

Abstract
We survey DNA sequence polymorphisms at the Sod locus and four neighboring regions of Drosophila melanogaster, spanning 55,513 base pairs (bp), in 15 strains from a natural population, plus one reference laboratory strain and one strain of Drosophila simulans. Our objective is to characterize a proposed selective sweep that occurred at a locus close to Sod in D. melanogaster and to characterize the strength of the selection event, its time, and the size of the hitchhiked region. Two regions, 1819 and 6kbr3r, show a pattern of polymorphism very similar to the one of Sod, implying that they have been affected by the same evolutionary process that impacted Sod. A third fragment, 2021 seems unaffected by the event. A fourth one, 4039, on the opposite flank of Sod in relation to 2021, is only partially affected. We estimate that the length of the chromosomal segment impacted by the selective sweep is 41–54 kb, the age of the selective sweep is 2,600–22,000 years, and the selective advantage is 0.020 < s < 0.103.
Genetic hitchhiking is the increase in frequency of an allele (or nucleotide variant) attributable to positive natural selection acting on another site linked to it. This phenomenon was theoretically described almost three decades ago (1). Experimental evidence has started to accumulate only in recent years (reviewed in ref. 2), but a comprehensive description of it is lacking. Very few studies have attempted to establish the boundaries (i.e., size) of the regions affected (3, 4), which would make it possible, based on mathematical models, to calculate both the age and the strength of selection associated with the selective sweep (5). The Sod locus, coding for the CuZn-superoxide dismutase (SOD) was one of the first found cases of a gene showing a pattern of genetic hitchhiking in chromosomal regions with standard levels of recombination (6). Here we report on the pattern of polymorphism at this locus and its surrounding regions. We have found upper limits for the region affected, both upstream and downstream from Sod, and from these we infer the age and strength of the past selective event.
SOD prevents the accumulation of free oxygen radicals by catalyzing the dismutation of the superoxide anion (O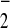 ) to H2O2 and O2 (7). In Drosophila melanogaster, the Sod locus is approximately in the middle of the left arm of chromosome 3 (68A7 in the polytene chromosome; www.fruitfly.org) and codes for a homodimeric metalloenzyme whose monomer is 151 aa long in the functional enzyme (8). Electrophoretic surveys have shown that many populations of D. melanogaster segregate for two common alleles at this locus: Sod-Slow (SodS) and Sod-Fast (SodF) (e.g., refs. 9 and 10). SODS and SODF only differ in residue 96 (lysine in SODS and asparagine in SODF; ref. 11). Several lines of investigation indicate that this polymorphism is maintained by natural selection. First, biochemical studies have shown that the two forms differ in their specific activity and thermal stability (12). Second, the observation of linkage disequilibrium built up between Sod and the closely linked Est-6 in three out of four large laboratory populations (10), as well as the presence of linkage disequilibrium between the two loci in nature (10, 13), suggest selective interactions between the two loci. [No third-chromosome inversion polymorphisms exist in the populations studied (10).] Third and more important, in laboratory populations the frequencies of the SodS and SodF alleles change consistently in the presence of ionizing radiation (14) or in flies selected for postponed senescence (15).
) to H2O2 and O2 (7). In Drosophila melanogaster, the Sod locus is approximately in the middle of the left arm of chromosome 3 (68A7 in the polytene chromosome; www.fruitfly.org) and codes for a homodimeric metalloenzyme whose monomer is 151 aa long in the functional enzyme (8). Electrophoretic surveys have shown that many populations of D. melanogaster segregate for two common alleles at this locus: Sod-Slow (SodS) and Sod-Fast (SodF) (e.g., refs. 9 and 10). SODS and SODF only differ in residue 96 (lysine in SODS and asparagine in SODF; ref. 11). Several lines of investigation indicate that this polymorphism is maintained by natural selection. First, biochemical studies have shown that the two forms differ in their specific activity and thermal stability (12). Second, the observation of linkage disequilibrium built up between Sod and the closely linked Est-6 in three out of four large laboratory populations (10), as well as the presence of linkage disequilibrium between the two loci in nature (10, 13), suggest selective interactions between the two loci. [No third-chromosome inversion polymorphisms exist in the populations studied (10).] Third and more important, in laboratory populations the frequencies of the SodS and SodF alleles change consistently in the presence of ionizing radiation (14) or in flies selected for postponed senescence (15).
Hudson et al. (6) sequenced Sod from 41 D. melanogaster lines (homozygous for the third chromosome): three laboratory strains plus 38 lines sampled from three natural populations (Culver City and El Rio Vineyard from California, ≈550 km apart, and Barcelona, Spain). The observed pattern of variation was significantly different from the pattern expected under a neutral model. A total of 28 sequences, including 19 SodS and 9 SodF alleles showed no variation at all over the 1,428 bp sequenced except for the one nucleotide accounting for the S/F polymorphism. The remaining 13 F sequences were made up of nine different haplotypes, which did not significantly deviate from neutral expectations. We concluded that the F/S SOD polymorphism is recent, but we did not provide any estimate of the time when the S allele arose (6). Moreover, we hypothesized that such a pattern of genetic variability, in which approximately half of the F and all S alleles exhibit no silent polymorphism, could be due to a recent selective sweep at Sod or a tightly linked site.
The present investigation seeks to identify the boundaries of the hitchhiked region as well as the target of selection during the selective sweep. This should help to estimate the age of the sweep, the strength of the selection involved, and the approximate location of the selected site, which should be near the middle of the swept region. We incorporate and extend our previous results (16) and include a survey of an additional fifth region, chosen to determine the upper bounds for the above sweep parameters, with both approximate upstream and downstream limits for the region affected. Additionally, we further support the hypothesis of a selective sweep in the region with comparisons between D. melanogaster and Drosophila simulans for the five studied segments, ruling out that the two bordering regions acquired standard neutral patterns of variation because of their higher mutation rate.
Materials and Methods
The Five Studied Regions.
We have sequenced five regions from each strain in the sample (Fig. 1). Some oligonucleotides used for amplification are given in ref. 17; all, as well as additional information concerning the material and methods used, can be obtained from the authors at a.saez@ic.ac.uk.
Figure 1.

The Sod region with location and size of the five DNA fragments investigated. The DNA fragments are depicted as boxes on a line representing the section of the third chromosome of D. melanogaster where they are located. Base pair lengths are shown above. The two exons of Sod are represented as black stripes and numbered 1 and 2. The thicker line from 1819 to 2021 represents clone 112. 4039 is closest to the telomere.
Drosophila Strains.
Fifteen lines of D. melanogaster, homozygous for the third chromosome, derive from flies collected in October 1991 in El Rio Vineyard (Lockeford, San Joaquin County, CA). This collection was characterized for the Slow/Fast SOD allozyme polymorphism (methods of ref. 18). 11 SodF and 4 SodS lines were chosen at random from the corresponding allozymic classes. For interspecific comparisons, we use an isofemale line from D. simulans (10F) derived from a collection made in October 1991 in Irvine (Orange County, CA).
DNA Extraction, Amplification, and Sequencing.
Genomic DNA was obtained according to ref. 19. Standard procedures were used for amplification and sequencing (ref. 6 and a.saez@ic.ac.uk). The DNA sequences have been deposited in the European Molecular Biology Laboratory (EMBL) database, accession nos. AJ519553–AJ519619.
Analyses.
Four homozygous lines have the SodS allele and 11 have the SodF. For the population analyses at Sod only one SodS allele is included, namely 255S (the SodS population frequency is around 5%; no more than one S strain would be expected in a random sample; see ref. 6). The other four regions are analyzed for the 16 lines sequenced. To confirm that our results were not biased owing to the four SodS lines, we repeated most of our analyses, and specifically those of Tables 1–3, with only one SodS line (521S), obtaining the same conclusions (data not shown).
Table 1.
P values for the five sequenced regions using the haplotype test (6)
| Regions and sequences |
Nr
|
|||
|---|---|---|---|---|
| 0.000 | 0.001 | 0.010 | 0.038 | |
| 4039 (n = 16, L = 1130, S = 36) | ||||
| 6 sequences with 2 substitutions | 0.410 | 0.336 | 0.034 | 0.00002* |
| 7 sequences with 4 substitutions | 0.457 | 0.367 | 0.031 | 0.00000* |
| 8 sequences with 6 substitutions | 0.450 | 0.351 | 0.024 | 0.00000* |
| 1819 (n = 16, L = 894, S = 27) | ||||
| 12 sequences with 5 substitutions | 0.059 | 0.037 | 0.00013* | 0.00000* |
| 13 sequences with 7 substitutions | 0.057 | 0.035 | 0.00009* | 0.00000* |
| 14 sequences with 10 substitutions | 0.058 | 0.036 | 0.00002* | 0.00000* |
| 6kbr3r (n = 16, L = 722, S = 20) | ||||
| 7 sequences with 0 substitutions | 0.141 | 0.115 | 0.013 | 0.00002* |
| 10 sequences with 1 substitution | 0.053 | 0.037 | 0.001* | 0.00000* |
| 11 sequences with 2 substitutions | 0.057 | 0.040 | 0.0007* | 0.00000* |
| Sod (n = 13, L = 1373, S = 44) | ||||
| 9 sequences with 5 substitutions | 0.034 | 0.016 | 0.00000* | 0.00000* |
| 10 sequences with 8 substitutions | 0.037 | 0.017 | 0.00000* | 0.00000* |
| 11 sequences with 14 substitutions | 0.056 | 0.027 | 0.00000* | 0.00000* |
| 2021 (n = 16, L = 1088, S = 49) | ||||
| 2 sequences with 0 substitutions | 0.996 | 0.995 | 0.935 | 0.500 |
| 4 sequences with 12 substitutions | 1.000 | 1.000 | 0.971 | 0.280 |
| 5 sequences with 20 substitutions | 1.000 | 1.000 | 0.998 | 0.568 |
Nr is the product of the effective population size (N) and the rate of recombination (r) between adjacent pairs per generation and genome; n is the size of the sample, L is nucleotide length, and S is the number of polymorphic sites. P values are based on 100,000 random samples.
Values below the Bonferroni critical value for 0.05%.
Table 3.
Nucleotide variability for the various regions in the Sod region
| Region | Different haplotypes | Length, bp | S | π | θ | Tajima's D | K | π/K |
|---|---|---|---|---|---|---|---|---|
| 4039 | 11 | 1,130 | 36 | 0.0089 | 0.0096 | −0.3045 | 0.0456 | 0.19 |
| 1819 | 9 | 894 | 27 | 0.0060 | 0.0091 | −1.3994 | 0.0693 | 0.09 |
| 6kbr3r | 8 | 722 | 20 | 0.0070 | 0.0084 | −0.6578 | 0.0959 | 0.07 |
| Sod intron | — | 694 | 29 | 0.0104 | 0.0135 | −0.9846 | 0.0722 | 0.14 |
| Sod synonymous | — | 105.67 | 4 | 0.0107 | 0.0122 | — | 0.1100 | 0.10 |
| Sod entire | 9 | 1,373 | 44 | 0.0075 | 0.0103 | −1.2136 | 0.0533 | 0.14 |
| 2021 | 15 | 1,088 | 49 | 0.0163 | 0.0136 | 0.8406 | 0.0807 | 0.20 |
π, nucleotide diversity; S, the number of polymorphic sites; θ, the population rate of nucleotide substitution per site; K, the average number of differences between D. simulans and the D. melanogaster sequences.
Sequences were edited and manually aligned with lasergene software (DNASTAR, Madison, WI) and esee (20); the total number of mutations was minimized, and substitutions were favored rather than gaps when both were present in equal numbers. mega (21) was used to help edit the alignments, calculate distances, and produce the phylogenetic trees. We used dnasp Versions 2.52 and 3.0 (22) for calculating the parameters of variability, for performing most of the neutrality tests, and for calculating linkage disequilibrium at 2021. Measures of inter- and intraspecific variation and of length were obtained after excluding positions that had gaps in the alignment. The “haplotype test” was applied as in Hudson et al. (6). We identified for every number of sequence subsets larger than one (i.e., from the two-sequences subset to the [n − 1]-sequences subset, where n is the sample size of each region, i.e., 2–15, or 2–12 at Sod) the subset holding the minimum number of substituted sites. Then, using the haplotype test, we calculated the probability of each subset, given the number of polymorphic sites, recombination rate, and length of the considered region, based on 100,000 simulated replicates. In Table 1 we show, for each locus, the three subsets with smallest P values. We applied a Bonferroni correction for multiple tests (suggested in ref. 6), which are as many as the number of analyzed subsets, i.e., 14, except for Sod, i.e., 11. The Bonferroni critical value at the 0.05% level turned out to be 0.0047 [=1 − (1 − 0.05)1/11] at Sod; and 0.0037 [=1 − (1 − 0.05)1/14] at the other four regions (ref. 23, p. 241). ORFs were obtained by using www.ncbi.nlm.nih.gov/gorf/gorf.html, and repetitive sequences by using compare and dotplot from the Wisconsin Package (Genetics Computer Group, Madison, WI). We searched for D. melanogaster sequences homologous to those of 4039, 1819, 6kbr3r, and 2021 according to www.fruitfly.org/blast/.
Results and Discussion
Variability at 4039, 1819, 6kbr3r, Sod, and 2021.
Fig. 2 shows the polymorphic sites found at 4039; for the other regions, see figure 4 in ref. 16. Three of the five sequenced fragments (Sod, 6kbr3r, and 1819) exhibit a pattern of variation similar to the one previously described for Sod in which a majority of the chromosomes exhibit identical nucleotide sequences (6). (See description of haplotype differences in ref. 16, but note that the 6kbr3r sequence of line 357F, absent from that report, is identical to 565F, increasing by one the number of sequences in the homogeneous subset.) In contrast, in 2021 only two sequences are identical to each other, whereas 4039 exhibits an intermediate pattern: one haplotype appears four times, two sequences differ from it at one site each, and two differ at two sites whereas all others are fairly heterogeneous (but two of these haplotypes are, each of them, represented twice). In contrast with our previous survey (6), several alleles differ from the most common sequence in one or very few nucleotides. About 75% of the sampled chromosomes at Sod, 6kbr3r, and 1819 show identical or very similar sequences; at 4039 the proportion of highly similar (or identical) is 50%, whereas at 2021 the sequences are very heterogeneous.
Figure 2.

Polymorphic sites at 4039. The numbers at the top of each polymorphic site indicate its position in the alignment of all of the listed sequences, including D. simulans (SIM). A dot signifies the nucleotide shown in the top sequence. The sample includes 15 lines from El Rio Vineyard (F and S refer to the Fast/Slow SOD polymorphism), plus clone BACR48O03 (BAC).
We use recomb-rate Version 1.0 (24) to estimate the recombination rate in D. melanogaster females at Sod: 3.8 × 10−8 per site and generation. If n = 106 (25), Nr (the effective population size times the rate of recombination between adjacent pairs per generation) is expected to be 0.038 (= 106 × 3.8 × 10−8). When applying the haplotype test (6), the higher the recombination rate, the lower the P values turn out to be; therefore, we have considered lower recombination levels for this test. However, by using the method of Hudson (26), Nr = 0.013 was obtained for 2021.
Table 1 shows that the hypothesis of neutral equilibrium is rejected, after a Bonferroni correction, in all regions except 2021 for Nr = 0.038. With Nr = 0.01, based on a lower but still standard level of recombination (27), the neutral equilibrium model is rejected only for the three central regions, after the Bonferroni correction. If there is no recombination, or when Nr = 0.001, the neutral model is not rejected for any region (after taking into account the correction), although most P values for the three central regions, 1819, 6kbr3r, and Sod, remain low.
A noticeable signal of recombination is provided by a largely destroyed association between regions in the haplotypes (see figure 4 of ref. 16). For example, one of the two most divergent lines at Sod (498F) belongs to the most common set of sequences at 6kbr3r (with one substitution difference from most other alleles). 581F exhibits, in the 5′ side, eight substitution differences from the most common Sod, but it is identical to it in the rest of the Sod sequence; at 6kbr3r (downstream from Sod; Fig. 1), 581F is highly divergent. Among the five most differentiated lines at 6kbr3r, only 968F is among the two most divergent lines at 1819; the other one is clone 112 (or BACR48O03), which belongs to the homogeneous subset at 6kbr3r and Sod. Recombination appears to have shuffled common and rare haplotypes also between 4039 and 1819, and it has broken the subset of S sequences (255, 438, 510, and 521), which remains homogeneous only at Sod.
To corroborate the results from the haplotype test (16), we use a similar method, the Sliding-Window Strobeck Test (28). It calculates the probability of obtaining, under neutral equilibrium, tracts or windows comprising all of the sequences (but not all sites), containing a minimum number of haplotypes. Moreover, it corrects for multiple tests and for arbitrarily chosen window sizes. Our results are shown in Table 2. The only relevant difference with respect to Table 1 is the lack of rejection of the neutral equilibrium model for 6kbr3r when Nr = 0.01. This may be due to the fact that this region may have been acquiring polymorphisms (after the selective sweep) faster than the others because of a higher content of noncoding sequences and repetitive tracts (see below).
Table 2.
P values using the Sliding-Window Strobeck Test (28)
| Regions |
Nr
|
|||
|---|---|---|---|---|
| 0.000 | 0.001 | 0.010 | 0.038 | |
| 4039 | 0.66 | 0.68 | 0.20 | 0.0036 |
| 1819 | 0.065 | 0.056 | 0.0074 | 0.00006 |
| 6kbr3r | 0.65 | 0.61 | 0.19 | 0.004 |
| Sod | 0.005 | 0.004 | 0.0001 | 0.00000 |
| 2021 | 0.69 | 0.78 | 0.81 | 0.34 |
P values based on 100,000 random samples. In bold, P < 0.05%.
The levels of nucleotide polymorphism for the various regions are shown in Table 3. The number of haplotypes is highest for 2021 and intermediate for 4039, consistent with hitchhiking acting at the other three regions. Nucleotide diversity per site (π) (29) at 4039, 1819, 6kbr3r, and Sod is in the range of average values for D. melanogaster (30): 0.004, 0.013, and 0.011 for “total” coding, silent, and noncoding regions, respectively. 2021 has the highest value of π, somewhat higher than the average of π at silent positions. The neutral parameter per site (θ), based on the number of polymorphic sites (S) (31), also yields values within the averages of ref. 30: 0.004, 0.014, and 0.011, for coding, silent, and noncoding regions, respectively. Again, the 2021 region has the highest value. A possible explanation of this high value is that this region may have “recovered” from the selective sweep because of a higher mutation rate. But this explanation is not supported by the observation that interspecific divergence between D. melanogaster and D. simulans is lower for 2021 than for 6kbr3r, which has a pattern of polymorphism similar to Sod and 1819. Also, 4039, despite presenting more haplotypes than the three central regions (Fig. 2 and Table 3), cannot account for this difference as a consequence of a higher mutation rate (this is the slowest evolving region, see Table 3). Accordingly, the ratio π/K (intraspecific diversity vs. interspecific divergence) is highest for 4039 and 2021 (Table 3). The Hudson–Kreitman–Aguade test (32) yields consistency with those ratios (not shown; but see below).
Tajima (33) and Fu and Li (34) tests do not reject the neutral null hypothesis for any of the five regions studied (Table 3 and data not shown), which was also the case for Sod (6). We apply these tests here as tools toward a statistical description of the sequences. All tests give negative values for 1819, Sod, and 6kbr3r (with one exception for this last region: Fu and Li's D with an outgroup) and positive values for 2021. The values obtained for 6kbr3r are always closer to zero than values obtained for Sod and 1819. 4039 statistics are positive and negative, and are generally the closest to zero, which indicates that the distribution in the frequency of substitutions at this region is the most in accordance with the standard neutral model. We conclude that there is an excess of singletons or low frequency substitutions in 1819, Sod, and 6kbr3r compared with 2021 and 4039, and that 2021 seems to have a larger incidence of high frequency substitutions compared with 4039. This last observation could be due to balancing selection acting tightly linked to 2021. However, a Sliding-Window analysis of this region leads to rejecting that hypothesis: the levels of interspecific and intraspecific variation clearly run in parallel to each other (data not shown), in contrast to what would be expected under the influence of a balance polymorphism linked to it (35).
The five regions seem to combine low and highly constrained sequences in different proportions. 6kbr3r is the most freely evolving; interspecific divergence (K) is higher than for the other regions (Table 3), and there is a tandem duplication between coordinates 263 and 399. (We found no repetitive sequences in other regions.) We found no coding sequence from the flybase matching 6kbr3r and only a short tract of this region as an ORF. Despite its overall high variability, 2021 manifests several ORFs and moderate levels of variation within them. It contains also a segment with 88% identity to cDNA clone RE43692. 1819 and Sod are made of transcribed and nontranscribed regions, with substantially fewer indels than 6kbr3r and 2021. 1819 contains a section of locus JIL-1. 4039 is the slowest evolving region and apparently has the largest proportion of coding regions, including gene Mocs1 and cDNA GH12095.
Specific Observations About Sod.
Our Sod survey exhibits a pattern of variation similar to the one described in the initial survey (6). However, the homogeneous subset is ≈75% in our sample, rather than only 50%, which may be a consequence of a higher number in the present sample of haplotypes from North America, colonized later than Europe (36). A second difference is the presence of a large percentage of similar (but not identical) sequences to Fast A or Slow in the El Rio population (the predominant haplotype, called Fast A in the first survey, is here represented by lines 5F, 94F, 357F, 521F, and 483F). In our first report (6), all sequences belonging to the homogeneous subset (Fast A and Slow haplotypes) were identical except for the Slow/Fast nonsynonymous substitution.
Our current results from El Rio (CA) confirm that the high incidence of Fast A in ref. 6. is not a local phenomenon restricted to the Barcelona population. A previous conclusion was that the Slow allele had emerged quite recently from the Fast A allele (6). The evidence was that 5 Slow lines from Barcelona, 10 from Culver City, 3 from El Rio Vineyard, and 1 from Davis (CA) were all identical at Sod (1,410 bp); these differed from Fast A only at the S/F site. We have now found one synonymous substitution in each of two of the four Slow lines. The time lag between the rise of Fast A and the subsequent rise of the Slow allele, although short, must be longer than previously thought.
Boundaries and Timing of the Selective Sweep.
We have noted above that 2021 appears to be beyond the 5′ end of the swept fragment, whereas the 3′ end is likely to be close to or in 4039. The other three regions (Sod, 6kbr3r, and 1819) are clearly included within the sweep: Figs. 3 and 4 show that ≈75% of the sequences are identical or very similar for each region. Sod, 6kbr3r, and 1819 are similar to one another in that a majority of comparisons indicate zero or very few differences. 4039 and 2021 yield more differences, although 4039 shares proportionally more cases with none or few differences than 2021. We take into account the greater length of 2021 in Fig. 4.
Figure 3.

Neighbor-joining trees of 4039, 1819, 6kbr3r, Sod, and 2021. The trees are based on pairwise differences per site (p-distance), excluding positions with gaps in any one of the D. melanogaster lines. Bootstrap values are shown at the nodes, based on 1,000 replications. D. simulans branches were added by using the average values of K (Table 3) (shown above the D. simulans branch).
Figure 4.

Distribution of pairwise distances at 4039, 1819, 6kbr3r, Sod, and 2021. The number of differences between all possible pairs of lines is plotted against the number of pairs. Sites with gaps are only excluded for the pairwise comparisons where they are present. We have excluded 510S, 438S, and 521S from the set of Sod lines (see Materials and Methods). The analysis of the 2021 alignment is performed in toto as well as separately for the 5′ and 3′ halves (596 and 597 bp, respectively).
We seek a determination of the target of selection with the following observations. Notice that 6kbr3r has fewer highly related sequences (11 out of 16, 69%) than 1819 (13 out of 16, 81%), even though it is shorter. Also, Tajima's (33) and Fu and Li's (34) tests suggest that 6kbr3r is less affected by the sweep than 1819. Nevertheless, 6kbr3r seems to be recovering from the sweep faster than 1819, because it is more divergent from D. simulans than 1819 and thus less constrained (Table 3). Notice also that 1819 seems to be evolving faster than Sod as manifested by greater divergence from D. simulans (Table 3), which may be an indication that 1819 has been more affected by the sweep than Sod. Consistent with this is that π/K (Table 3) decreases from Sod (0.14) to 6kbr3r (0.07) and 1819 (0.09). The Hudson–Kreitman–Aguade test probabilities are much higher between 6kbr3r and 1819 (0.93) than between them and Sod (0.34 and 0.31), consistent with a more similar π/K between the first two regions than between them and Sod. We tentatively conclude that the locus of selection is between 1819 and 6kbr3r, but closer to 1819 than to Sod. The selective episode, accordingly, encompasses the span Sod to 1819; it has a downstream outer boundary (in cytological orientation) at 2021 and an upstream boundary near 4039. Therefore, the upper limit estimates for the size of the swept region are 41 kb (from 4039 to Sod) and 54 kb (from 4039 to the proximal end of 2021). With these distances, we estimate the selection coefficient s = 0.020–0.027 (half-length of the swept fragment × r/0.01 = 20,500 or 27,000 × 10−8/0.01). With a more likely recombination rate, r = 3.8 × 10−8 (see above; ref. 24), s = 0.078–0.103.
Five transcripts have been assigned in clone BACR48O03 to the region of 15,206 bp between 6kbr3r and 1819, where, according to our inference, the target of selection is located; two of them are very close to 6kbr3r, and three are intermediate between 1819 and 6kbr3r: GH03576, LD31387, and GH10915 (37). One of these might represent the target of selection.
Using a recombination rate specific for the Sod region (24), r = 3.8 × 10−8, we estimate the age of the selective sweep, t, is 2,600 years (38). In ref. 16. we estimated that t was at least 5,000 years old, with an upper boundary of 31,000 years. This last value was solved from μt Σ(n′L′)i = S′, where μ is the neutral mutation rate at noncoding and silent sites, n′ is the number of observed sequences in the homogeneous subset at each region (i), L′ is the number of silent and noncoding sites taken from those subsets and regions, and S′ is the observed number of polymorphic sites in the homogeneous sets of sequences at all regions after excluding those likely to have arisen by gene conversion. We can use again this method with the current data set, which includes eight new homogeneous sequences from 4039 and one more 6kbr3r sequence, from line 357F. We consider n′ equal to 8, 13, 11, and 13, for 4039, 1819, 6kbr3r, and Sod, respectively (Fig. 2; and Fig. 4 from ref. 16). We estimate that ≈80% of 4039 consists of coding sequence (based on a combination of blast searches and ORF analyses, and therefore its coding length would be ≈0.8 × 1,130 bp = 904 bp, from which ≈226 sites (25%) would be silent; the rest of 4039, ≈20%, or 226 bp, would be noncoding, for a total of 452 bp of silent and noncoding sites (L′). For 1819 (894 bp) and assuming 25% to be coding (≈224 bp), we get L′ (1819) ≈ 731 bp. At 6kbr3r we estimate a large percent (90%) of noncoding sites (≈650 out of 722 bp), and then L′ (6kbr3r) ≈ 668 bp. The number of nonsynonymous sites at Sod is 331 bp; therefore, L′ (Sod) is ≈1373 − 331 bp = 1,042 bp. We count now the number of polymorphisms in the homogeneous sets that are not found among the heterogeneous sequences or that are not in unconsidered sites (i.e., sites with polymorphic indels): at 4039, sites 86 and 1029 (Fig. 2); at 1819, sites 185–187 (counted as one substitution, because it is likely due to a single mutation), 371, 454, and 716; at 6kbr3r, sites 96 and 242; and at Sod, 141, 310, 1109, and 1426. In total, S′ = 12 polymorphisms. Finally, substituting in the above equation, μt(8 · 452 + 13 · 731 + 11 · 668 + 13 · 1,042) = 12, we obtain t ≈ 22,000 years. This estimate is 9,000 years more recent than ref. 16. This difference is largely due to several polymorphisms that we have not now considered: 185–187 at 1819 is now taken as one substitution; polymorphisms from 521F, also at 1819, are not included because we think that this haplotype is part of the heterogeneous subset; and site 1369 from Sod is ignored because it is affected by an indel in two strains from D. melanogaster. In any case, our previous and current estimates (this one to a lesser extent) are likely to be biased upward, owing to an unknown number of polymorphisms included in the calculations that might have arisen by gene conversion or recombination rather than by mutation (16). The mutation rate assumed for silent sites (16 × 10−9) may also be an underestimate, which would overestimate the age of the selection episode. We conclude that the actual age is between 2,600 and 22,000 years. The selective sweep may have resulted from an adaptation to non-African environments after the migration of a D. melanogaster population from Africa 10,000–15,000 years ago (36).
An investigation of molecular variation at Est-6 in a set of homozygous lines very similar to ours (refs. 13 and 38; see also ref. 10) has shown a pattern of polymorphism like the one we have observed for Sod, 1819, or 6kbr3r (13, 38), as well as evidence of linkage disequilibrium between Sod and Est-6. Some hypotheses that are advanced to account for these observations involve concerted evolution of Sod and Est-6. Est-6 and Sod are separated by ≈1,000 kb. Assuming a lower typical level of recombination in the region, r = 10−8, the probability of recombination between the two loci is 0.01, which becomes quite high over thousands of generations. Moreover, in view of the fact that region 2021 is between Sod and Est-6, it is reasonable to conclude that linkage disequilibrium between the two loci cannot be a consequence of the selective sweep impacting the Sod locus, but rather it would have arisen through epistatic interactions or some other process. It may be that selective sweeps are frequent across the D. melanogaster genome and that they can be the consequence of adaptation to newly colonized environments (2, 39), arisen during D. melanogaster's recent migrations to new continents (36).
Several alternative (or complementary) scenarios, other than a selective sweep, can be explored (6, 16). The existence of inversion polymorphisms suppressing or reducing reciprocal crossing over in the Sod region could account for the presence in our sample of haplotype “families.” However, no third-chromosome inversions have been found in large samples from El Rio (10). In any case, chromosomal inversions alone could not readily account for the large differences in amounts of polymorphism between the heterogeneous and homogeneous subsets. They could account for the reduction of genetic exchange between haplotype groups at regions close to the boundaries of those inversions (40) but not for the very different levels of polymorphism among sequences in a region, especially when the less variable group of sequences is at the same time the more frequent. We have examined the possibility that two alleles very divergent for Sod or 1819 could have come from different populations and would have later combined with each other and with other alleles (16). This possibility cannot be unequivocally discarded but is contradicted by the absence of well differentiated lines at 4039 or 2021. Nevertheless, population expansions, geographic subdivisions, and genetic admixtures likely had a substantial imprint on the extant populations of D. melanogaster, especially outside of Africa (41). This may be the reason that 2021 contains a considerable fraction of paired sites in linkage disequilibrium: 163 out of 820 by the χ2 test (20%), or 113 by the Fisher's exact test (13.8%), for P < 0.05. However, we do not think that nonequilibrium processes can account for the different patterns observed, say, between 2021 and Sod, in the same set of flies, isolated from the same population, and so closely linked in the genome. Future tests able to distinguish demographic vs. selectionist scenarios will be of great help to discern between the two possibilities.
Acknowledgments
We thank Richard Hudson for early contributions to this investigation, S. Qin and J. W. Jacobs for the use of an automatic sequencer, E. S. Balakirev for a physical map of clone 112, and S. Ramos-Onsins for the haplotype-test software. A.G.S. was supported by a postdoctoral fellowship from the Spanish Ministry of Education and Science and by the European Union through Training and Mobility of Researchers Network Contract ERBFMRX-CT97-0113 CODENET. Research was supported by National Institutes of Health Grant GM42397 (to F.J.A.).
Footnotes
References
- 1.Maynard-Smith J, Haigh J. Genet Res. 1974;23:23–35. [PubMed] [Google Scholar]
- 2.Nurminsky D I. Cell Mol Life Sci. 2001;58:125–134. doi: 10.1007/PL00000772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kirby D A, Stephan W. Genetics. 1995;141:1483–1490. doi: 10.1093/genetics/141.4.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benassi V, Depaulis F, Meghlaoui G K, Veuille M. Mol Biol Evol. 1999;16:347–353. doi: 10.1093/oxfordjournals.molbev.a026115. [DOI] [PubMed] [Google Scholar]
- 5.Kaplan N L, Hudson R R, Langley C H. Genetics. 1989;123:887–899. doi: 10.1093/genetics/123.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hudson R R, Bailey K, Skarecky D, Kwiatowski J, Ayala F J. Genetics. 1994;136:1329–1340. doi: 10.1093/genetics/136.4.1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fridovich I. J Biol Chem. 1997;272:18515–18517. doi: 10.1074/jbc.272.30.18515. [DOI] [PubMed] [Google Scholar]
- 8.Lee Y M, Friedman D J, Ayala F J. Arch Biochem Biophys. 1985;241:577–589. doi: 10.1016/0003-9861(85)90583-1. [DOI] [PubMed] [Google Scholar]
- 9.Singh R S, Hickey D A, David J. Genetics. 1982;101:235–256. doi: 10.1093/genetics/101.2.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smit-McBride Z, Moya A, Ayala F J. Genetics. 1988;120:1043–1051. doi: 10.1093/genetics/120.4.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee Y M, Ayala F J. FEBS Lett. 1985;179:115–119. doi: 10.1016/0014-5793(85)80203-9. [DOI] [PubMed] [Google Scholar]
- 12.Lee Y M, Misra H P, Ayala F J. Proc Natl Acad Sci USA. 1981;78:7052–7055. doi: 10.1073/pnas.78.11.7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Balakirev E S, Balakirev E I, Rodríguez-Trelles F, Ayala F J. Genetics. 1999;153:1357–1369. doi: 10.1093/genetics/153.3.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peng T X, Moya A, Ayala F J. Proc Natl Acad Sci USA. 1986;83:684–687. doi: 10.1073/pnas.83.3.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tyler R H, Brar H, Singh M, Latorre A, Graves J L, Mueller L D, Rose M R, Ayala F J. Genetica. 1993;91:143–149. doi: 10.1007/BF01435994. [DOI] [PubMed] [Google Scholar]
- 16.Hudson R R, Sáez A G, Ayala F J. Proc Natl Acad Sci USA. 1997;94:7725–7729. doi: 10.1073/pnas.94.15.7725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kwiatowski J, Patel M, Ayala F J. Nucleic Acids Res. 1989;17:1264. doi: 10.1093/nar/17.3.1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ayala F J, Powell J R, Tracey M L, Mourao C A, Perez-Salas S. Genetics. 1972;70:113–139. doi: 10.1093/genetics/70.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Palumbi, S. R., Martin, A. P., Romano, S., Macmillan, W. O. & Stice, L. (1991) Special Publ. Dept. Zoology (Univ. of Hawaii, Honolulu), No. 13, p. 271.
- 20.Cabot E L, Beckenbach A T. Comput Appl Biosci. 1989;5:233–234. doi: 10.1093/bioinformatics/5.3.233. [DOI] [PubMed] [Google Scholar]
- 21.Kumar S, Tamura K, Nei M. Comput Appl Biosci. 1994;10:189–191. doi: 10.1093/bioinformatics/10.2.189. [DOI] [PubMed] [Google Scholar]
- 22.Rozas J, Rozas R. Bioinformatics. 1999;15:174–175. doi: 10.1093/bioinformatics/15.2.174. [DOI] [PubMed] [Google Scholar]
- 23.Sokal R R, Rohlf F J. Biometry: The Principles and Practice of Statistics in Biological Research. New York: Freeman; 1995. [Google Scholar]
- 24.Comeron J M, Kreitman M, Aguadé M. Genetics. 1999;151:239–249. doi: 10.1093/genetics/151.1.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andolfatto P, Przeworski M. Genetics. 2000;156:257–268. doi: 10.1093/genetics/156.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hudson R R. Genet Res. 1987;50:245–250. doi: 10.1017/s0016672300023776. [DOI] [PubMed] [Google Scholar]
- 27.Chovnick A, Gelbart W, McCarron M. Cell. 1977;11:1–10. doi: 10.1016/0092-8674(77)90312-9. [DOI] [PubMed] [Google Scholar]
- 28.Andolfatto P, Wall J D, Kreitman M. Genetics. 1999;153:1297–1311. doi: 10.1093/genetics/153.3.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nei M, Li W H. Proc Natl Acad Sci USA. 1979;76:5269–5273. doi: 10.1073/pnas.76.10.5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moriyama E N, Powell J R. Mol Biol Evol. 1996;13:261–277. doi: 10.1093/oxfordjournals.molbev.a025563. [DOI] [PubMed] [Google Scholar]
- 31.Watterson G A. Theor Popul Biol. 1975;7:256–276. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- 32.Hudson R R, Kreitman M, Aguadé M. Genetics. 1987;116:153–159. doi: 10.1093/genetics/116.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tajima F. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fu Y X, Li W H. Genetics. 1993;133:693–709. doi: 10.1093/genetics/133.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kreitman M, Hudson R R. Genetics. 1991;127:565–582. doi: 10.1093/genetics/127.3.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.David J R, Capy P. Trends Genet. 1988;4:106–111. doi: 10.1016/0168-9525(88)90098-4. [DOI] [PubMed] [Google Scholar]
- 37.Stapleton M, Liao G, Brokstein P, Hong L, Carninci P, Shiraki T, Hayashizaki Y, Champe M, Pacleb J, Wan K, et al. Genome Res. 2002;12:1294–1300. doi: 10.1101/gr.269102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ayala F J, Balakirev E S, Sáez A G. Gene. 2002;300:19–29. doi: 10.1016/s0378-1119(02)00852-1. [DOI] [PubMed] [Google Scholar]
- 39.Schlötterer C, Vogl C, Tautz D. Genetics. 1997;146:309–320. doi: 10.1093/genetics/146.1.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hasson E, Eanes W F. Genetics. 1996;144:1565–1575. doi: 10.1093/genetics/144.4.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Begun D J, Aquadro C F. Nature. 1993;365:548–550. doi: 10.1038/365548a0. [DOI] [PubMed] [Google Scholar]