cannot (Sommer et al., 1993; Rehrauer et al., 1998). The only other mutation that enhances inhibition of homologous recombination is T95R, which is at a position available to contact RecA in UmuD′2 but buried in UmuD2. A low resolution picture of UmuD′–RecA interactions based on cryo-electron microscopy shows that UmuD′2C binds in the deep helical groove of the RecA::ssDNA nucleoprotein filament, although no details of the orientation of UmuD′2 can be determined (Frank et al., 2000). Based on our model, one attractive possibility is that the N-terminal arms of UmuD′2 might wrap around the filament, holding the complex where it could competitively inhibit binding of dsDNA for homologous recombination.
Contacts between UmuD′2 and RecA are also required for translesion synthesis (Tang et al., 1998), but the exact nature of these interactions is unknown. Many of the UmuD′ mutants that more strongly inhibit homologous recombination also exhibit deficiencies in SOS mutagenesis, with the magnitude of the two effects being roughly correlated (Sommer et al., 2000). This suggests that tighter binding to RecA::ssDNA nucleoprotein filaments may reduce the availibility of UmuD′2 for translesion synthesis. The E35K mutant, which bound RecA most strongly, is the only inhibitor of recombination with a large enough defect in mutagenesis to be identified in non-mutability screens (Ohta et al., 1999). The remaining mutations deficient in SOS mutagenesis, which are distinct from those that affect recombination (McLenigan et al., 1998; Ohta et al., 1999), lie in the dimerization interface of UmuD′2 or on the surface of the core near the base of the N-terminal arms, a region that is buried by the extended N-termini in UmuD2. The latter mutations could affect any number of interactions with UmuD′2 that are critical for translesion synthesis. Besides contacting RecA, UmuD′2 also interacts with UmuC and the α-subunit of DNA pol III (Reuven et al., 1999; Sutton et al., 1999; Tang et al., 1999). The regions of UmuD′2 involved in these interactions have not been identified, nor is it known whether contacts with all of these proteins are made simultaneously. Considering the small size of UmuD′2, it is possible that different interactions are required at different stages of translesion synthesis.
Different contacts with DNA pol III enable UmuD′2 to promote translesion synthesis and UmuD2 to effect a DNA damage checkpoint
Both UmuD′2 and UmuD2 interact with the catalytic (α), proofreading (ε) and processivity (β) subunits of pol III (Sutton et al., 1999), but they do so in distinctive fashions that result in different biological outcomes. In a comparison of the affinities of UmuD2 and UmuD′2 for each subunit of pol III, UmuD′2 interacted most strongly with the α-subunit, while UmuD2 bound most tightly to the β sliding clamp (Sutton et al., 1999). Although the regions of contact have not yet been identified, the distinct binding patterns of UmuD′2 and UmuD2 are likely to be due to the differences in their surfaces. Binding to UmuD′2 might involve the extended N-termini or residues that are buried in UmuD2; UmuD2 contacts could utilize the folded down N-terminal arm and adjacent sections of the core (M.Sutton and G.Walker, submitted). Exploring the details of such interactions will be an interesting area for further investigation.
RecA could bring together the active site for cleavage of UmuD
When UmuD2 interacts with the RecA::ssDNA nucleoprotein filament, it is cleaved to UmuD′2. Since the extended N-terminal arms of UmuD2 fold over near the catalytic cleft, the cleavage site of UmuD2 (the C24–G25 amide bond) could lie near the catalytic site, ready to be cleaved. Cleavage of one monomer of UmuD2 by the other is supported by the findings that cleavage can be inter-molecular, both in vitro and in vivo (Ferentz et al., 1997; McDonald et al., 1998), and that it can occur within a dimer (McDonald et al., 1999). Yet in the solution structure, the catalytic residues S60 and K97 are not poised for cleavage (Figure 3B). In contrast, within the crystal, the terminal amino group of K97 points directly at the hydroxyl of S60, as if ready to deprotonate it. The difference between these conformations is that in the crystal the two sides of the catalytic cleft have been pushed together relative to the NMR structure. It is quite possible that crystal packing may mimic one role of RecA in facilitating self-cleavage of UmuD, that of squeezing the catalytic residues together.
Existing data on UmuD–RecA interactions support such a model. Cross-linking studies of UmuD with the RecA::ssDNA filament have indicated that residues 57, 67, 81 and 112 of UmuD lie near the RecA interface (Lee and Walker, 1996). More recently, mutational studies have shown the importance of residues 101 and 102 in UmuD cleavage (Sutton et al., 2001). These residues all lie in solvent-accessible loops of UmuD and thus could interact with other proteins (Figure 7). This suggests that UmuD might be cradled within the RecA::ssDNA nucleoprotein filament with contacts on both sides of the catalytic cleft but not within it (no cross-linking was observed to residues 19, 24 or 60). RecA contacts could squeeze together the two sides of the cleft, bringing K97 and S60 together. If the cleavage site of UmuD were already near the active site, cleavage could proceed immediately upon deprotonation of K97, an event that may also be induced by RecA. This contrasts with the previous proposal that interactions with the RecA filament recruit the otherwise mobile N-terminal arm of UmuD to the active site (McDonald et al., 1999).
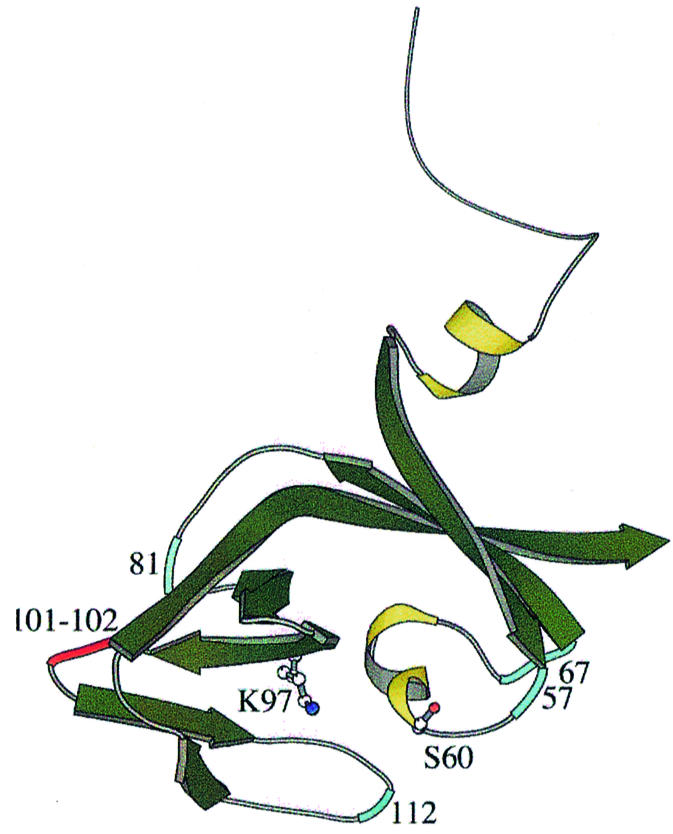
Fig. 7. Contacts between UmuD and RecA. Residues that cross-link to RecA are indicated in blue, while sites of mutations defective in UmuD cleavage are in red.
Although the serine–lysine catalytic dyad found in the UmuD/LexA family of proteins is also used by the signal peptidases, the latter enzymes do not require RecA for cleavage. The signal peptidases are considerably larger than UmuD, yet the recent structure of the E.coli signal peptidase shows that its catalytic domain strongly resembles that of UmuD′ (Paetzel et al., 1998; Paetzel and Strynadka, 1999). Intriguingly, the residues of UmuD implicated in contacts with RecA are all at sites occupied by extra residues or domains of the signal peptidase. These regions of the protein might hold together the active site, eliminating the need for RecA. The additional residues in the signal peptidase also provide a highly hydrophobic environment for the catalytic lysine that lowers its pKa, keeping it deprotonated and activated for cleavage. Meanwhile, the active site serine is attached to the rest of the enzyme by a loop, as in UmuD′. Flexibility in this region could help accommodate a range of substrates and might also attenuate the activity of a catalytic site that would otherwise be poised for instant cleavage at all times.
A model for the N-terminal arm of UmuD and the LexA family linker
Although the C24–G25 amide bond of UmuD must be located at the catalytic serine during cleavage, the exact orientation of the cleavage site within the catalytic cleft has not been established. A reasonable model for the N-terminal arms of UmuD2 could offer insights into interactions of UmuD2 with other proteins, the cleavage reaction to form UmuD′, and serve as a model for the linker regions of the homologous repressors.
The inter-monomer NOEs observed in UmuD′–UmuD identify the region of the core occluded by the N-terminal arm of UmuD, but do not allow the arm to be positioned precisely. Since the bases of the arms are the top of UmuD′2 as viewed in Figure 6A, the simplest model would have the N-terminal arm of one UmuD monomer folding over into the catalytic cleft of the opposite monomer, leaving the N-terminus projecting from the bottom of the dimer. The other possibility is that the arm folds down near the dimerization interface and then forms a tight turn to come up through the catalytic cleft (Figure 6B). There are several reason to favor the perhaps less intuitive second model. First, in this case, the orientation of the cleavage site would agree with the model that Paetzel and Strynadka (1999) have proposed based on the orientation of an inhibitor bound to the active site of the E.coli signal peptidase. Secondly, this orientation of the arm would account for why the UmuD/LexA family has highly conserved residues N-terminal to the cleavage site (residues 22 and 24 in UmuD) that are important for cleavage: these residues would fit into the catalytic cleft, while residue 23, which is frequently charged, could point out into the solvent. If the arm pointed in the opposite direction, these residues would not contact the dimer core, so there would be no apparent reason for them to be conserved. Thirdly, in this model, the two highly conserved prolines (P27 and P29) C-terminal to the cleavage site are perfectly positioned to bend the peptide chain up into the catalytic cleft (Figure 6B). Finally, this model would require that the α1 helices be reoriented so that the arm could extend far enough down to approach the catalytic cleft from below, and the NMR data suggest that there must be some such rearrangement.
It is quite possible that this picture of the N-terminal arm of UmuD may be relevant to the linker regions of the LexA family of repressors. These proteins are all dimers, and a recent crystal structure of the dimerization domain of λ repressor indicates that it uses the same dimerization interface as UmuD′2 (Bell et al., 2000). Most repressor mutations that affect dimerization map onto the dimerization interface of the UmuD′2 solution structure (Ferentz et al., 1997), but a recent study of 434 repressor has revealed that several mutations near the catalytic cleft also affect dimerization (Donner et al., 1998). These mutations correspond to residues 53 and 55 of UmuD′ and to an extra loop that precedes residue 93 in UmuD′, and thus are all in the region of the core that is buried by the N-terminal arm of UmuD. Analogous contacts between the linker of 434 and its dimerization domain could explain why this region of the repressor is important in mediating dimerization.
Structural basis for regulation of UmuD and UmuD′ levels in vivo
Perhaps as critical as regulation of the production of UmuD and UmuD′ during the SOS response is regulation of the proteins’ removal from the cell at the end of the response. The Lon and ClpXP proteases provide tight control of SOS mutagenesis by degrading UmuD and UmuD′, respectively, but there is no physical model for how the proteases discriminate between these targets. Our model for the N-terminus of UmuD provides a possible picture of the unique recognition of UmuD by the Lon protease. Lon recognizes two specific sites within the N-terminus of UmuD, a primary site between residues 15 and 18 (FPLF) and an auxiliary site between residues 26 and 29 (FPSP) (Gonzalez et al., 1998). If the UmuD N-terminus contacts the core of UmuD′ with the cleavage site near the active site, residues 15–18 would lie just beyond the catalytic cleft, while residues 26–29 would be at the other end of the cleft (Figure 6B). Thus both sites would be on the same side of UmuD2, in a fixed orientation relative to one another, and at least the auxiliary site would have a defined structure. Lon might recognize the primary site, test whether the auxiliary site is in the right place and is shaped correctly, and then degrade the protein.
In contrast to Lon, ClpXP degrades UmuD′ not UmuD, and only within the context of the UmuD′–UmuD heterodimer (Frank et al., 1996). During the course of investigating ClpXP degradation, Gonzalez et al. (2000) discovered (i) that residues 9–12 of UmuD were required for proteolysis and (ii) that a peptide comprising the N-terminal 24 amino acids of UmuD is able to earmark UmuD′2 specifically for degradation. The latter observation could be explained by our model for the N-terminal arm of UmuD: residues ∼19–24 would be expected to interact with the core of UmuD′2, mimicking the N-terminal arm of UmuD and providing the recognition site on UmuD that targets UmuD′ for degradation.
Conclusions
The solution structure of UmuD′2 and the mapping of the UmuD′–UmuD interface, along with the model of the N-terminus of UmuD, provide a structural basis for visualizing many phases of the SOS response of E.coli to DNA damage. UmuD and UmuD′ serve as components of several different protein machines throughout the SOS response, and it is the plasticity of their structures, as we have observed in solution, that enables them to function in so many roles. Initially, autocleavage of LexA, a repressor with homology to UmuD, in the presence of RecA::ssDNA nucleoprotein filaments initiates the entire response. By analogy to UmuD2 cleavage, RecA might bring together the serine–lysine catalytic dyad for cleavage of LexA. Once LexA repression of the umuDC operon is relieved, UmuD2 is expressed with its N-termini folded over near the catalytic cleft. After RecA-induced autocleavage of UmuD removes the 24 N-terminal residues producing UmuD′, the new N-terminus of UmuD′ is released from the active site and becomes mobile. This results in major differences between the structures of UmuD2 and UmuD′2 that affect their interactions with other proteins and thus their biological roles. Contacts to the free arms of UmuD′2 or to the region of UmuD′2 that was buried by the extended UmuD2 N-termini can be used to distinguish UmuD′2 from UmuD2. Such interactions make UmuD′2 uniquely capable of inhibiting homologous recombination through interactions with RecA. Distinctive interactions between UmuD′2 and UmuD2 and subunits of DNA pol III enable UmuD′2 to participate in translesion synthesis and UmuD2 to effect a DNA damage checkpoint. The exact nature of the protein–protein interactions leading to the unique biological roles of UmuD′2 and UmuD2 will be an intriguing area of further investigation.
Materials and methods
Sample preparation
UmuD′ and UmuD were overexpressed in E.coli BL21(DE3) and purified as described previously (Lee et al., 1994; Ferentz et al., 1997). Isotopically enriched samples were purified from cells grown on M9 minimal medium containing 0.5 g/l [15N]NH4Cl and 2 g/l [13C]glucose and a suitable D2O:H2O ratio. Samples were dialyzed against 150 mM NaCl, 10 mM sodium phosphate pH 6.0, 1 mM dithiothreitol (DTT), 0.1 mM EDTA and concentrated to 1–4 mM in monomer. UmuD′–UmuD heterodimer samples were prepared by adding unlabeled UmuD to labeled UmuD′ in equimolar quantities. The presence of heterodimer was confirmed by 15N-HSQC spectra. Heterodimer samples were 1 mM in UmuD′.
NMR spectroscopy
Assignment of the UmuD′ homodimer has been reported previously (Ferentz et al., 1997). Distance constraints for structure calculations were obtained from 70 and 120 ms 15N-NOESY-HSQC spectra on a 15N-labeled sample, a 120 ms 15N-NOESY-HSQC on a 50% 2H-, 100% 15N-labeled sample, a 120 ms 15N-NOESY-HSQC on a mixture of 100% 2H, 15N-labeled UmuD′ and unlabeled UmuD′, an 80 ms 13C-NOESY-HSQC spectrum and an 80 ms 2D NOESY in D2O, all of which were acquired at 30°C on a Varian UnityPlus750 spectrometer.
Spectra of the UmuD′–UmuD heterodimer were acquired at 30°C on Varian Inova 500, Inova 750 and Bruker Avance 500 spectrometers. Backbone resonance assignments for UmuD′ within the UmuD′–UmuD heterodimer were obtained from HNCA, HN(CO)CA, HN(CA)CB, HN(COCA)CB, HNCO and HN(CA)CO spectra on a sample containing 2H-, 15N-, 13C-labeled UmuD′ and unlabeled UmuD (Yamazaki et al., 1994; Matsuo et al., 1996). Partial side chain assignments were obtained from CC(CO)NH-TOCSY and HCC(CO)NH-TOCSY experiments (Lin and Wagner, 1999) on a sample containing 70% 2H-, 100% 15N-, 13C-labeled UmuD′ and unlabeled UmuD. Inter-monomer NOEs were obtained as described previously using samples containing 100% 2H-, 15N-labeled UmuD′ mixed with unlabeled UmuD and a 120 ms 15N-NOESY-HSQC (Talluri and Wagner, 1996).
Partial backbone and side chain assignments of UmuD within the UmuD′–UmuD heterodimer were obtained similarly from HNCA, HN(CO)CA, HN(CA)CB, HN(COCA)CB, HNCO, CC(CO)NH-TOCSY, CCNH-TOCSY and HCC(CO)NH-TOCSY spectra, with inter-monomer NOEs being identified from a 200 ms 15N-NOESY-HSQC on a sample containing 100% 2H-, 15N-labeled UmuD mixed with unlabeled UmuD′.
Data were processed with FELIX (Molecular Simulations, Inc.) and analyzed using XEASY (Bartels et al., 1995).
Structure calculations
Distance restraints were derived from NOESY data by integrating the cross-peaks in the 70 ms spectra using XEASY, either manually or with the peakint program, and using CALIBA (Güntert et al., 1997) to convert the volumes to distances. 15N-NOESY-HSQC peak volumes were referenced to dαN(i,i + 1) distances of 2.3 Å within β-strands, while 2D NOESY and 13C-NOESY-HSQC spectra were referenced to Hδ–Hε distances in aromatic rings and to Hα–Hα distances between antiparallel β-strands. Cross-peaks visible only at longer mixing times were classified as 5.0 Å distance constraints (Wüthrich, 1986). NOEs observed in the mixing experiment were assigned unambiguously as inter-monomer restraints (Ferentz et al., 1997). Other NOEs involving residues at the dimerization interface were treated as ambiguous unless examination of preliminary sets of structures clearly indicated that they could be assigned unambiguously as inter- or intra-monomer. Distance restraints were supplemented with hydrogen bond restraints within β-sheets and helices. Restraints are summarized in Table I.
Dihedral angle restraints for φ and χ1 were obtained from analysis of HNHA (Vuister and Bax, 1993), and HNHB (Archer et al., 1991) and 15N-NOESY-HSQC spectra, respectively. Additional φ and ψ angle restraints were derived using TALOS (Cornilescu et al., 1999).
Structures were calculated with X-PLOR 3.851 on Silicon Graphics R10000 workstations (Brünger, 1987). Starting structures were generated by randomizing the φ and ψ dihedral angles in a monomer chain, duplicating those coordinates and translating the second set of coordinates 100 Å from the original position. Each pair of monomers was then subjected to simulated annealing (50 000 steps of 5 fs at 3000 K and 50 000 cooling steps) using NMR-derived restraints and generated symmetry restraints for residues 40–139 (Nilges, 1993). The resulting structures were viewed with InsightII (Molecular Simulations, Inc.) and the quality of the structures was assessed by PROCHECK (Morris et al., 1992). R.m.s. deviations were calculated using Superpose (Diamond, 1992). Figures were created using InsightII, Molscript (Kraulis, 1991) and GRASP (Nicholls, 1993). Atomic coordinates and distance restraints are deposited in the Protein Data Bank (PDB ID 1I4V).
Acknowledgments
Acknowledgements
We thank Yingxi Lin, Hiroshi Matsuo and Mark Sutton for many helpful discussions. This work was supported by grants from the National Institutes of Health to G.C.W. (CA21613) and G.W. (GM47467) and by fellowships to A.E.F. from the American Cancer Society and the Bunting Institute of Radcliffe College.
References
- Archer S.J., Ikura,M., Torchia,D.A. and Bax,A. (1991) An alternative 3D NMR technique for correlating backbone 15N with side chain Hb resonances in larger proteins. J. Magn. Reson., 95, 636–641. [Google Scholar]
- Bartels C., Xia,T., Billeter,M., Güntert,P. and Wüthrich,K. (1995) The program XEASY for computer-supported NMR spectral analysis of biological molecules. J. Biomol. NMR, 6, 1–10. [DOI] [PubMed] [Google Scholar]
- Battista J.R., Ohta,T., Nohmi,T., Sun,W. and Walker,G.C. (1990) Dominant negative umuD mutations decreasing RecA-mediated cleavage suggest roles for intact UmuD in modulation of SOS mutagenesis. Proc. Natl Acad. Sci. USA, 87, 7190–7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell C.E., Frescura,P., Hochschild,A. and Lewis,M. (2000) Crystal structure of the λ repressor C-terminal domain provides a model for cooperative operator binding. Cell, 101, 801–811. [DOI] [PubMed] [Google Scholar]
- Boudsocq F., Campbell,M., Devoret,R. and Bailone,A. (1997) Quantitation of the inhibition of Hfr × F– recombination by the mutagenesis complex UmuD′C. J. Mol. Biol., 270, 201–211. [DOI] [PubMed] [Google Scholar]
- Brünger A. (1987) X-PLOR Version 3.1: A System for X-ray Crystallography and NMR. Yale University Press, New Haven, CT.
- Burckhardt S.E., Woodgate,R., Scheuermann,R.H. and Echols,H. (1988) UmuD mutagenesis protein of Escherichia coli: overproduction, purification, and cleavage by RecA. Proc. Natl Acad. Sci. USA, 85, 1811–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh J., Fairbrother,W., Palmer,A.I. and Skelton,N. (1996) Protein NMR Spectroscopy Principles and Practice. Academic Press, San Diego, CA.
- Cornilescu G., Delaglio,F. and Bax,A. (1999) Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR, 13, 289–302. [DOI] [PubMed] [Google Scholar]
- Courcelle J., Khodursky,A., Peter,B., Brown,P. and Hanawalt,P. (2001) Comparative gene expression profiles following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics, 158, 41–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond R. (1992) On the multiple simultaneous superposition of molecular structures by rigid body transformation. Protein Sci., 1, 1279–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donner A.L., Paa,K. and Koudelka,G.B. (1998) Carboxyl-terminal domain dimer interface mutant 434 repressors have altered dimerization and DNA binding specificities. J. Mol. Biol., 283, 931–946. [DOI] [PubMed] [Google Scholar]
- Ferentz A., Opperman,T., Walker,G. and Wagner,G. (1997) Dimeriz ation of the UmuD′ protein in solution and its implications for regulation of SOS mutagenesis. Nature Struct. Biol., 4, 979–983. [DOI] [PubMed] [Google Scholar]
- Fesik S.W. and Zuiderweg,E.R.P. (1988) Heteronuclear three dimensional NMR spectroscopy. A strategy for the simplification of homonuclear two dimensional NMR spectra. J. Magn. Reson., 78, 588–593. [Google Scholar]
- Frank E.G., Ennis,D.G., Gonzalez,M., Levine,A.S. and Woodgate,R. (1996) Regulation of SOS mutagenesis by proteolysis. Proc. Natl Acad. Sci. USA, 93, 10291–10296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank E.G., Cheng,N., Do,C.C., Cerritelli,M.E., Bruck,I., Goodman,M.F., Egelman,E.H., Woodgate,R. and Steven,A.C. (2000) Visualization of two binding sites for the Escherichia coli UmuD′2C complex (DNA pol V) of RecA–ssDNA filaments. J. Mol. Biol., 297, 585–597. [DOI] [PubMed] [Google Scholar]
- Gonzalez M., Frank,E.G., Levine,A.S. and Woodgate,R. (1998) Lon-mediated proteolysis of the Escherichia coli UmuD mutagenesis protein: in vitro degradation and identification of residues required for proteolysis. Genes Dev., 12, 3889–3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez M., Rasulova,F., Maurizi,M.R. and Woodgate,R. (2000) Subunit-specific degradation of the UmuD/D′ heterodimer by the ClpXP protease: the role of trans recognition in UmuD′ stability. EMBO J., 19, 5251–5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Güntert P., Mumenthaler,C. and Wüthrich,K. (1997) Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol., 273, 283–298. [DOI] [PubMed] [Google Scholar]
- Kay L.E., Ikura,M., Tschudin,R. and Bax,A. (1990) Three-dimensional triple-resonance NMR spectroscopy of isotopically enriched proteins. J. Magn. Reson., 89, 496–514. [DOI] [PubMed] [Google Scholar]
- Kay L.E., Xu,G.-Y., Singer,A.U., Muhandiram,D.R. and Forman-Kay,J.D. (1993) A gradient-enhanced HCCH-TOCSY experiment for recording side-chain 1H and 13C correlations in H2O samples of proteins. J. Magn. Reson., B101, 333–337. [Google Scholar]
- Kraulis P.J. (1991) Molscript—a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- Lee M.H. and Walker,G.C. (1996) Interactions of Escherichia coli UmuD with activated RecA analyzed by cross-linking UmuD monocysteine derivatives. J. Bacteriol., 178, 7285–7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.H., Ohta,T. and Walker,G.C. (1994) A monocysteine approach for probing the structure and interactions of the UmuD protein. J. Bacteriol., 176, 4825–4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y. and Wagner,G. (1999) Efficient side-chain and backbone assignment in large proteins: application to tGCN5. J. Biomol. NMR, 15, 227–239. [DOI] [PubMed] [Google Scholar]
- Little J.W. (1993) LexA cleavage and other self-processing reactions. J. Bacteriol., 175, 4943–4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maor-Shoshani A., Reuven,N., Tomer,G. and Livneh,Z. (2000) Highly mutagenic replication by DNA polymerase V (UmuC) provides a mechanistic basis for SOS untargeted mutagenesis. Proc. Natl Acad. Sci. USA, 97, 565–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo H., Li,H. and Wagner,G. (1996) A sensitive HN(CA)CO experiment for deuterated proteins. J. Magn. Reson., B110, 112–115. [DOI] [PubMed] [Google Scholar]
- McDonald J.P., Frank,E.G., Levine,A.S. and Woodgate,R. (1998) Intermolecular cleavage by UmuD-like mutagenesis proteins. Proc. Natl Acad. Sci. USA, 95, 1478–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald J.P., Peat,T.S., Levine,A.S. and Woodgate,R. (1999) Intermolecular cleavage by UmuD-like enzymes: identification of residues required for cleavage and substrate specificity. J. Mol. Biol., 285, 2199–2209. [DOI] [PubMed] [Google Scholar]
- McLenigan M., Peat,T., Frank,E., McDonald,J., Gonzalez,M., Levine,A., Hendrickson,W. and Woodgate,R. (1998) Novel Escherichia coli umuD′ mutants: structure–function insights into SOS mutagenesis. J. Bacteriol., 180, 4658–4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montelione G.T. and Wagner,G. (1990) Triple resonance experiments for establishing conformation-independent sequential NMR assignments in isotope-enriched polypeptides. J. Magn. Reson., 87, 183–188. [Google Scholar]
- Morris A.L., MacArthur,M.W., Hutchinson,E.G. and Thornton,J.M. (1992) Stereochemical quality of protein structure coordinates. Proteins, 12, 345–364. [DOI] [PubMed] [Google Scholar]
- Nicholls A.J. (1993) GRASP Manual. Columbia University, New York, NY.
- Nilges M. (1993) A calculation strategy for the structural determination of symmetric dimers by 1H NMR. Proteins, 17, 297–309. [DOI] [PubMed] [Google Scholar]
- Nohmi T., Battista,J.R., Dodson,L.A. and Walker,G.C. (1988) RecA-mediated cleavage activates UmuD for mutagenesis: mechanistic relationship between transcriptional derepression and posttranslational activation. Proc. Natl Acad. Sci. USA, 85, 1816–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta T., Sutton,M.D., Guzzo,A., Cole,S., Ferentz,A.E. and Walker,G.C. (1999) Mutations affecting the ability of the Escherichia coli UmuD′ protein to participate in SOS mutagenesis. J. Bacteriol., 181, 177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opperman T., Murli,S., Smith,B.T. and Walker,G.C. (1999) A model for a umuDC-dependent prokaryotic DNA damage checkpoint. Proc. Natl Acad. Sci. USA, 96, 9218–9223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paetzel M. and Strynadka,N.C. (1999) Common protein architecture and binding sites in proteases utilizing a Ser/Lys dyad mechanism. Protein Sci., 8, 2533–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paetzel M., Dalbey,R.E. and Strynadka,N.C.J. (1998) Crystal structure of a bacterial signal peptidase in complex with a β-lactam inhibitor. Nature, 396, 186–190. [DOI] [PubMed] [Google Scholar]
- Peat T.S., Frank,E.G., McDonald,J.P., Levine,A.S., Woodgate,R. and Hendrickson,W.A. (1996) Structure of the UmuD′ protein and its regulation in response to DNA damage. Nature, 380, 727–730. [DOI] [PubMed] [Google Scholar]
- Perry K.L., Elledge,S.J., Mitchell,B., Marsh,L. and Walker,G.C. (1985) umuDC and mucAB operons whose products are required for UV light- and chemical-induced mutagenesis: UmuD, MucA, and LexA proteins share homology. Proc. Natl Acad. Sci. USA, 82, 4331–4335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehrauer W.M., Bruck,I., Woodgate,R., Goodman,M.F. and Kowalczykowski,S.C. (1998) Modulation of RecA nucleoprotein function by the mutagenic UmuD′C protein complex. J. Biol. Chem., 273, 32384–32387. [DOI] [PubMed] [Google Scholar]
- Reuven N.B., Arad,G., Maor-Shoshani,A. and Livneh,Z. (1999) The mutagenesis protein UmuC is a DNA polymerase activated by UmuD′, RecA, and SSB and is specialized for translesion replication. J. Biol. Chem., 274, 31763–31766. [DOI] [PubMed] [Google Scholar]
- Shinagawa H., Iwasaki,H., Kato,T. and Nakata,A. (1988) RecA protein-dependent cleavage of UmuD protein and SOS mutagenesis. Proc. Natl Acad. Sci. USA, 85, 1806–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slilaty S.N. and Little,J.W. (1987) Lysine-156 and serine-119 are required for LexA repressor cleavage: a possible mechanism. Proc. Natl Acad. Sci. USA, 84, 3987–3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer S., Bailone,A. and Devoret,R. (1993) The appearance of the UmuD′C protein complex in Escherichia coli switches repair from homologous recombination to SOS mutagenesis. Mol. Microbiol., 10, 963–971. [DOI] [PubMed] [Google Scholar]
- Sommer S., Coste,G. and Bailone,A. (2000) Specific amino acid changes enhance the anit-recombination activity of the UmuD′C complex. Mol. Microbiol., 35, 1443–1453. [DOI] [PubMed] [Google Scholar]
- Sutton M.D. and Walker,G.C. (2001) umuDC-mediated cold sensitivity is a manifestation of functions of the UmuD2C complex involved in a DNA damage checkpoint control. J. Bacteriol., 183, 1215–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton M.D., Opperman,T. and Walker,G.C. (1999) The Escherichia coli SOS mutagenesis proteins UmuD and UmuD′ interact physically with the replicative DNA polymerase. Proc. Natl Acad. Sci. USA, 96, 12373–12378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton M.D., Smith,B.T., Godoy,V.G. and Walker,G.C. (2000) The SOS response: recent insights into umuDC-dependent mutagenesis and DNA damage tolerance. Annu. Rev. Genet., 34, 479–497. [DOI] [PubMed] [Google Scholar]
- Sutton M.D., Kim,M. and Walker,G.C. (2001) Genetic and biochemical characterization of a novel umuD mutation: insights into a mechanism for UmuD self-cleavage. J. Bacteriol., 183, 347–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talluri S. and Wagner,G. (1996) An optimized 3D NOESY-HSQC. J. Magn. Reson., B112, 200–205. [DOI] [PubMed] [Google Scholar]
- Tang M., Bruck,I., Eritja,R., Turner,J., Frank,E., Woodgate,R., O’Donnell,M. and Goodman,M. (1998) Biochemical basis of SOS-induced mutagenesis in Escherichia coli: reconstitution of in vitro lesion bypass dependent on the UmuD′2C mutagenic complex and RecA protein. Proc. Natl Acad. Sci. USA, 95, 9755–9760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M., Shen,X., Frank,E.G., O’Donnell,M., Woodgate,R. and Goodman,M.F. (1999) UmuD′2C is an error-prone DNA polymerase, Escherichia coli pol V. Proc. Natl Acad. Sci. USA, 96, 8919–8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M., Pham,P., Shen,X., Taylor,J., O’Donnell,M., Woodgate,R. and Goodman,M. (2000) Roles of E.coli DNA polymerases IV and V in lesion-targeted and untargeted SOS mutagenesis. Nature, 404, 1014–1018. [DOI] [PubMed] [Google Scholar]
- Vuister G.W. and Bax,A. (1993) Quantitative J correlations: a new approach for measuring homonuclear three-bond J(HNHa) coupling constants in 15N-enriched proteins. J. Am. Chem. Soc., 115, 7772–7777. [Google Scholar]
- Woodgate R., Rajagopalan,M., Lu,C. and Echols,H. (1989) UmuC mutagenesis protein of Escherichia coli: purification and interaction with UmuD and UmuD′. Proc. Natl Acad. Sci. USA, 86, 7301–7305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wüthrich K. (1986) NMR of Proteins and Nucleic Acids. Wiley, New York, NY.
- Yamazaki T., Lee,W., Arrowsmith,C.H., Muhandiram,D.R. and Kay,L.E. (1994) A suite of triple resonance NMR experiments for the backbone assignment of 15N, 13C, 2H labeled proteins with high sensitivity. J. Am. Chem. Soc., 116, 11655–11666. [Google Scholar]