

Abstract
The rare inherited human genetic disorder Cockayne syndrome (CS) is characterized by developmental abnormalities, UV sensitivity and premature aging. The cellular and molecular phenotypes of CS include increased sensitivity to UV-induced and oxidative DNA lesions. Two genes are involved: CSA and CSB. The CS group B (CSB) protein has roles in transcription, transcription-coupled repair, and base excision repair. It is a DNA stimulated ATPase and remodels chromatin in vitro. Here, we have analyzed wild-type (wt) and motif II, V and VI mutant CSB proteins. We find that the mutant proteins display different degrees of ATPase activity deficiency, and in contrast to the in vivo complementation studies, the motif II mutant is more defective than motif V and VI CSB mutants. Furthermore, CSB wt ATPase activity was studied with different biologically important DNA cofactors: DNA with different secondary structures and damaged DNA. The results indicate that the state of DNA secondary structure affects the level of CSB ATPase activity. We find that the CSB protein is phosphorylated in untreated cells and that UV irradiation leads to its dephosphorylation. Importantly, dephosphorylation of the protein in vitro results in increased ATPase activity of the protein, suggesting that the activity of the CSB protein is subject to phosphorylation control in vivo. These observations may have significant implications for the function of CSB in vivo.
INTRODUCTION
Nucleotide excision repair (NER) is a complicated process, which removes a broad spectrum of DNA lesions. Two subpathways of NER exist: (i) the global genome repair pathway and (ii) the transcription-coupled repair (TCR) pathway, and TCR is an important factor in Cockayne syndrome (CS) (1). CS is a premature aging syndrome with complex symptoms, including developmental abnormalities, neurologic dysfunction and short average life span. Cellular characteristics include hypersensitivity to UV light, and failure of RNA synthesis to recover to normal rates following UV irradiation. Two genes have been shown to be involved: CSA and CSB (2). The 168 kDa CS group B (CSB) protein contains an acidic amino acid stretch, a glycine-rich region, two putative nuclear localization signal (NLS) sequences, and seven conserved ATPase motifs (3). Members of the SNF2-like family of DNA-dependent ATPases, including CSB, contain seven characteristic motifs, I, Ia and II–VI, that are also present in DNA and RNA helicases (4). Helicase activity has not been demonstrated for any members of the SNF2-like family, which is part of Superfamily 2 (SF2), but in general they have the ability to destabilize protein–DNA interactions (5). The CSB protein displays DNA-dependent ATPase activity, which is better stimulated by double-stranded DNA (dsDNA) than single-stranded DNA (ssDNA) (6–8), and CSB is able to remodel chromatin in vitro (9).
So far, only helicases, and thus no proteins of the SNF2-like family, have been crystallized. However, structures derived from the crystallized SF1 helicases Rep and PcrA reveal striking similarity to the distantly related SF2 RNA helicase NS3, and the seven conserved helicase motifs form a nucleotide binding (NTB) pocket (10). In particular, the conserved aspartate and glutamate residues of motif II (Walker B) coordinate the Mg2+ ion and probably also the attacking water molecule during ATP hydrolysis (11,12). Motif V seems to contact the oligonucleotide, while motif VI is directly or indirectly involved in NTP binding (11,12).
The integrity of the SNF2-like ATPase domain is critical for most functions of CSB in vivo. CSB cDNA with point mutations in motifs Ia, II, III, V and VI, as opposed to wild-type (wt) CSB cDNA, do not complement the deficiencies of the SV40-transformed CS-B cell line, CS1AN.S3.G2 (13,14). In contrast, both a deletion of the acidic region and a point mutation in a putative NTB motif do not interfere with the ability of CSB to complement CSB-deficient cells (14–16).
CS-B cells and cell extracts are generally deficient in incision at 8-oxoguanine (8-oxoG) and 8-oxoadenine lesions (13,17–19). The reduced 8-oxoG activity can be complemented by transfection with wt CSB and partially complemented by motif II (E646Q) mutant cDNA, but not with motif V (T912/913V) or VI (Q942E) CSB mutants (13,20). Furthermore, the 8-oxoG repair capacity of wt and mutant transfected cells correlates with mRNA and protein levels of the 8-oxoG DNA glycosylase (OGG1) responsible for the removal of 8-oxoG (17,18). This suggests that the general base excision repair (BER) defect is associated with a subtle transcription deficiency, and that wt and motif II but not motif V and VI mutant CSB proteins are able to support efficient OGG1 expression. There also appears to be a functional interaction between OGG1 and CSB (21). To better understand the biological functions of the mutant CSB proteins, we report here the functional analysis of purified, recombinant CSB wt and motif II, V and VI mutant proteins. We find that the ATPase activity of the motif II CSB mutant is nearly abolished whereas motif V and VI mutant proteins retain residual catalytic activity. DNA binding of CSB mutant proteins is similar to the CSB wt protein. The motif V and VI mutant proteins show slightly reduced ATP binding while the motif II mutant protein exhibits increased ATP binding. Also, CSB wt and mutant protein tagged with enhanced cyano fluorescent protein (ECFP) all display homogenous nuclear distribution. These results suggest that deficiencies in functions other than DNA binding or DNA-stimulated ATP hydrolysis detected in vitro may be responsible for the defects of CS-B null cell lines transfected with either motif V or motif VI CSB mutant genes. Furthermore, CSB wt ATPase activity was studied with different biologically important DNA cofactors, and the results indicate that the CSB wt ATPase activity is maximally stimulated by DNA substrates containing a small loop or bubble.
Although the CSB protein is involved in RNA polymerase II (RNAPII) elongation, it does not affect the transcription of all genes, suggesting that the activity of the CSB protein is regulated in vivo (13,22). Control of phosphorylation status is an important factor in both transcription and repair (23–25), and phosphorylation affects subcellular localization as well as activity of different members of the SNF2 family (26,27). Phosphorylation of target proteins is an integral part of the DNA damage response, and the p53 protein, which interacts with CSB, is phosphorylated in response to DNA damage (28,29). The extent of phosphorylation of the CSB protein has not previously been investigated and we were interested in this parameter and, particularly, how phosphorylation status might affect its activities and how it might change during DNA repair. Here we present evidence that CSB activity is controlled by phosphorylation. Notably, we document that CSB ATPase activity is stimulated by dephosphorylation, and that UV irradiation results in dephosphorylation of CSB in vivo.
MATERIALS AND METHODS
Plasmid constructs
CSB wt containing an N-terminal hemagglutinin antigen (HA) epitope and a C-terminal histidine (His6) cloned in the pFASTBAC vector, pFB-CSBwt (8), was kindly provided by Alan Lehmann. A PflM1 fragment of pcDNA3.1-CSB containing mutations resulting in either E646Q, T912/913V or Q942E substitutions (13,14), was exchanged with the corresponding fragment in pFB-CSBwt using the rapid ligation kit according to the manufacturer’s instructions (Roche), thus generating pFB-CSBE646Q, pFB-CSBT912/913V and pFB-CSBQ942E.
Baculovirus production and infection
Bacmids and virus from pFB-CSBwt, pFB-CSBE646Q, pFB-CSBT912/913V and pFB-CSBQ942E were produced using the BAC to BAC Baculovirus Overexpression System (LifeTechnologies, Inc.) essentially as described in the accompanying manual. Suspension cultures of HiFive cells were infected with the recombinant baculoviruses at a multiplicity of infection of 10. Three days post-infection cells were collected from a 200 ml culture and washed twice in ice-cold phosphate-buffered saline (PBS).
CSB protein purification
Recombinant proteins were purified using a modification of a method described by Citterio et al. (8). Pellet was lysed in ice-cold lysis buffer 1 (25 mM Tris–HCl pH 9.0, 1 mM EDTA, 10% glycerol, 1% NP-40, 0.3 M KCl, 1 mM β-mercaptoethanol, 0.1 mM PMSF, 2 µM each of chymostatin, leupeptin, antipain, pepstatin A). The lysate was centrifuged at 12000 g for 15 min. For purification the filtrated supernatant was diluted with 2 vol buffer 2 (buffer 1 at pH 6.8 and without NP-40 and EDTA). A Ni2+–nitriloacetic acid–agarose column (Qiagen) equilibrated with buffer A (25 mM HEPES–KOH pH 7, 0.01% NP-40, 10% glycerol, 1 mM β-mercaptoethanol, 0.1 mM PMSF, 0.3 M KCl) and 5 mM imidazole was loaded with the diluted lysate. The column was washed with buffer A and 20 mM imidazole. Proteins were eluted with buffer A and a 20 ml linear gradient of 20–250 mM imidazole and CSB eluted at ∼60 mM imidazole. A HiTrap heparin column (Amersham Pharmacia Biotech) equilibrated with buffer A was directly loaded with the CSB containing Ni2+–nitriloacetic acid fractions. The column was washed with buffer A, and the proteins were eluted with buffer A and 0.8 M KCl. Protein was stored in aliquots at –80°C. Protein concentration was determined in the flow-through, wash and selected fractions of both the Ni2+–nitriloacetic acid–agarose column and the heparin column by Bradford analysis. Purity of CSB was checked by 7% acrylamide Tris–acetate SDS–PAGE (Invitrogen) and by immunoblotting using a Penta-His antibody (Qiagen). Furthermore, specific activity of different Ni2+–nitriloacetic agarose fractions and heparin fractions was tested, and the activity corresponded well with the pooled fractions.
CSB ATPase activity
Standard reactions (10 µl) were performed with 150 ng of DNA cofactor, supercoiled (>90%) pUC19 plasmid, 10 ng of protein, and 1 µCi of [γ-32P]ATP (3000 Ci/mmol; Hartmann Analytic) in buffer B (20 mM Tris–HCl pH 7.5, 4 mM MgCl2, 50 µM ATP, 40 µg/ml BSA, 1 mM DTT). Reactions were incubated for 1 h at 30°C and stopped by the addition of 5 µl of 0.5 M EDTA. Samples (1 µl) were analyzed on a polyethylenimine-cellulose thin layer chromatography (PEI-TLC) plate developed in 0.75 M KH2PO4. Plates were exposed on screen and ATP hydrolysis was analyzed using a Molecular Imager. For activity determinations, either the amount of protein or DNA cofactor was varied. For determination of kinetic parameters the amount of CSB protein was kept constant, while the amount of cold substrate was varied between 5 and 100 µM, or ATP concentration was kept constant and the time was varied (5, 10, 20, 30, 40, 60 min). Less than 20% of the ATP was hydrolyzed during the incubations. Results from multiple experiments are reported as mean values with standard deviations. Differences were assessed by one-way analysis of variance followed by Student’s t-test, and P < 0.05 was considered to be significant.
Cofactor DNA substrates
Initially, pUC19 plasmid was purified using Qiagen maxiprep (protocol supplied by manufacturer). CsCl-gradient analysis showed that >90% was in a supercoiled form.
UV damage. The pUC19 plasmid was irradiated with 300 J/m2 on ice resulting in approximately 10 lesions per plasmid (30).
Methylene blue (MB). Fifty micrograms of plasmid DNA were diluted with 0.30 ml of H2O and 0.3 ml of 6 µM MB was added. The diluted sample was irradiated with visible light (18.7 kJ/m2/s) for 3 min, resulting in approximately 10 8-oxoG per plasmid (31).
Cisplatin. Forty micrograms of pUC19 plasmid were incubated overnight at 37°C with 4.8 µM cisplatin in 3 mM NaCl, 0.5 mM Na2HPO4, 0.5 mM NaH2PO4, resulting in 10 adducts per plasmid (32). The reaction was stopped by addition of NaCl to 60 mM.
Methyl methane sulfonate (MMS). Ten micromolar MMS with 100 µM DNA (∼1000 bp) in 10 mM Tris pH 7.8 gives approximately 1 adduct per 1000 nt (33). pUC19 (40 µg) was incubated overnight at 37°C with 0.48 mM MMS and 10 mM Tris pH 7.8.
The DNA was precipitated, washed, redissolved and diluted to 150 ng/µl, and checked on an agarose gel. The synthetic oligonucleotides used (DNA Technology, Denmark), were FPLC-purified, and are shown in Table 1.
Table 1. DNA substrates used as cofactors in ATPase assays.

DNA binding assay
A 166 bp DdeI pUC19 fragment was labeled using the Klenow fragment (Invitrogen) and 5 µCi [α-32P]ATP (300 mCi/mmol; Amersham Pharmacia Biotech). Nitrocellulose filters were pre-wetted in binding buffer (25 mM HEPES–KOH pH 8.3, 40 mM MgCl2, 10 mM EDTA, 450 µg/ml BSA, 10 mM DTT, 10% glycerol) and indicated amounts of CSB proteins were incubated with 1 ng of DNA in binding buffer with or without 3 mM ATP-γS or ADP. Samples were incubated for 30 min at room temperature, and 100 µl of binding buffer was added. Samples were passed through a nitrocellulose filter, washed twice with 0.5 ml of binding buffer, and exposed on a screen. Background radioactivity bound in the absence of protein was subtracted from total radioactivity bound to the filter and percent bound DNA of total DNA was determined. A Hill plot was used to analyze the data (34).
ATP binding assay
CSB wt and mutant proteins (25, 50, 100 and 200 nM) were incubated with 10 nM DNA, 83 nM ATP-γ35S in buffer B for 5 min at 30°C. Samples were applied to a pre-wetted nitrocellulose filter (1× buffer B without BSA) and washed 10× with ATP wash buffer (50 mM Tris pH 7.9, 0.1 M EDTA, 5 mM MgCl2, 2.5% glycerol, 800 mM KCl). Filters were exposed to screen. Background radioactivity bound in the absence of protein was subtracted from total radioactivity bound to the filter and the data from at least three independent experiments were normalized against results obtained for 25 nM CSB wt protein.
Localization of ECFP-tagged CSB
CSB wt cDNA was cloned upstream of the ECFP gene in pMC-ECFP (kindly provided by Christian Mielke), which was constructed from pMC-2P (35), to give rise to the vector pMC-CSB-ECFP. The ECFP-tagged CSB E646Q, Q942E and T912/913V mutant vectors were produced by replacing the 1300 bp NsiI fragment of pMC-CSB-ECFP with each of the mutant cDNA sequences obtained by PCR amplification from pcDNA3.1-CSB-derived plasmids containing the respective mutations (13,14). All PCR-amplified sequences were verified by full-length sequencing. CS1AN.S3.G2 cells, grown in DMEM supplemented with 10% fetal calf serum (FCS) (Gibco BRL, Life Technologies), 100 U/ml penicillin, and 100 µg/ml streptomycin, were transfected with the resulting plasmids using Lipofectamine (Gibco BRL, Life Technologies) according to the manufacturer’s instructions. Stably transfected cell lines were selected and maintained in medium containing 0.2 µg/ml puromycin. pMC-CSB-ECFP-transfected cells were further selected for UV resistance by treatment with 4 J/m2 UV-C on 3 consecutive days. The cellular distribution of ECFP-tagged proteins was analyzed employing an epifluorescence microscope (Olympus) and pictures were captured using a Cooled CCD camera.
In vitro phosphorylation of CSB
Whole-cell extracts for in vitro phosphorylation were prepared by lysing 2–5 × 106 cells with radioimmunoprecipitation (RIPA) buffer (150 mM NaCl, 1% Triton, 0.1% SDS, 10 mM Tris pH 8, 0.5% sodium deoxycholate, 0.2 mM PMSF, 20 µg/ml aprotinin, 10 µg/ml leupeptine, 1 mM Na-orthovanadate, 10 U/ml DNase) supplemented with phosphatase inhibitors (Sigma, 1:100). The lysate was microcentrifuged at 16 000 g for 10 min at 4°C. In vitro phosphorylation reactions were carried out with kinase buffer (20 mM Tris–HCl pH 7.5, 50 mM KCl, 10 mM MgCl2, 25 µM ATP) and 5 µCi of [γ-32P]ATP. GTP, which is also a phosphoryl donor of casein kinase II (CKII), was added when indicated. The reactions (10 µl) were initiated by the addition of CKII (New England Biolabs) or whole-cell extracts and carried out at 30°C for 15 min. The resulting products were analyzed by electrophoresis on a 7% acrylamide Tris–acetate gel (Invitrogen). The gel was washed and visualized by a Molecular Imager. Alternatively, phosphorylation reactions were performed without [γ-32P]ATP, and the products analyzed by immunoblotting.
Dephosphorylation of CSB
CSB protein was incubated with the indicated amount of protein phosphatase 1 (PP1; New England Biolabs) in phosphatase buffer (50 mM Tris–HCl pH 7.0 at 25°C, 0.1 mM EDTA, 5 mM DTT, 0.01% Brij 35, 1 mM MnCl2 and 3 mM ATP) for 15 min at 30°C. The products were analyzed by immunoblotting or used in standard ATPase reactions as described above.
In vivo phosphorylation of CSB
Human diploid fibroblasts, GM38B, were seeded and grown for 48 h. Cells (2–5 × 106) were incubated with phosphate-free DMEM (Gibco) supplemented with 5% dialyzed FCS for 1 h to exhaust the pool of endogenous phosphate, and then irradiated with 0 or 6 J/m2 UV. The cells were supplemented with fresh phosphate-free medium containing 100 µCi 32P inorganic phosphate (Pi) (Hartmann Analytic) for 4 h. Radiolabeling was terminated by washing twice with ice-cold PBS, cells were lysed with RIPA buffer, and the lysate was centrifuged at 12 000 g for 10 min at 4°C. The supernatant was pre-cleared with Protein A magnetic beads (New England Biolabs), and incubated with 5 µg of polyclonal anti-ERCC6 (A. F. Schützdeller, Germany) for 1 h at 4°C. Protein A magnetic beads were incubated with immune complexes for 1 h at 4°C, and beads were collected using the accompanying magnet. The beads were washed three times with RIPA buffer and proteins were analyzed by SDS–PAGE and visualized by a Molecular Imager and silver stain.
RESULTS
CSB wt and mutant protein activity
Site-directed mutations in ATPase motifs II, V and VI (Fig. 1A) were subcloned into the pFB-CSBwt vector, thus enabling purification of these mutants as recombinant proteins. CSB cDNA containing any of these mutations, in contrast to wt CSB, cannot complement most of the defects of CS1AN.S3.G2 cells (Fig. 1B) (13,20). Figure 2A shows that the CSB wt and CSB mutant proteins were of similar purity as seen by silver staining, and had the expected size of ∼168 kDa. The introduced mutations are minor conserved substitutions, which are not expected to exhibit significant structural perturbations. We observed that CSB wt and mutant proteins showed the same behavior in solution and during chromatography and aggregated at high concentrations in solution.
Figure 1.

Diagram showing the CSB protein. (A) The recombinant CSB protein contains a HA epitope; a highly acidic region, A; a glycine rich region, G; two serine phosphorylation signals, S; preceded by NLSs, L; the seven conserved Swi/Snf ATPase motifs, I, IA, II–VI; a NTB fold, NTB; and finally the C-terminal histidine tag, His. Introduced N- and C-terminal tags are shown in gray. Site-directed mutations in ATPase motifs II, V and VI are indicated. (B) Complementation characteristics of the wt and mutant proteins in CS1AN.S3.G2 cells (13,14,18).
Figure 2.

CSB purification and ATPase activity. (A) Comparison of 100 ng of each of the purified proteins by silver stain and immunoblot using His antibody. wt, CSB wt; E, CSBE646Q; Q, CSBQ942E; T, CSBT912/913V. (B) [γ-32P]ATP was incubated with 10 ng of recombinant CSB wt protein in the presence or absence of the indicated DNA cofactors under standard reaction conditions but without 50 µM cold ATP (see Materials and Methods), and analyzed on PEI-TLC. (C) Quantification of ATP hydrolysis after incubation with 10 ng of recombinant CSB wt, 50 µM cold ATP, and the indicated amounts of supercoiled, undamaged pUC19 plasmid DNA. Release of Pi was quantified using a Molecular Imager. Error bars represent standard deviations of at least three independent experiments.
The CSB protein is a DNA-stimulated ATPase, with preference for dsDNA (8), and CSB wt protein purified in our laboratory showed the same behavior (Fig. 2B). A single-stranded pre-melted 60mer stimulated the CSB ATPase activity weakly when compared to a double-stranded 60mer or plasmid DNA, whereas very little or no activity was seen with a (dT)16 oligonucleotide or no DNA. In all subsequent ATPase assays, 50 µM cold ATP was included to keep the ATP hydrolysis below 20% and thereby allow activity determinations. The CSB ATPase shows a clear dose response upon addition of increasing amounts of pUC19 cofactor DNA (Fig. 2C). CSB ATPase activity showed a plateau above 50 ng of pUC19 DNA (Fig. 2C). Thus, to ensure that the ATPase activity is not limited by availability of cofactor DNA, 150 ng of DNA was used in subsequent reactions. Lineweaver–Burke analysis of the data yielded a Keffector value for pUC19 DNA of 3.1 ± 0.9 nM (55 ± 16 ng).
The extent of ATP hydrolysis was observed to be proportional to the amount of CSB wt present (Fig. 3). The conservative substitution E646Q in motif II of the SNF2-like domain resulted in complete abolishment of the ATPase function of the protein. In contrast, it was seen that the two other conservative substitutions T912/913V and Q942E in motifs V and VI, respectively, retained a low level of ATPase activity (Fig. 3). Since the CSB wt and mutant proteins were purified by the same procedure and are at a similar level of purity and concentration (Fig. 2A), the ATPase activity is not due to any contaminating species, but is a property of the purified proteins. Furthermore, we performed simultaneous analysis of the specific activity, purity of CSB wt and mutant proteins, and the amount of CSB present in the flow-through, wash and different fractions of both the Ni2+–nitriloacetic acid–agarose column and the heparin column. This analysis showed that the differences in activity are not due to loss of active enzyme during the purification procedure (data not shown). The protein titrations are also consistent with an impairment of ATPase activity of the mutant proteins, and even large quantities of mutant enzyme, compared to wt, do not overcome the deficiency.
Figure 3.

ATPase activity of CSB wt and mutant proteins. [γ-32P]ATP hydrolysis after incubation with 0–150 ng of recombinant CSB wt and mutant proteins, 50 µM cold ATP and 150 ng of plasmid DNA under standard reaction conditions. Error bars represent standard deviations of at least three independent experiments.
Km was calculated in five independent experiments with constant protein concentration, supercoiled pUC19 plasmid and an ATP concentration ranging from 5 to 100 µM. A Km value for ATP of ∼13 µM with dsDNA as the cofactor and a Km of ∼23 µM with ssDNA was calculated for the CSB wt protein (Table 2). kcat was calculated from ATPase kinetics analysis with time ranging from 5 to 60 min. kcat was ∼31 min–1 with dsDNA as the cofactor and with ssDNA cofactor kcat was ∼5 min–1 for the CSB wt protein (Table 2), and these results are quite similar to results reported earlier (6,8). CSBQ942E has a 4.6-fold increase in Km accompanied by a 3.8-fold decrease in kcat compared to CSB wt protein. The CSBT912/913V protein has a 9.2-fold increase in Km and, as the CSBQ942E protein, a 3.8-fold reduction in kcat when compared to CSB wt. These data indicate that mutations of conserved residues in motifs V and VI impair ATP binding as well as ATP hydrolysis itself.
Table 2. Kinetic parameters of CSB wt and mutant ATPase.
| CSB wt | CSBE646Q | CSBQ942E | CSBT912/913V | |||
|---|---|---|---|---|---|---|
| This study | Selby and Sancar (22) | Citterio et al. (8) | This study | This study | This study | |
| ATPase kcat (min–1) | ||||||
| dsDNA | 31 ± 6 | 45–53 | 27–33 | nd | 8 ± 2 | 8 ± 3 |
| ssDNA | 5.1 ± 0.6 | 3.7–6 | ||||
| ATPase Km (µM) | ||||||
| dsDNA | 13 ± 1 | nd | 60 ± 10 | 120 ± 20 | ||
| ssDNA | 23 ± 5 |
DNA effectors were supercoiled pUC19 plasmid (dsDNA) or preboiled single-stranded 60mer (ssDNA). For kcat determinations, 10 ng of CSB, 50 µM ATP and 150 ng of DNA were used and the time of incubation was varied (0, 5, 10, 20, 30, 40 and 60 min). For Km determinations, 10 ng of CSB, 150 ng of DNA, and varying concentrations of ATP (5–100 µM) were incubated for 1 h. Values represent mean ± standard deviations of at least three independent experiments; nd, Pi release was undetectable, thus no parameters could be determined.
In addition to the ATPase activity, we also examined ATP and DNA binding of the different mutant CSB proteins, using a nitrocellulose filter-binding assay. In support of the kinetic data, ATP binding of the motif V and VI mutants was reduced compared to the CSB wt protein (Fig. 4, data are normalized against ATP binding of 25 nM CSB wt protein). In contrast, the motif II mutant exhibited increased ATP binding compared to CSB wt protein (Fig. 4). The DNA binding analysis showed that CSB wt protein exhibited DNA binding in the absence of nucleotide and reduced DNA binding in the presence of ADP or the weakly hydrolyzable ATP analog ATPγS (Fig. 5A). To determine the apparent dissociation constant, the data were analyzed by a Hill plot. In the absence of nucleotide, Kd was 0.27 mM, while the presence of ATPγS increased Kd to 1.5 mM. In the presence of ADP, Kd was found to be 2.9 mM. Purified CSB mutant proteins all exhibited DNA binding with Kd at wt levels both in the absence (Fig. 5B) and presence of nucleotide (data not shown).
Figure 4.

ATP binding of CSB wt and mutant proteins. CSB wt protein was incubated with ATP-γ35S (83 nM). Samples were applied to nitrocellulose membranes, washed and exposed on screen. Binding was analyzed on a Molecular Imager and data were normalized against 25 nM CSB wt. Error bars represent standard deviations of at least three independent experiments.
Figure 5.

DNA binding of CSB wt and mutant proteins. (A) Indicated amounts of CSB wt protein with or without 3 mM ATPγS or ADP was incubated with ∼1 ng of radioactively labeled 166 bp DdeI pUC19 fragment for 30 min, bound to a nitrocellulose filter, and bound DNA was analyzed on a Molecular Imager. Points represent the mean of three independent experiments. (B) CSB wt and mutant proteins without nucleotide were incubated as indicated above. Points represent the mean of three independent experiments.
Localization of CSB wt and mutant proteins
To determine whether differences in in vivo complementation data could be attributed to a deficiency in subcellular localization, which may arise as a result of misfolding due to the amino acid substitutions, CSB wt and mutant proteins were tagged with ECFP and expressed in CS1AN.S3.G2 cells. The analysis showed that ECFP alone localized all over the cell, whereas the CSB wt-ECFP fusion protein localized to the nucleus and rescued the CS1AN cells from UV sensitivity (Fig. 6, and data not shown). ECFP fusions with the three different mutants also showed predominant nuclear localization (Fig. 6). The tagged CSB proteins were homogeneously distributed in the nucleus with ∼20% of the cells showing additional brighter fluorescent foci.
Figure 6.
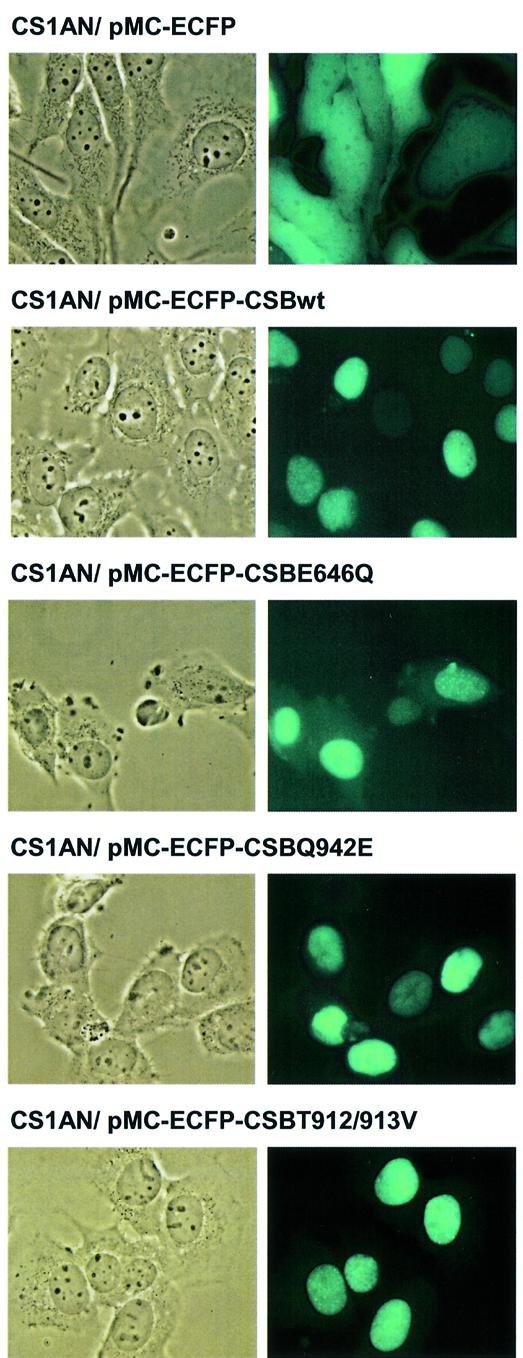
Cellular localization of ECFP-tagged CSB wt and mutant protein. CS1AN.S3.G2 cells stably transfected with N-terminal fusion of ECFP to CSB wt and mutant proteins. Images were captured using epifluorescence microscopy.
CSB ATPase activity dependence on secondary structures of the DNA cofactor and on substrates containing DNA damage
It was next investigated whether CSB ATPase activity is affected by cofactor DNA substrates with different secondary structure related to NER or transcription arrest (bubble of 15 nt), or transcription elongation (bubble of 35 nt) (36,37). The structures tested are shown in Table 1. Interestingly, the ATPase activity was significantly increased (1.6-fold) with DNA cofactor substrates containing a loop of 16 thymines and with bubble substrates containing 15 thymines in the bubble (loop and b15 in Fig. 7A). In contrast, cofactor DNA containing double-strand (ds) to single-strand (ss) transitions in non-rigid structures [ds–ss and splayed arm (sa) and a bubble of 35 thymines (b35)] did not increase the CSB ATPase activity significantly above that of a double-stranded 31mer. To test if DNA damage directly can stimulate the CSB ATPase, pUC19 plasmid DNA was damaged by UV irradiation, photoactivated MB, cisplatin or MMS. No damaged DNA had an effect, yet a consistent, but statistically insignificant, stimulation was observed with cisplatin-treated DNA (Fig. 7B). Since the activity of other SNF2-like family members is affected by the presence of interacting proteins (5), we also investigated whether CSB ATPase activity was influenced by some of its known interaction partners. However, neither XPA nor p53 had any effect on CSB ATPase activity (data not shown).
Figure 7.
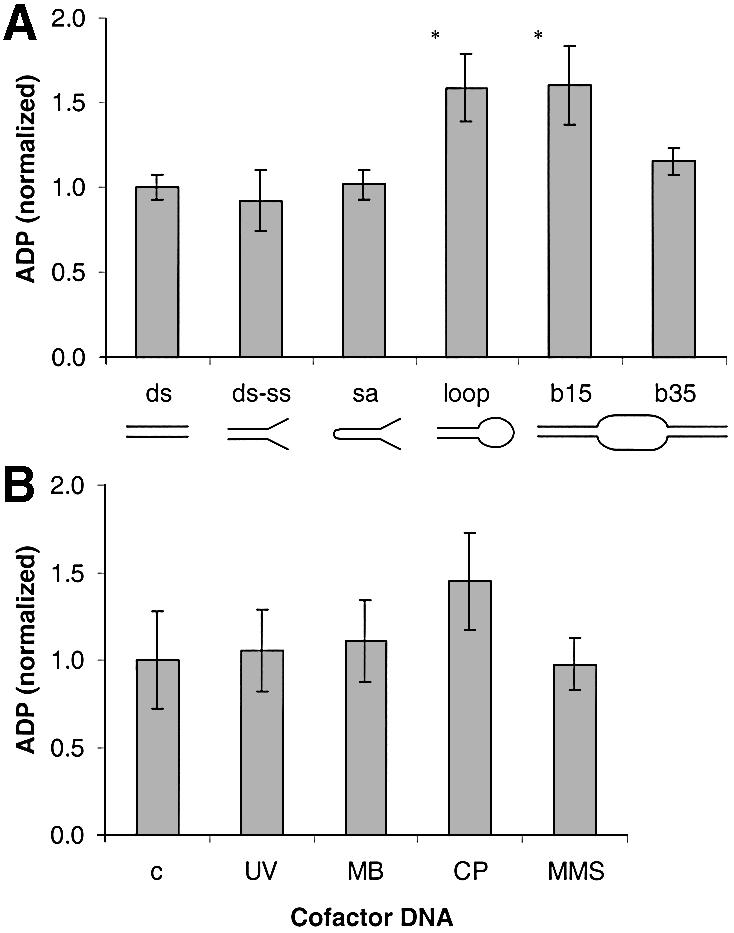
CSB ATPase activity stimulated by different cofactor DNA substrates. Reaction conditions were as described in Figure 2 with 150 ng DNA. (A) CSB wt ATPase activity in the presence of synthetic oligonucleotides with the indicated secondary structures. (B) CSB wt ATPase activity in the presence of pUC19 DNA damaged with UV radiation: MB, methylene blue and visible light; CP, cisplatin; MMS, methyl methane sulfonate, as described in Materials and Methods. Values were normalized against control DNA; *P < 0.05, statistically significantly different from control DNA.
Phosphorylation of the CSB protein in vitro
When the CSB gene was first cloned, it was reported to harbor two putative CKII serine phosphorylation sites in the vicinity of putative NLSs (3). It has not previously been shown whether CSB is post-translationally modified by phosphorylation. We performed a database search, which resulted in the identification of several other possible CKII phosphorylation sites, a number of protein kinase C phosphorylation sites and two tyrosine kinase phosphorylation sites. We then investigated whether the CSB protein is phosphorylated. Initially, recombinant CSB wt protein was incubated with purified CKII in the presence of [γ-32P]ATP, and the products were analyzed by SDS–PAGE. As seen in the top panel in Figure 8A, CKII is able to phosphorylate CSB in vitro. When cold GTP, which is a substrate for the CKII kinase and not for other kinases, was included in the reaction, the radioactive labeling of CSB was diminished (compare lanes 2–4, top panel, in Fig. 8B). The bottom panel shows the Coomassie stain of the gel and verifies equal loading of the CSB protein. In addition, whole-cell extracts prepared from human primary fibroblasts, GM38B cells, could phosphorylate CSB, and this phosphorylation was out competed by addition of cold GTP as well (compare lanes 6–8, top panel, Fig. 8B), thus substantiating the potential role of CKII.
Figure 8.

Phosphorylation of the CSB protein in vitro. (A) Recombinant CSB wt protein was incubated with 50 U of purified CKII in the presence of [γ-32P]ATP for 0, 10 or 20 min. CSB phosphorylation was detected by SDS–PAGE, exposure on screen and the use of a Molecular Imager. Coomassie stain of the same gel indicates equal loading. (B) As (A) with the indicated amounts of either purified CKII, or whole-cell extract (WCE) and GTP as indicated.
CSB is phosphorylated in vivo
Recombinant proteins purified from insect cells are often phosphorylated. Western blotting and phospho-specific antibodies were used to test whether this is also the case for the CSB protein. The results shown in Figure 9A indicate that purified recombinant CSB wt was heavily phosphorylated at threonine residues but not at serine or tyrosine. Incubation with CKII increased the threonine phosphorylation slightly, but also induced phosphorylation at tyrosine and serine (Fig. 9A, lanes 2 and 3). This is somewhat in conflict with the substrate specificity of CKII, which mainly phosphorylates threonine and serine residues, and could be explained by cross-reactivity of the antibody. Furthermore, it was tested if PP1 was able to remove the phosphorylation of the recombinant CSB. As shown in Figure 9A, lanes 4 and 5, PP1 reduced CSB phosphorylation significantly.
Figure 9.
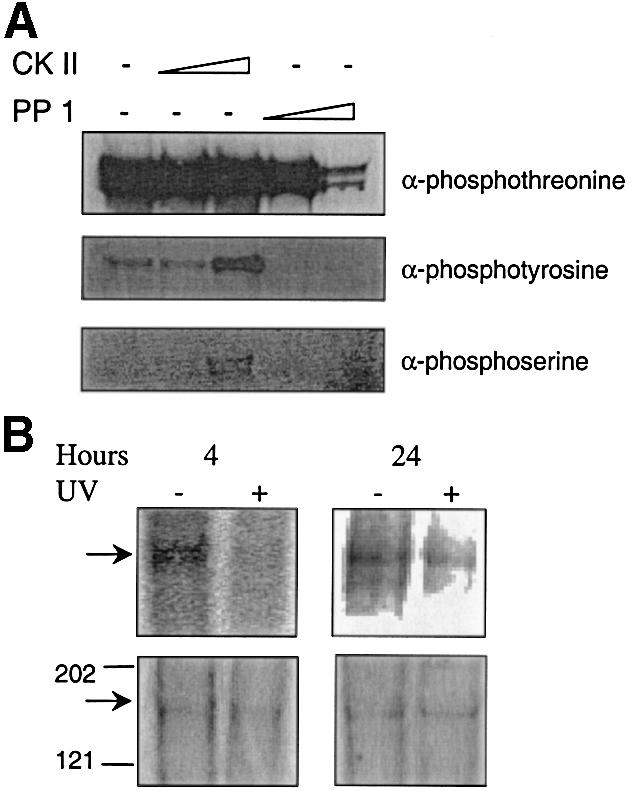
Phosphorylation of the CSB protein in vivo. (A) Purified recombinant CSB wt protein (1 µg) was phosphorylated with 50 or 200 U of CKII or dephosphorylated with 100 or 500 mU of PP1. After immunoblotting, phosphorylation was detected using phosphospecific antibodies as indicated. (B) CSB is phosphorylated in vivo. GM38B cells were irradiated either with 0 or 6 J/m2 and incubated for 4 or 24 h with 32P Pi phosphate at 15 µCi/ml. The cells were lysed and immunoprecipitated with antibody against CSB. Immunoprecipitated proteins were resolved using 7% acrylamide Tris–acetate SDS–PAGE. The gel was washed and visualized with a Molecular Imager. Lower panel shows the silver staining of the same gel to confirm that equal amounts of protein were loaded. Arrow points to CSB protein; size (in kDa) of a protein marker is indicated.
We next analyzed CSB phosphorylation in human cells in vivo. Normal human diploid fibroblasts (GM38B) were radioactively labeled with inorganic phosphate, proteins were isolated and CSB was precipitated by the use of a CSB-specific antibody. The lower panels of Figure 9B show the silver staining of precipitated proteins analyzed by SDS–PAGE, and indicate that similar amounts of CSB were precipitated in untreated cells and cells irradiated with 6 J/m2. CSB is clearly phosphorylated in unirradiated cells (top panels, Fig. 9B) and, interestingly, UV radiation abolishes CSB phosphorylation after 4 h, while phosphorylation of CSB is partially recovered 24 h after UV (Fig. 9, results were confirmed in three independent biological experiments).
CSB ATPase activity is regulated by phosphorylation
Having concluded that CSB is phosphorylated in vivo, we next investigated whether phosphorylation might affect the catalytic activity of CSB. ATPase activity was analyzed after dephosphorylating the recombinant CSB protein. As shown in Figure 10, dephosphorylation with 100 mU of PP1 significantly (P < 0.01) increased the ATPase activity of the purified protein by 38%. PP1 had no ATPase activity and heat-inactivated (HI) PP1 did not result in a similar increase, confirming that increased CSB ATPase activity was a result of dephosphorylation. We were also curious to see if the ATPase activity of the mutant proteins was altered by dephosphorylation and found that it increased by ∼40% as a result of dephosphorylation (data not shown). However, compared to CSB wt protein, the activity remained very low for the motif V and VI mutants and almost undetectable for the motif II mutant after dephosphorylation (data not shown).
Figure 10.
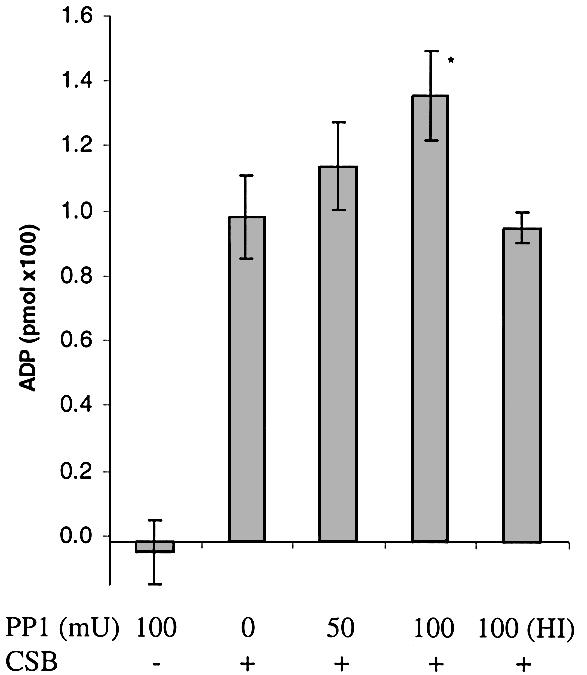
CSB ATPase activity is increased after dephosphorylation. Recombinant CSB wt protein (100 ng) was incubated with 50, 100 mU of PP1 or 100 mU of HI PP1. After dephosphorylation, 1/10 of the reaction (10 ng of CSB) was transferred to ATPase reactions and incubated for a further 1 h. Error bars represent standard deviations of at least three independent experiments and values were normalized against untreated CSB. *P < 0.01, statistically significantly different from untreated CSB.
DISCUSSION
In this study, purified CSB wt, motif II, motif V and motif VI recombinant mutant proteins were characterized. In motif II the conserved glutamate was substituted with glutamine (CSBE646Q), in motif V the two conserved threonines were substituted with valines (CSBT912/913V) and, finally, the conserved glutamine of motif VI was substituted with glutamate (CSBQ942E). Citterio et al. (8) reported that dsDNA is the preferred DNA effector for the CSB wt protein and our results are consistent with this. We found a kcat of ∼31 min–1 with dsDNA and ssDNA effector only supported CSB ATP hydrolysis to a level of ∼5 min–1.
The ATPase activity of the motif II E646Q mutant studied here was undetectable. This is comparable to a K538R motif I mutant reported to be completely devoid of ATPase activity (8). These results are consistent with a role of the lysine of motif I and the glutamate of motif II in ATP binding and hydrolysis deduced from the structural analysis of the SF1 and SF2 proteins crystallized so far (10). Also, a mutation in the eIF4A RNA helicase, corresponding to the E646Q mutation in CSB, abolished the ATPase activity of eIF4A completely (38). Despite the similar reduction in ATPase activity, RNA synthesis recovery of CS-B cells was partially complemented by the motif I mutant but not the motif II mutant (8,13). The difference may reside in the types of assays used for quantifying RNA synthesis recovery [grain count of CSB microinjected cells versus 3H-uridine incorporation in CSB-transfected cells (8,13)]. However, it may also suggest that while the ATPase activity of the two mutant proteins was reduced to a similar extent, the two motifs do not display totally overlapping functions in vivo.
In contrast to motif I and II mutants, the CSBT912/913V and CSBQ942E mutants showed residual ATPase activity with ∼4-fold reduction of kcat in the presence of dsDNA as the cofactor, and the kinetic data indicate impairment of both ATP hydrolysis and binding (Fig. 3 and Table 2). In support of this, a filter-binding analysis of the non-hydrolyzable ATP analog ATPγ35S also showed reduced ATP binding by the motif V and VI mutant proteins (Fig. 5). Furthermore, DNA binding of the mutant proteins was at the wt level (Fig. 6). These results are comparable to results obtained for mutational analysis of two different virus RNA helicases. A motif V mutant plum pox potyvirus RNA helicase (T305S resembling the CSBT912/913V mutant studied here) displayed significantly reduced ATPase activity and abrogated RNA helicase activity, while RNA binding remained intact (39). Similarly, a motif VI (Q1486H) mutation of the NS3 helicase was shown to abrogate ATPase and helicase activity completely, while single-stranded RNA binding was increased (40). The CSBQ942E and CSBT912/913V mutants are very comparable to vector alone in genetic complementation of survival, RNA synthesis recovery and apoptosis after UV treatment and also sensitivity toward 4-nitroquinoline-1-oxide (14). Thus, the data presented here suggest that residual ATP hydrolysis does not result in even partial complementation, and that efficient ATP hydrolysis is an essential requirement for CSB to function in vivo in TCR and RNA synthesis recovery. The differential requirement of motif II (15) and motifs V and VI (21) in 8-oxoG repair can thus not be explained by their involvement in either ATP hydrolysis or DNA binding, but must reside in other functions such as the ability to change DNA topology or interactions with other proteins. In addition, we found similar nuclear localization of both CSB wt and mutant proteins. Therefore, we can conclude that the complementation defects of the mutant proteins are not caused by a deficiency in localization.
To investigate the regulation of CSB protein activity, we studied the effect of different cofactor DNA substrates on CSB wt ATPase activity. We find that substrates containing a loop or a small bubble stimulate CSB ATPase activity significantly, while no increase is observed with substrates where the double-strand to single-strand transition is sterically less strained. This suggests that the CSB ATPase activity is stimulated by a sharp bend in the DNA backbone. Although these data are in contrast to previous findings of stimulation by a splayed arm cofactor (8), the results presented here were obtained under conditions of <20% ATP hydrolysis, thereby allowing an actual kinetic analysis. RNAPII stalling leads to transcription bubble retraction (37), and it could be speculated that CSB encounters this DNA substrate when present in a stalled RNAPII complex and that the CSB ATPase activity is subsequently stimulated.
The presence of DNA damage per se did not stimulate the CSB ATPase activity above that of undamaged DNA, however, cisplatin-treated DNA consistently resulted in increased ATPase activity although not statistically significant. This may be explained by the damage spectra offered by these different agents. The main cisplatin lesion, d(GpG) 1,2-intrastrand cross-links, induces a very prominent bend of the DNA helix (41). In contrast, the UV induced cis–syn cyclobutane pyrimidine dimers and (6–4) photoproducts (42), 8-oxoG, 4,6-diamino-5-formamidopyrimidine and 2,6-diamino-4-hydroxy-5-formaidopyrimidine lesions induced by photoactivated MB (43), and methylated bases induced by MMS (33), result in less bending of the DNA helix. Thus, the increase in ATPase activity may be related to the degree of helix distortion introduced by the cisplatin lesions, and we are currently investigating this possibility in further detail.
The response to DNA damage involves the regulation of protein activity by phosphorylation of a large number of downstream targets. In addition, phosphorylation of the SNF2-like family proteins hBRM and BRG-1 leads to exclusion of the proteins from condensed chromatin during mitosis, whereas phosphorylated ATRX, which is also a SNF2-like family member, associates with condensed chromatin (26,27). Thus, we decided to analyze the potential phosphorylation of the CSB protein. Post-translational modification has not previously to our knowledge been investigated for the CSB protein and could play a major role in its function and regulation. Interestingly, CKII seems to play a role in the DNA damage checkpoint. CKII cDNA overexpression partially complements the UV sensitivity of xeroderma pigmentosum group D and C cells, and mutation of a subunit of the yeast homolog of CKII results in deficient adaptation to double-strand breaks and consequently irreversible arrest (44,45). We find that both purified CKII and human cell extracts can phosphorylate recombinant CSB protein in vitro, and that GTP inhibits this phosphorylation. Furthermore, purified recombinant CSB protein is heavily phosphorylated. Importantly, the CSB protein is found to be phosphorylated in vivo in human cells. Four hours after UV irradiation, CSB phosphorylation is absent, while CSB phosphorylation approaches initial levels 24 h after UV. This indicates that dephosphorylation of the protein occurs during the DNA repair process (NER).
Control of phosphorylation status seems to be essential to achieve competent repair. Inhibitors of protein phosphatases cause a dramatic reduction of repair activity in cell extracts (46), i.e. excessive phosphorylation can inhibit repair. In support of this, the cdk-activating kinase subcomplex of TFIIH inhibits repair in the presence of an ATP-generating system (25). Furthermore, chromatin remodeling, which CSB is known to be involved in (9), can also be regulated by phosphorylation. Sif et al. (47) reported that a mitotically inactivated SWI/SNF complex containing phosphorylated BRG-1 is deficient in chromatin remodeling, and regains nucleosome remodeling activity upon dephosphorylation in vitro. Accordingly, we observed that CSB dephosphorylation in vitro resulted in an increase in ATPase activity. Our combined observations therefore suggest that CSB activity in vivo may also be controlled by phosphorylation, and that CSB activity is activated during the NER process via decreased phosphorylation. We are currently investigating how phosphorylation affects the chromatin remodeling function of CSB, and the interaction of CSB with different NER proteins. Extracts from UV-irradiated CS cells were shown to lack hypophosphorylated, initiation competent RNAPII (48). This may indicate that both CSB and stalled RNAPII are dephosphorylated upon UV irradiation in wt cells, and that CSB is important for the release of hypophosphorylated RNAPII for transcription initiation.
In summary, the analyzed residues of motifs II, V and VI are essential for CSB ATPase activity as well as biological activity and residual ATPase activity is insufficient to complement the UV-sensitive phenotypes of CS cells. Results presented here indicate that CSB senses DNA secondary structure and thereby is activated. As a model of CSB function in elongation, it can be speculated that when RNAPII is stalled reversibly at intrinsic signals, due to RNA secondary structure or low nucleotide pools, CSB may help push the polymerase past the impediment as a result of DNA structure-dependent activation of CSB. CSB may stimulate elongation both by changing the conformation of nascent DNA and by acting on chromatin structure in the proximity of the polymerase (9). However, when the polymerase cannot be stimulated to elongate, due to the nature of the blockage, TFIIH is recruited, the presence of damage is verified and NER (or possibly BER) proceeds. We find that CSB is dephosphorylated in response to UV and that dephosphorylation increases CSB ATPase activity. This suggests that phosphorylation status is involved in controlling the level of CSB activity in vivo. Whether CSB phosphorylation is controlled in a cell-cycle-dependent manner, after oxidative damage, and perhaps by RNAPII stalling are important questions that await further studies.
Acknowledgments
ACKNOWLEDGEMENTS
Ulla Henriksen and Inger Bjørndal are acknowledged for excellent technical assistance. Morten Sunesen is thanked for his help on CSB localization studies. Jean-Philippe Laine and Jeff Stuart are thanked for their critical reading of the manuscript. The project was supported by Danish Research Council, and The European Community (QLK6-CT-1999-02002).
REFERENCES
- 1.Venema J., Mullenders,L.H., Natarajan,A.T., van Zeeland,A.A. and Mayne,L.V. (1990) The genetic defect in Cockayne syndrome is associated with a defect in repair of UV-induced DNA damage in transcriptionally active DNA. Proc. Natl Acad. Sci. USA, 87, 4707–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nance M.A. (2000) GeneReviews at GeneTests-GeneClinics: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle, 1997–2001. Available at http://www.geneclinics.org or http://www.genetests.org, Vol. 2002.
- 3.Troelstra C., van Gool,A., de Wit,J., Vermeulen,W., Bootsma,D. and Hoeijmakers,J.H. (1992) ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne’s syndrome and preferential repair of active genes. Cell, 71, 939–953. [DOI] [PubMed] [Google Scholar]
- 4.Eisen J.A., Sweder,K.S. and Hanawalt,P.C. (1995) Evolution of the SNF2 family of proteins: subfamilies with distinct sequences and functions. Nucleic Acids Res., 23, 2715–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pazin M.J. and Kadonaga,J.T. (1997) SWI2/SNF2 and related proteins: ATP-driven motors that disrupt protein–DNA interactions? Cell, 88, 737–740. [DOI] [PubMed] [Google Scholar]
- 6.Selby C.P. and Sancar,A. (1997) Human transcription-repair coupling factor CSB/ERCC6 is a DNA-stimulated ATPase but is not a helicase and does not disrupt the ternary transcription complex of stalled RNA polymerase II. J. Biol. Chem., 272, 1885–1890. [DOI] [PubMed] [Google Scholar]
- 7.Tantin D., Kansal,A. and Carey,M. (1997) Recruitment of the putative transcription-repair coupling factor CSB/ERCC6 to RNA polymerase II elongation complexes. Mol. Cell. Biol., 17, 6803–6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Citterio E., Rademakers,S., van der Horst,G.T., van Gool,A.J., Hoeijmakers,J.H. and Vermeulen,W. (1998) Biochemical and biological characterization of wild-type and ATPase-deficient Cockayne syndrome B repair protein. J. Biol. Chem., 273, 11844–11851. [DOI] [PubMed] [Google Scholar]
- 9.Citterio E., Van Den Boom,V., Schnitzler,G., Kanaar,R., Bonte,E., Kingston,R.E., Hoeijmakers,J.H. and Vermeulen,W. (2000) ATP-dependent chromatin remodeling by the Cockayne syndrome B DNA repair-transcription-coupling factor. Mol. Cell. Biol., 20, 7643–7653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hall M.C. and Matson,S.W. (1999) Helicase motifs: the engine that powers DNA unwinding. Mol. Microbiol., 34, 867–877. [DOI] [PubMed] [Google Scholar]
- 11.Velankar S.S., Soultanas,P., Dillingham,M.S., Subramanya,H.S. and Wigley,D.B. (1999) Crystal structures of complexes of PcrA DNA helicase with a DNA substrate indicate an inchworm mechanism. Cell, 97, 75–84. [DOI] [PubMed] [Google Scholar]
- 12.Kim J.L., Morgenstern,K.A., Griffith,J.P., Dwyer,M.D., Thomson,J.A., Murcko,M.A., Lin,C. and Caron,P.R. (1998) Hepatitis C virus NS3 RNA helicase domain with a bound oligonucleotide: the crystal structure provides insights into the mode of unwinding. Structure, 6, 89–100. [DOI] [PubMed] [Google Scholar]
- 13.Selzer R.R., Nyaga,S., Tuo,J., May,A., Muftuoglu,M., Christiansen,M., Citterio,E., Brosh,R.M.,Jr and Bohr,V.A. (2002) Differential requirement for the ATPase domain of the Cockayne syndrome group B gene in the processing of UV-induced DNA damage and 8-oxoguanine lesions in human cells. Nucleic Acids Res., 30, 782–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muftuoglu M., Selzer,R., Tuo,J., Brosh,R.M.,Jr and Bohr,V.A. (2002) Phenotypic consequences of mutations in the conserved motifs of the putative helicase domain of the human Cockayne syndrome group B gene. Gene, 283, 27–40. [DOI] [PubMed] [Google Scholar]
- 15.Brosh R.M. Jr, Balajee,A.S., Selzer,R.R., Sunesen,M., Proietti De Santis,L. and Bohr,V.A. (1999) The ATPase domain but not the acidic region of Cockayne syndrome group B gene product is essential for DNA repair. Mol. Biol. Cell, 10, 3583–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sunesen M., Selzer,R.R., Brosh,R.M.,Jr, Balajee,A.S., Stevnsner,T. and Bohr,V.A. (2000) Molecular characterization of an acidic region deletion mutant of Cockayne syndrome group B protein. Nucleic Acids Res., 28, 3151–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dianov G., Bischoff,C., Sunesen,M. and Bohr,V.A. (1999) Repair of 8-oxoguanine in DNA is deficient in Cockayne syndrome group B cells. Nucleic Acids Res., 27, 1365–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stevnsner T., Nyaga,S., de Souza-Pinto,N.C., van der Horst,G.T.J., Gorgels,T.G.M.F., Hogue,B.A., Thorslund,T. and Bohr,V.A. (2002) Mitochondrial repair of 8-oxoguanine is deficient in Cockayne syndrome group B. Oncogene, 21, 8675–8682. [DOI] [PubMed] [Google Scholar]
- 19.Tuo J., Jaruga,P., Rodriguez,H., Dizdaroglu,M. and Bohr,V.A. (2002) The cockayne syndrome group B gene product is involved in cellular repair of 8-hydroxyadenine in DNA. J. Biol. Chem., 277, 30832–30837. [DOI] [PubMed] [Google Scholar]
- 20.Tuo J., Muftuoglu,M., Chen,C., Jaruga,P., Selzer,R.R., Brosh,R.M.,Jr, Rodriguez,H., Dizdaroglu,M. and Bohr,V.A. (2001) The Cockayne syndrome group B gene product is involved in general genome base excision repair of 8-hydroxyguanine in DNA. J. Biol. Chem., 276, 45772–45779. [DOI] [PubMed] [Google Scholar]
- 21.Tuo J., Chen,C., Zeng,X., Christiansen,M. and Bohr,V.A. (2002) Functional crosstalk between hOGG1 and the helicase domain of Cockayne syndrome group B protein. DNA Repair, 1, 913–927. [DOI] [PubMed] [Google Scholar]
- 22.Selby C.P. and Sancar,A. (1997) Cockayne syndrome group B protein enhances elongation by RNA polymerase II. Proc. Natl Acad. Sci. USA, 94, 11205–11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Svejstrup J.Q., Vichi,P. and Egly,J.M. (1996) The multiple roles of transcription/repair factor TFIIH. Trends Biochem. Sci., 21, 346–350. [PubMed] [Google Scholar]
- 24.Tijsterman M., Tasseron-de Jong,J.G., Verhage,R.A. and Brouwer,J. (1998) Defective Kin28, a subunit of yeast TFIIH, impairs transcription-coupled but not global genome nucleotide excision repair. Mutat. Res., 409, 181–188. [DOI] [PubMed] [Google Scholar]
- 25.Araujo S.J., Tirode,F., Coin,F., Pospiech,H., Syvaoja,J.E., Stucki,M., Hubscher,U., Egly,J.M. and Wood,R.D. (2000) Nucleotide excision repair of DNA with recombinant human proteins: definition of the minimal set of factors, active forms of TFIIH and modulation by CAK. Genes Dev., 14, 349–359. [PMC free article] [PubMed] [Google Scholar]
- 26.Muchardt C., Reyes,J.C., Bourachot,B., Leguoy,E. and Yaniv,M. (1996) The hbrm and BRG-1 proteins, components of the human SNF/SWI complex, are phosphorylated and excluded from the condensed chromosomes during mitosis. EMBO J., 15, 3394–3402. [PMC free article] [PubMed] [Google Scholar]
- 27.Berube N.G., Smeenk,C.A. and Picketts,D.J. (2000) Cell cycle-dependent phosphorylation of the ATRX protein correlates with changes in nuclear matrix and chromatin association. Hum. Mol. Genet., 9, 539–547. [DOI] [PubMed] [Google Scholar]
- 28.Bartek J. and Lukas,J. (2001) Pathways governing G1/S transition and their response to DNA damage. FEBS Lett., 490, 117–122. [DOI] [PubMed] [Google Scholar]
- 29.Yu A., Fan,H.Y., Liao,D., Bailey,A.D. and Weiner,A.M. (2000) Activation of p53 or loss of the Cockayne syndrome group B repair protein causes metaphase fragility of human U1, U2 and 5S genes. Mol. Cell, 5, 801–810. [DOI] [PubMed] [Google Scholar]
- 30.Athas W.F., Hedayati,M.A., Matanoski,G.M., Farmer,E.R. and Grossman,L. (1991) Development and field-test validation of an assay for DNA repair in circulating human lymphocytes. Cancer Res., 51, 5786–5793. [PubMed] [Google Scholar]
- 31.Anson R.M., Croteau,D.L., Stierum,R.H., Filburn,C., Parsell,R. and Bohr,V.A. (1998) Homogenous repair of singlet oxygen-induced DNA damage in differentially transcribed regions and strands of human mitochondrial DNA. Nucleic Acids Res., 26, 662–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Batty D., Rapic’-Otrin,V., Levine,A.S. and Wood,R.D. (2000) Stable binding of human XPC complex to irradiated DNA confers strong discrimination for damaged sites. J. Mol. Biol., 300, 275–290. [DOI] [PubMed] [Google Scholar]
- 33.Encell L., Shuker,D.E., Foiles,P.G. and Gold,B. (1996) The in vitro methylation of DNA by a minor groove binding methyl sulfonate ester. Chem. Res. Toxicol., 9, 563–567. [DOI] [PubMed] [Google Scholar]
- 34.Brosh R.M. Jr and Matson,S.W. (1996) A partially functional DNA helicase II mutant defective in forming stable binary complexes with ATP and DNA. A role for helicase motif III. J. Biol. Chem., 271, 25360–25368. [DOI] [PubMed] [Google Scholar]
- 35.Mielke C., Tummler,M., Schubeler,D., von Hoegen,I. and Hauser,H. (2000) Stabilized, long-term expression of heterodimeric proteins from tricistronic mRNA. Gene, 254, 1–8. [DOI] [PubMed] [Google Scholar]
- 36.Wakasugi M. and Sancar,A. (1998) Assembly, subunit composition and footprint of human DNA repair excision nuclease. Proc. Natl Acad. Sci. USA, 95, 6669–6674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fiedler U. and Timmers,H.T. (2001) Analysis of the open region of RNA polymerase II transcription complexes in the early phase of elongation. Nucleic Acids Res., 29, 2706–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pause A., Methot,N., Svitkin,Y., Merrick,W.C. and Sonenberg,N. (1994) Dominant negative mutants of mammalian translation initiation factor eIF-4A define a critical role for eIF-4F in cap-dependent and cap-independent initiation of translation. EMBO J., 13, 1205–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fernandez A., Guo,H.S., Saenz,P., Simon-Buela,L., Gomez de Cedron,M. and Garcia,J.A. (1997) The motif V of plum pox potyvirus CI RNA helicase is involved in NTP hydrolysis and is essential for virus RNA replication. Nucleic Acids Res., 25, 4474–4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim D.W., Kim,J., Gwack,Y., Han,J.H. and Choe,J. (1997) Mutational analysis of the hepatitis C virus RNA helicase. J. Virol., 71, 9400–9409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gelasco A. and Lippard,S.J. (1998) NMR solution structure of a DNA dodecamer duplex containing a cis-diammineplatinum(II) d(GpG) intrastrand cross-link, the major adduct of the anticancer drug cisplatin. Biochemistry, 37, 9230–9239. [DOI] [PubMed] [Google Scholar]
- 42.Kim J.K., Patel,D. and Choi,B.S. (1995) Contrasting structural impacts induced by cis–syn cyclobutane dimer and (6–4) adduct in DNA duplex decamers: implication in mutagenesis and repair activity. Photochem. Photobiol., 62, 44–50. [DOI] [PubMed] [Google Scholar]
- 43.Boiteux S., Gajewski,E., Laval,J. and Dizdaroglu,M. (1992) Substrate specificity of the Escherichia coli Fpg protein (formamidopyrimidine-DNA glycosylase): excision of purine lesions in DNA produced by ionizing radiation or photosensitization. Biochemistry, 31, 106–110. [DOI] [PubMed] [Google Scholar]
- 44.Teitz T., Eli,D., Penner,M., Bakhanashvili,M., Naiman,T., Timme,T.L., Wood,C.M., Moses,R.E. and Canaani,D. (1990) Expression of the cDNA for the beta subunit of human casein kinase II confers partial UV resistance on xeroderma pigmentosum cells. Mutat. Res., 236, 85–97. [DOI] [PubMed] [Google Scholar]
- 45.Toczyski D.P., Galgoczy,D.J. and Hartwell,L.H. (1997) CDC5 and CKII control adaptation to the yeast DNA damage checkpoint. Cell, 90, 1097–1106. [DOI] [PubMed] [Google Scholar]
- 46.Ariza R.R., Keyse,S.M., Moggs,J.G. and Wood,R.D. (1996) Reversible protein phosphorylation modulates nucleotide excision repair of damaged DNA by human cell extracts. Nucleic Acids Res., 24, 433–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sif S., Stukenberg,P.T., Kirschner,M.W. and Kingston,R.E. (1998) Mitotic inactivation of a human SWI/SNF chromatin remodeling complex. Genes Dev., 12, 2842–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rockx D.A., Mason,R., van Hoffen,A., Barton,M.C., Citterio,E., Bregman,D.B., van Zeeland,A.A., Vrieling,H. and Mullenders,L.H. (2000) UV-induced inhibition of transcription involves repression of transcription initiation and phosphorylation of RNA polymerase II. Proc. Natl Acad. Sci. USA, 97, 10503–10508. [DOI] [PMC free article] [PubMed] [Google Scholar]