

Abstract
The deliberate inhibition of expression of one of the protein subunits (Rpp38) of human nuclear RNase P is achievable by using external guide sequence (EGS) technology. Both the protein product and the mRNA are greatly reduced 24 h after transient transfection with a gene coding for an appropriate EGS. Control experiments indicated that four other protein subunits of RNase P and their RNAs are also inhibited with no external manipulation. The remaining RNase P proteins, their mRNAs, and the RNA subunit of RNase P all are unchanged. Several short nucleotide sequences adjacent to the ORFs for the inhibited genes are similar and could be targets for transcriptional repression. The explanation of coordinate inhibition of the expression of the product of one particular gene by the transfection of an EGS (or RNA interference) requires some care in terms of interpreting phenotypic effects because, in our case, several gene products that are not targeted are also inhibited.
Keywords: external guide sequences‖upstream sequences‖downstream sequences‖Rpp38
The regulation of expression of single genes in human cells by external manipulation is an important problem. RNA interference, the use of antisense RNA, and similar methods (1, 2) have been used with moderate success on cells in tissue culture. One other method, which uses RNase P as the agent that cleaves targeted RNAs, is also quite promising (3, 4). In the past, this mechanism has been used, for example, to decrease the amount of viral infection by regulating certain essential viral genes (5). This method, which uses external guide sequences (EGSs), has now been used to regulate expression of a gene that encodes one of the protein subunits of RNase P.
Human nuclear RNase P has at least 10 protein subunits and one RNA subunit and is localized in the nuclei of cells (6–8). While using EGS action to study the expression of one of the subunits, Rpp38, we did not know whether any of the other proteins would also be affected. In fact, the mechanism for the control of several genes simultaneously in human cells is a diverse and puzzling problem. After surveying cells after transient transfection with a plasmid that harbored EGSRpp38 by Western and Northern blots, we found an expected decrease in both the mRNA and translation product of Rpp38. As controls, we also explored the amounts of mRNA and protein made by other subunits of RNase P. Four other genes were inhibited, whereas the rest were unchanged. This finding led to an examination of what factors in terms of upstream and downstream DNA and RNA sequences were involved with this phenomenon and whether there could be a common regulatory feature.
Materials and Methods
Construction of EGSs and Plasmids.
A DNA fragment that contains the Rpp38 cDNA under control of a T7 promoter was obtained by PCR using the full-length Rpp38 cDNA clone F3053 in PCR-Script-Amp (kindly provided by C. C. Liew, University of Toronto, Toronto) as a template. Primer oligonucleotides were P38F5 5′-TAA TAC GAC TCA CTA TAG GGC TCA AGT TATC-3′ and P38BHIR 5′-GGATCCTTACTTTGGAGTAGC-3′. The PCR fragment was subcloned into pUC19 digested with SmaI. A 3/4 EGSRpp38 (9) was obtained by PCR using a plasmid encoding the gene for ptRNATyr from Escherichia coli as a template with primer oligonucleotides P38EGS2A 5′-CTG CAG TTC AGC AGA CTC TAA ATC-3′ and P38EGS2B 5′-GGA TCC GGT ACC TTT AAA AAT GGT GAC GTC GGA AGG ATT CGA ACC-3′ (9). A control EGS, 3/4 EGS-TLRpp38 was obtained by PCR using pmU6-EF1-TL (5) as a template with primer oligonucleotides P38EGS2A and P38TLMU6R 5′-GGA TCC GGT ACC TTT AAA AAT GGT GTA AGA CGA AGG CGG ATC CCCT-3′. Both the EGSRpp38 and EGS-TLRpp38 PCR fragments were digested with PstI and BamHI and subcloned into pUC19 under T7 promoters for transcription in vitro. The same PCR fragments were digested with PstI and KpnI and subcloned into pmU6 (kindly provided by R. Reddy, Baylor College of Medicine, Waco, TX) for experiments in vivo.
A second set of plasmids was constructed with the drug resistance marker, neoR, incorporated in the plasmids. For this purpose, a neoR DNA fragment was obtained by digestion of pLXSN (10) with EcoRI and AccI, and the fragment was subcloned into pmU6, pmU6-EGS-TLRpp38, and pmU6EGSRpp38. Thus, a method of guaranteeing that all cells that we examined were transfected was available.
Other aspects of the EGS-accessible sites in mRNA were elucidated by using methods as described (9).
Transcription in Vitro.
Rpp38 mRNA under the control of the T7 promoter was transcribed from pUC19 harboring these genes after digestion of the plasmid with HpaI or NcoI. EGSRpp38 and EGS-TLRpp38 RNA was prepared from the appropriate plasmid DNA (see above) that had been linearized by treatment with DraI. pSupS1, the precursor to the suppressor tRNASer from Schizosaccharomyces pombe, was prepared as described (11). The transcription reactions were carried out as described (12), and transcripts were purified by using Sephadex G-50 Quick Spin columns (Roche Applied Science).
Assay for Cleavage by RNase P.
Assays were performed in 10 λ reactions in 50 mM Tris⋅HCl, pH 7.5, 10 mM MgCl2, 100 mM NH4Cl with 2,000–4,000 cpm (0.2 pmol) of radiolabeled Rpp38 mRNA fragments and EGS RNA (0.4–0.8 pmol) in the presence of human RNase P (2 μl) as described (12). Samples were electrophoresed in 5% polyacrylamide/7 M urea gels. Human RNase P was partially purified through the DEAE Sepharose step from HeLa cells according to established protocols (13).
Cell Cultures and Transfection.
HeLa cells were maintained in DMEM supplemented with 10% FBS. Cells at 50–60% confluence on 150-mm diameter petri plates were transfected with 20 μg of the appropriate plasmid DNA by using the SuperFect reagent (Qiagen, Chatsworth, CA) according to the manufacturer's protocol.
Northern, Southern, and Western Analysis.
HeLa cell S16 extracts were prepared from monolayer cells that were grown in 150-mm-diameter petri plates. For Northern analysis, 10 μg of total RNA, prepared from the extracts by the SDS/phenol method, was loaded onto an 2% agarose gel. Northern hybridizations were performed as described (14). Oligonucleotides complementary to Rpp38 mRNA (5′-AAG TGC ATA TTC TCG CTC TCC-3′), Rpp25 mRNA (5′-GGA CTA CCG GAA GCG GTG GCGG-3′), Rpp40 (5′-TGA CAA CGG GAC GAT GGT TTA CCC-3′), EGSRpp38 (5′-GAC GTC GGA AGG ATT CGA AGCT-3′), human nuclear mature tRNALeu and 5S rRNA (15), human RNase P RNA (positions 319–340), and human RNase MRP RNA (positions 245–265; 16) were used. All oligonucleotides were end-labeled with T4 polynucleotide kinase (New England Biolabs) and [γ-32P]ATP (Amersham Pharmacia).
For Southern analysis total RNA obtained from HeLa cell S16 extracts was purified by using an RNeasy Mini Kit (Qiagen). cDNA was reverse-transcribed from this total RNA (10 μg) with random hexamer primers (Roche Applied Science) by using Superscript II reverse transcriptase (GIBCO/BRL) and was cleaned with QIAquick columns (Qiagen) before labeling with the Megaprime DNA labeling system (Amersham Pharmacia). Labeling reaction was carried out with 0.5 μg of cDNA in the presence of random nonamer primers, [α-32P]dCTP of specific activity 6,000 Ci/mmol, and Klenow DNA polymerase. Labeled cDNA was heat-denatured and hybridized to the membrane containing cDNA gene fragments of RNase P subunits, which were obtained by PCR from the corresponding plasmids. β-Actin cDNA was kindly provided by the laboratory of Mark Mooseker (Yale University). Quantitation of the spots on the hybridized membrane were performed by counting the hybridized spots with a Fuji PhosphorImager and adjusting the signals by using adjacent blank spots as background.
For Western analysis, HeLa cells were detached from petri dishes with 2× trypsin-EDTA, washed in 1× PBS, pelleted, and boiled for 5 min in gel loading buffer. These samples were separated in a 12.5% polyacrylamide/SDS gel, transferred to a nitrocellulose membrane (Schleicher & Schuell), and incubated with antibodies against RNase P subunits as described (6).
Indirect Immunofluorescence, Microscopy, and Imaging.
HeLa cells were grown overnight on coverslips (22 × 22 mm) to ≈20% confluent, transiently transfected with 2 μg of plasmid, and after another 24 h of growth fixed with 2% paraformaldehyde, and then treated according to the protocol (17). Rabbit polyclonal antibodies against Rpp38, Rpp25 (1:400), and Rpp21 (1:600) were added for 1 h. Alexa 568 goat anti-rabbit IgG conjugate (1:2,000) (Molecular Probes) was also added for 30 min. Confocal fluorescence microscopy of fixed cells was performed by using a Bio-Rad MRC-1024 laser scanner mounted on a 2FL reflector slider on a Zeiss Axiovert equipped with differential interference contrast optics (PlanApo ×100, 1.4 numerical aperture oil immersion objective; Zeiss). Fluorescent images were acquired with a Texas red filter and processed with lasersharp software (Bio-Rad). Digital processing and color adjustment of images were done by using programs from Lemke Software (Peine, Germany) and photoshop (Adobe Systems, Mountain View, CA).
Sequence Comparisons.
All of the sequences upstream and downstream of the genes encoding human RNase P proteins we examined were from GenBank. These sequences included the genomic sequences encoding ORFs, the sequences coding for upstream and downstream UTRs, and DNA sequences further upstream and downstream. Standard computer programs collated these sequences and searched for common sequences of perfect matches of six or more nucleotides. Details of the method are available from the authors.
Results
Design and Testing of EGSs.
An EGS against Rpp38 was designed by using the general methods as reported (9). Partial RNase T1 digestion of Rpp38 mRNA yielded several cleavage sites, and one was chosen for EGS-directed RNase P cleavage. Plasmid pmU6, which contained an RNA polymerase III promoter, was cloned harboring a gene for the EGS. This plasmid and another with no EGS and EGS-TL were used to transiently transfect HeLa cells. Transfection was complete (see below) because the extracts we sampled gave no results that cells were uninfected as judged by the results of Western blots (Fig. 1 C and D) and Northern blots (Fig. 1 A and B; Table 1). We note also that between 60 and 72 h after transfection cells begin to lose plasmids and normal growth and RNase P activity resumes (Fig. 1D). Before that time, the cells with a transfected EGS gene do not grow for about 48 h or grow so slowly that they appear to be nongrowing. Cells transfected without the EGS gene grow at normal rates. RNase P activity, assayed in whole-cell extracts, is ≈10% (per total amount of protein) in the EGS-containing cells with respect to cells with no EGS (data not shown).
Figure 1.

Expression of EGSRpp38 in HeLa cells leads to the reduction of Rpp38 mRNA and Rpp38 protein. HeLa cells were transiently transfected with 10 μg of empty (no EGS) pmU6 or pmU6- EGSRpp38 plasmid. Samples of total RNA and total protein were prepared from these HeLa cells 0, 12, 24, 48, and 60 h after transfection. For Northern blot analysis of Rpp38 mRNA and EGSRpp38, RNA samples (10 μg) of total RNA were hybridized with end-labeled antisense oligonucleotide that corresponded to Rpp38 mRNA (A) and EGSRpp38 RNA (B). Lane H, sample of total RNA, prepared from nontransfected HeLa cells. Lane C, 0.1 pmol of Rpp38 mRNA fragment (A) and 0.1 pmol of EGSRpp38 RNA (B), transcribed in vitro. For Western blot analysis, samples (10 μg) of total protein were immunoblotted with a rabbit polyclonal antibodies against Rpp38 (1:100 dilution) (C and D). The lanes in C and D represent cells harvested after the given number of hours after transfection.
Table 1.
Relative amounts of mRNA and proteins of RNase P subunits after transfection with pmU6 or pmU6EGSRpp38
| Gene product | pmU6/pmU6-EGSRpp38
|
||
|---|---|---|---|
| cDNA | Western blot | Northern blot | |
| Rpp14 | 1.2 | ||
| Rpp20 | 0.2 | 1 | |
| Rpp21 | 0.14 | 0.4 | |
| Rpp25 | 0.09 | 0.4 | 0.4 |
| Rpp29 | 0.17 | 0.6 | |
| Rpp30 | 1 | 1 | |
| Rpp38 | 0.12 | 0.4 | 0.2 |
| Rpp40 | 0.5 | 1 | 1 |
| hpop5 | 0.18 | ||
| hpop1 | 0.25 | ||
| Lamin A | 0.5 | ||
| β-actin | 1.1 | 1 | |
| H1 RNA | 1.1 | 1 | |
| MRP RNA | 1 | ||
| 5S RNA | 1 | ||
| pre-tRNALeu/tRNALeu | 0.8 | ||
cDNA results were quantitated by using data obtained from the controls in Fig. 5 (see Materials and Methods). A result of at least <0.2 was regarded as reliably lower than the control of no change. In each column, the results were normalized to controls. Western blots (Fig. 2) were also quantitated but the results are not as reliable as shown for some Northerns (Figs. 1 and 3). The data shown, however, are quite clear for the bands that show a decrease with respect to unchanged bands. A Northern blot was quantitated for precursor and tRNA (data not shown) to make a rough estimate of their amounts.
If neomycin resistance in the plasmids was used as a means of assuring that all cells we examined were transfected, the results appeared identical to those without the drug resistance marker.
Western Blots of Transfected Cells.
S16 extracts of transfected HeLa cells were displayed on a SDS/polyacrylamide gel (see Materials and Methods) and blotted with polyclonal antibodies (Fig. 1). The data, shown in Figs. 1 C and D and 2, and Table 1, indicate that Rpp38, Rpp29, Rpp25, and Rpp21 were decreased in expression 24 h after transfection whereas other proteins were not. Note in Fig. 1 C and D the kinetics of protein production were measured for 72 h after transient transfection. In the control cells (no EGS), no proteins tested were reduced in expression. Rpp29, Rpp25, and Rpp21 were decreased in intensity after 24 h (compare with EGSRpp38-TL; Fig. 2). Another nuclear protein, lamin A (18), was also inhibited in synthesis. β-Actin, a housekeeping gene, was unchanged.
Figure 2.

The inhibition of Rpp38 expression caused by EGSRpp38 in HeLa cells influences the expression of some other RNase P protein subunits in vivo. Samples of total protein (10 μg each lane) were prepared from the HeLa cells, transiently transfected by 10 μg of pmU6-EGSRpp38-TL plasmid for 24 h (lane 1) and 10 μg of pmU6-EGSRpp38 plasmid for 24 h (lane 2) and 72 h (lane 3). For Western blot analysis, these samples were separated in a 12.5% polyacrylamide/SDS gel, transferred to nitrocellulose filters, and immunoblotted with polyclonal antibodies against the indicated RNase P protein subunits (1:100 dilution), β-actin (1:500), and lamin A (1:500).
Northern Blots of Transfected Cells.
Protein-free extracts of total cells were also fractionated on an agarose gel and probed with radioactive oligonucleotides complementarily to certain RNAs in the cells. We note that the RNA for Rpp38 is decreased after 24 h after transfection (Fig. 1A and Table 1) as is that for the mRNA of Rpp25 (Fig. 3A), but not those coding for Rpp40 (Fig. 3B). We also show that the amounts of H1 RNA, the RNA subunit of RNase P, and RNase MRP RNA (Fig. 3 C and D), as well as that of ribosomal 5S RNA (data not shown), are unaltered. In the control cells, there is no change in mRNA levels.
Figure 3.

Expression of EGSRpp38 decreases the amount of Rpp25 mRNA and does not change the amount of Rpp40 mRNA in vivo. HeLa cells were transiently transfected by pmU6 or pmU6-EGSRpp38 plasmids and harvested 0, 12, 24, 48, and 60 h after transfection. Samples (10 μg) of total RNA isolated from these cells were hybridized with the end-labeled antisense oligonucleotide that corresponded to Rpp25 RNA (A), Rpp40 RNA (B), H1 RNA (C), and MRP RNA (D). Lane H, total RNA, isolated from nontransfected HeLa cells. Lane C, 0.05 pmol of H1 RNA (C) and 0.2 pmol of MRP RNA (D), transcribed in vitro.
The amounts of tRNALys and its precursor RNA were also probed by Northern blots. Although there was a decrease in the amount of mature tRNA and concomitant increase in precursor tRNA, the effect is rather small, ≈25% total reduction, over a 24-h period after transfection (gel data not shown; Table 1). Presumably, if assays had been performed in cells in which transfection was permanent, this effect would have been larger.
We also checked for the presence of Rpp21, Rpp25, and Rpp38 in the nuclei of cells transfected with EGSRpp38. These proteins disappeared from nuclei (Fig. 4) when observed 24 h after transient transfection.
Figure 4.
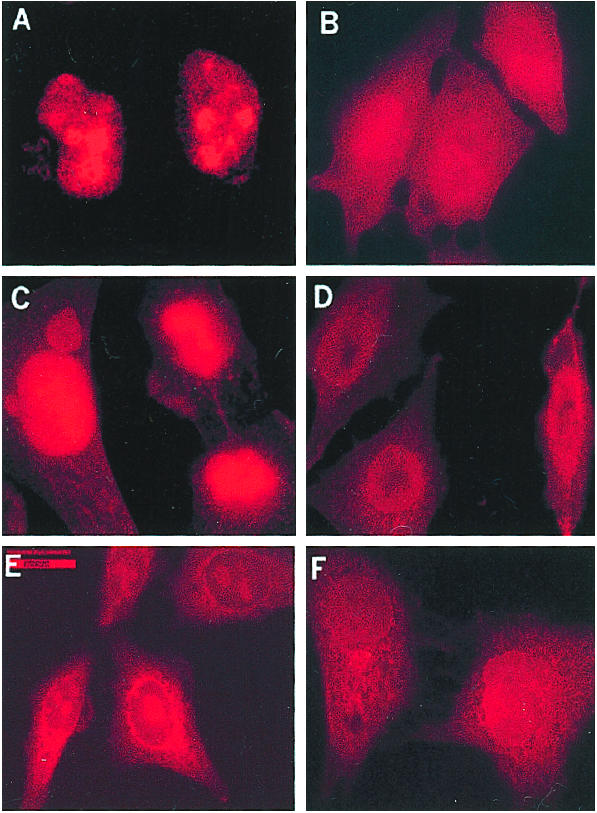
Rpp38, Rpp25, and Rpp21 are absent from the cell nucleolus after expression of EGSRpp38 in vivo. HeLa cells grown in six-well plates transfected with 2 μg of empty pmU6 plasmid (A, C, and E) or pmU6-EGSRpp38 plasmid (B, D, and F) were grown for 24 h and then examined for the presence of Rpp38 (A and B), Rpp25 (C and D), and Rpp21 (E and F) in indirect immunofluorescence analysis. All images were acquired during the same experimental observation. No specific signal was observed when control preimmune sera of rabbits were tested (data not shown). (Scale bar: 20 μm).
Southern Blots of Transfected Cells.
To complete the assays of mRNA levels, RNA was prepared from transiently transfected cells and cDNA was manufactured from this preparation (see Materials and Methods). A filter blot was prepared that contained DNA from all genes for protein subunits, as well as H1 RNA and β-actin mRNA. The filter was then probed with the cDNA and the probed membrane was quantitated (see Materials and Methods). The results are shown in Fig. 5 and Table 1. As indicated earlier, about half of the genes were obviously turned off in the experimental cells whereas there is no change in the control cells.
Figure 5.

Different expression level of RNase P protein subunits after disruption of Rpp38 in HeLa cells. Five hundred nanograms (A), 100 ng (B), 10 ng (C), and 0.1 ng (D) of cDNA corresponding to Rpp14 (lane 1), Rpp20 (lane 2), Rpp21 (lane 3), Rpp25 (lane 4), Rpp29 (lane 5), Rpp30 (lane 6), Rpp38 (lane 7), Rpp40 (lane 8), hPop1 (lane9), hPop5 (lane 10), β-actin (lane 11), and H1 RNA transcribed in vitro (lane 12) were dotted on nitrocellulose filters, cross-linked with Stratagene Stratalinker, and hybridized with labeled cDNA probe. To obtain the labeled cDNA probe, one 150-mm plate of HeLa cells transfected with 20 μg of pmU6-EGSRpp38-TL or pmU6-EGSRpp38 plasmid was then harvested 24 h posttransfection. Total RNA purified from these cells was used in the reverse transcription reaction. cDNA obtained from this reaction was labeled with [α-32P]dCTP.
Is There a Common Means of Gene Regulation?
The regulation of RNase P genes is unclear. In our particular case, the means of control is at the transcriptional level, as the data indicate. We examined upstream and downstream sequences in and adjacent to the RNase P protein coding genes (see Materials and Methods). We compared the upstream and downstream transcribed sequences separately of each mRNA (5′ UTR and 3′ UTR) and the upstream 1,000 nt beyond the 5′ UTR with each set of genes. The comparisons were done with the inhibited genes [hPop5, Rpp20, Rpp21, Rpp25 (Rpps25, a separate gene from Rpp25 found in the chromatographic peak eluates of RNase P activity), Rpp29, Rpp38, and hPop1] and the noninhibited genes (Rpp14, Rpp30, Rpp40, and hPop1) separately (Table 2). (The precise nature of hPop1 is not clear. Some calculations were made with it as an inhibited gene and others with it not.) Clearly, the upstream and downstream sequences of Rpp38 should, in principle, have correlated with the sequences bounding all of the other inhibited genes to determine whether there were several sequences that were common among these genes that were inhibited. In fact, there were many common sequences of 6 nt or larger (some identical and some almost identical; Table 3). Several isolated sequences of larger length that were shared between a few of the inhibited genes were similar to known enhancer/promoter sequences. For example, sequence AP-2 (activator protein 2, caCCCCggggcc; ref. 19), common to the 5′ UTR of some of our inhibited genes, was altered in Rpp38, Rpp29, Rpp25, and Rpp21. Other similar comparisons also showed up but did not have the same correlation with these four genes just mentioned, all of which were inhibited by the addition of EGSRpp38.
Table 2.
Noncoding sequences
| Protein | 5′ UTR + intron, nt | 5′ UTR intron, nt | 3′ UTR + intron, nt | 3′ UTR intron, nt |
|---|---|---|---|---|
| Rpp14 | 4,056 | 3,891 | 1,020 | — |
| hPop5 | 19 | — | 276 | — |
| Rpp20 | 1,684 | 516 | 230 | — |
| Rpp21 | 13 | — | 21 | — |
| Rpp25 | 62 | — | 679 | 125 |
| Rpps25 | 808 | 731 | 312 | — |
| Rpp29 | 25 | — | 423 | — |
| Rpp30 | 468 | 176 | 1,518 | — |
| Rpp38 | 5,930 | 5,895 | 92 | — |
| Rpp40 | 2,084 | 1,849 | 32 | — |
| hPop1 | 6,045 | 5,945 | 1,512 | — |
UTR, untranslated region of mRNA. The products of Rpp14, Rpp30, and Rpp40 (and, possibly, hPop1) are all not inhibited. The rest of the gene products are inhibited in expression.
Table 3.
Common sequences found adjacent to inhibited genes
| Sequence | Genes | Symbol |
|---|---|---|
| None | All | 5′ UTR |
| None | All | 3′ UTR |
| CTTTCT | All | US 5′ UTR |
| GGACCC | All | US 5′ UTR |
| TGTGAGG | All | US 5′ UTR |
| TACCCC | No hPop 5, hPop 1 | US 5′ UTR |
See text for genes. US 5′ UTR, upstream sequences at the 5′ side of an ORF and its 5′ UTR.
A large number of hexamers and some heptamers were also found in common among all of the upstream and downstream noncoding sequences adjacent to the noninhibited genes (Table 4). The precise function of these sequences, and those found separately in the inhibited genes, is not yet known and requires further experiments in vitro. The hexamers and heptamers did not fall in precisely the same position distant from the ORFs or the initiation of transcription. Clearly, these sequences occur too frequently in the nucleotide tracts we examined to be accounted for on a random basis unless many of them are clustered in only a few general locations in the genome.
Table 4.
Common sequences found adjacent to noninhibited genes
| Sequence | Genes | Symbol |
|---|---|---|
| AATCCT | All | 5′ UTR |
| AAGTAAG | All | 5′ UTR |
| TAACTA | All | 5′ UTR |
| GAAATG | All | 5′ UTR |
| AACTGG | All | 5′ UTR |
| GTCCAT | All | 5′ UTR |
| GACTTC | All | 5′ UTR |
| TTAACT | All | 5′ UTR |
| TAAGTT | All | 5′ UTR |
| GTTAATA | All | 5′ UTR |
| AACCAA | All | 5′ UTR |
| GTAAACAA | No hPop1 | 5′ UTR |
| TAAACAA | No hPop1 | 5′ UTR |
| AAATTA | All | 3′ UTR |
| TGATCA | All | US 5′ UTR |
| GTCCAT | All | US 5′ UTR |
| GAGAATT | All | US 5′ UTR |
| ATGGGT | All | US 5′ UTR |
| CTCAATA | All | US 5′ UTR |
| TGTTTGT | All | US 5′ UTR |
| AAAACAT | All | US 5′ UTR |
| GATCAG | no hPop1 | US 5′ UTR |
See text for genes. US 5′ UTR, upstream sequence at the 5′ side of an ORF and its 5′ UTR.
Discussion
The regulation of the protein subunits of mammalian RNase P is unknown. The RNA component, H1 RNA, is under polymerase III control whereas the 10 (or more) protein subunits are under polymerase II control. At this date, we have little idea whether mRNA level, promoter, or enhancer factors are involved in regulation. Our data indicate that some measure of mRNA control could be critical because when one mRNA is reduced, several others are reduced in level. Of those assayed, four of the protein subunits are down-regulated in response to the deliberate inhibition of expression and manipulation of a fifth subunit. Other subunits are not inhibited. No general physiological phenomenon is apparent. However, another nuclear gene, that for lamin A, was also repressed compared to the normal amount. The coordinate inhibition of these genes is a novel phenomenon and requires further investigation.
The use of an EGS to change the phenotype of certain cellular characteristics acts quickly and specifically with nonessential genes. We show here that the same is true for the mRNA of an essential gene, Rpp38, but the amount of protein in the cell for other “inhibited” genes is variable, even though the majority of RNase P function is lost rapidly. The data, showing differences in reduction of mRNAs and protein levels, regardless of the precise mode of regulation, may partly reflect natural half-lives of each species. The RNAs and proteins may have variable susceptibility to degradative enzyme action and/or to components of the regulatory process. One possible explanation for these data is the differences in the degradative half-lives of the various proteins. Rpp14, Rpp40, and hpop5 are localized throughout the total cell body when they are fused with GFP and the cells are examined for fluorescence after transfection with the appropriate clones. Some of the other proteins, like Rpp20 and Rpp30, show ≈50% localization in the nucleus or nucleoli and the rest of the proteins show complete nuclear and nucleolar localization (D.W. and J. Wolenski, unpublished work). Although the amounts of the mRNAs vary somewhat in our experiments, it is not clear whether they all are involved in the same mechanism or in totally different inhibited and noninhibited pathways.
The various sequences, either in DNA or UTR RNA, that were common to the inhibited genes and the noninhibited genes, could be recognition (or antirecognition) signals for regulatory proteins that interact with several genes and lead to nuclease degradation of mRNAs. The immediate topography of these sequences, nonexact serial copies, and other such facets have to be studied carefully. Several speculative models of gene inhibition can be drawn up that must be explored. For example, the proteins and their mRNAs may function only if they are part of the RNase P complex in vivo. In our case, the message of Rpp38 is attacked and the amount of Rpp38 protein in the enzyme complex begins to decrease. This decrease immediately leads to a decrease in the other inhibited proteins, and their mRNAs quickly are degraded, although the exact mechanism is not understood. A corollary of this mechanism is that when one of the inhibited mRNAs disappears, they all disappear. Suggestions such as this one can be tested only in vitro with affinity and other experiments for mRNAs and various cellular components. The noninhibited proteins may be protected when not in the RNase P enzyme complex by serving in other intracellular functions that require their activity alone or in complexes with non-RNase P proteins. We also note that EGSs directed at three other mRNAs of subunits of RNase P that are inhibited in expression also gave results similar to those reported here (Haifeng Zhang and S.A., unpublished experiments).
The various subunits may be involved in heat shock activities, other complex interactions with RNAs as in helicases, or functions that involve nucleolar formation or structure. Certainly, previous studies indicate that possibility strongly (20, 21).
Acknowledgments
We thank Dr. Taijiao Jiang for preparing blots for Southern analysis. Brian Quinlan gave expert technical assistance in analyzing sequences and Tae Hoon Kim (Harvard University, Cambridge, MA) provided aid in analyzing some enhancer and promoter sequences. We benefited greatly from discussions with various colleagues, in particular, Prof. Dan DiMaio (Yale University School of Medicine), Prof. Tom Maniatis (Harvard University), and members of our laboratory. This research was supported by U.S. Public Health Service Grant GM-194422 (to S.A.).
Abbreviation
- EGS
external guide sequence
References
- 1.Hannon G J. Nature. 2002;418:244–251. doi: 10.1038/418244a. [DOI] [PubMed] [Google Scholar]
- 2.Manoharan M. Antisense Nucleic Acid Drug Dev. 2002;12:103–128. doi: 10.1089/108729002760070849. [DOI] [PubMed] [Google Scholar]
- 3.Forster A C, Altman S. Science. 1990;249:783–786. doi: 10.1126/science.1697102. [DOI] [PubMed] [Google Scholar]
- 4.Gopalan V, Vioque A, Altman S. J Biol Chem. 2002;277:6759–6762. doi: 10.1074/jbc.R100067200. [DOI] [PubMed] [Google Scholar]
- 5.Plehn-Dujovich D, Altman S. Proc Natl Acad Sci USA. 1998;95:7327–7332. doi: 10.1073/pnas.95.13.7327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eder P, Kekuda R, Stolc V, Altman S. Proc Natl Acad Sci USA. 1997;94:1101–1106. doi: 10.1073/pnas.94.4.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jarrous N, Eder P, Wesolowski D, Altman S. RNA. 1999;5:153–157. doi: 10.1017/s135583829800185x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jarrous N, Altman S. Methods Enzymol. 2001;342:93–100. doi: 10.1016/s0076-6879(01)42538-9. [DOI] [PubMed] [Google Scholar]
- 9.Guerrier-Takada C, Altman S. Methods Enzymol. 1999;313:442–456. doi: 10.1016/s0076-6879(00)13028-9. [DOI] [PubMed] [Google Scholar]
- 10.Miller A D, Rosman G J. BioTechniques. 1989;7:980–990. [PMC free article] [PubMed] [Google Scholar]
- 11.Krupp G, Cherayil B, Frendeway D, Nishikawa S, Soll D. EMBO J. 1986;5:1697–1703. doi: 10.1002/j.1460-2075.1986.tb04413.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yuan Y, Hwang E-S, Altman S. Proc Natl Acad Sci USA. 1992;89:8006–8010. doi: 10.1073/pnas.89.17.8006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bartkiewicz M, Gold H, Altman S. Genes Dev. 1989;3:488–499. doi: 10.1101/gad.3.4.488. [DOI] [PubMed] [Google Scholar]
- 14.Guerrier-Takada C, Li Y, Altman S. Proc Natl Acad Sci USA. 1995;92:11115–11119. doi: 10.1073/pnas.92.24.11115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang T, Altman S. Proc Natl Acad of Sci USA. 2001;98:920–925. doi: 10.1073/pnas.021561498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee D Y, Clayton D A. Genes Dev. 1997;11:582–592. doi: 10.1101/gad.11.5.582. [DOI] [PubMed] [Google Scholar]
- 17.Jarrous N, Wolenski J S, Wesolowski D, Lee C, Altman S. J Cell Biol. 1999;146:559–571. doi: 10.1083/jcb.146.3.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burke B, Stewart C L. Nat Rev Mol Cell Biol. 2002;3:575–585. doi: 10.1038/nrm879. [DOI] [PubMed] [Google Scholar]
- 19.Clarke J H, Haridasse V, Glazer R I. Biochemistry. 2002;41:11847–11856. doi: 10.1021/bi025600k. [DOI] [PubMed] [Google Scholar]
- 20.Jiang T, Altman S. Proc Natl Acad Sci USA. 2002;99:5295–5300. doi: 10.1073/pnas.072083699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y, Altman S. Proc Natl Acad Sci USA. 2001;98:441–444. doi: 10.1073/pnas.021555498. [DOI] [PMC free article] [PubMed] [Google Scholar]