

Abstract
Synaptic vesicle fusion is catalyzed by assembly of synaptic SNARE complexes, and is regulated by the synaptic vesicle GTP-binding protein Rab3 that binds to RIM and to rabphilin. RIM is a known physiological regulator of fusion, but the role of rabphilin remains obscure. We now show that rabphilin regulates recovery of synaptic vesicles from use-dependent depression, probably by a direct interaction with the SNARE protein SNAP-25. Deletion of rabphilin dramatically accelerates recovery of depressed synaptic responses; this phenotype is rescued by viral expression of wild-type rabphilin, but not of mutant rabphilin lacking the second rabphilin C2 domain that binds to SNAP-25. Moreover, deletion of rabphilin also increases the size of synaptic responses in synapses lacking the vesicular SNARE protein synaptobrevin in which synaptic responses are severely depressed. Our data suggest that binding of rabphilin to SNAP-25 regulates exocytosis of synaptic vesicles after the readily releasable pool has either been physiologically exhausted by use-dependent depression, or has been artificially depleted by deletion of synaptobrevin.
Keywords: exocytosis, neurotransmitter release, synapse, synaptic plasticity, synaptobrevin
Introduction
Presynaptic terminals release neurotransmitters by synaptic vesicle exocytosis. Membrane fusion during exocytosis is catalyzed by the SNARE proteins synaptobrevin/VAMP, SNAP-25, and syntaxin 1, and by the SM-protein Munc18-1 (reviewed in Lin and Scheller, 2000; Jahn et al, 2003). However, only Munc18-1 but not synaptobrevin and SNAP-25 are absolutely required for synaptic membrane fusion. In synapses lacking synaptobrevin or SNAP-25, synaptic vesicle fusion still occurs spontaneously, and can still be evoked by action potentials or hypertonic sucrose, although at a reduced level (Schoch et al, 2001; Washbourne et al, 2002). In contrast, synapses lacking Munc18-1 exhibit no spontaneous or evoked release (Verhage et al, 2000). These findings suggest that other SNARE proteins are redundant with synaptobrevin and SNAP-25, and/or that fusion can occur without SNARE proteins.
Synaptic vesicles contain a family of GTP-binding proteins called Rab3A, B, C, and D that perform redundant functions in neurotransmitter release (Schlüter et al, 2004; reviewed in Darchen and Goud, 2000). Rab3 proteins are thought to act via two conserved GTP-dependent effector proteins: a cytosolic protein called rabphilin (Shirataki et al, 1993; Li et al, 1994), and active zone proteins called α-RIMs (Wang et al, 1997, 2000). Rabphilin belongs to a large protein family that includes granulophilin/exophilin 2/Slp4, Slp3/exophilin 6, and Slp5/exophilin 9 (reviewed in Izumi et al, 2003; Fukuda, 2005). These proteins are characterized by an N-terminal zinc-finger sequence that interacts with Rab3 and/or Rab27 (another exocytotic Rab protein), and two C-terminal C2 domains that at least in rabphilin bind Ca2+. Nerve terminals contain two α-RIMs (RIM1α and 2α; Wang and Südhof, 2003) that contain an N-terminal Rab3-binding zinc-finger sequence and two C-terminal C2 domains similar to rabphilin. However, the C2 domains of rabphilin bind Ca2+ (Ubach et al, 1999), whereas those of RIMs do not (Dai et al, 2005).
Genetic analyses in mice and Caenorhabditis elegans have provided insights into the functions of rab3 and α-RIMs, but were relatively uninformative for rabphilin. In mice, deletion of Rab3A alone caused a significant synaptic phenotype (Geppert et al, 1994, 1997; Castillo et al, 1997), while deletion of all four Rab3 isoforms is lethal (Schlüter et al, 2004). Consistent with a role in release, deletion of Rab3 in C. elegans (where there is only a single isoform) produced a synaptic phenotype (Nonet et al, 1997). In both mice and C. elegans, deletion of RIM1α severely impaired synaptic vesicle exocytosis due to a postdocking defect (Koushika et al, 2001; Schoch et al, 2002). In addition, the RIM1α deletion caused large changes in synaptic plasticity in mice (Castillo et al, 2002; Schoch et al, 2002). In contrast to deletions of rab3 and RIM1α, deletions of rabphilin produced no detectable effect in mice (Schlüter et al, 1999), and only a mild phenotype in C. elegans that was, however, dramatically enhanced by concurrent mutations in a synaptic SNARE protein (Staunton et al, 2001). The C. elegans data indicated that rabphilin, although not essential by itself, may contribute to SNARE function. However, the significance of this observation for neurotransmitter release remained unclear.
In the present study, we have examined the possibility that rabphilin may perform a function in exocytosis that is related to SNARE proteins, but would not become apparent in standard screens for a phenotype in rabphilin knockout (KO) mice. Our data demonstrate that in wild-type synapses, deletion of rabphilin dramatically increases release after the readily releasable pool (RRP) of vesicles has been exhausted, whereas in synaptobrevin-deficient synapses, deletion of rabphilin enhances all Ca2+-triggered release, presumably because the synaptobrevin deletion creates a continuous state of depletion of the RRP. We find that the ‘bottom', Ca2+-independent surface of the C2B domain of rabphilin directly binds to the SNARE protein SNAP-25, thus providing a mechanistic explanation for the action of rabphilin observed in our physiological experiments. These data indicate that rabphilin is a regulator of neurotransmitter release that functions in conjunction with plasma membrane SNARE proteins when the RRP has been depleted.
Results
Interaction of the SNARE complex with Rab3A via rabphilin
Using GST-pulldowns, we first tested whether rat brain rabphilin binds to SNARE proteins. We found that GST-SNAP-25 efficiently captured rabphilin, whereas GST-synaptobrevin and GST-syntaxin did not (Figure 1A). In contrast, GST-syntaxin bound to Munc18-1, its major brain binding partner (Hata et al, 1993), whereas GST-synaptobrevin and GST-SNAP-25 did not. The binding of rabphilin to SNAP-25, and of Munc18-1 to syntaxin, was specific as synaptophysin 1 did not bind to any SNARE protein. All three GST-SNARE proteins similarly captured complexins (Figure 1A), suggesting that all three GST-SNARE proteins nucleated the assembly of SNARE complexes because complexins only bind to assembled SNARE complexes (McMahon et al, 1995). Although rabphilin thus does not appear to bind to fully assembled SNARE complexes, it does bind to SNAP-25/syntaxin heterodimers (Supplementary Figure 1).
Figure 1.
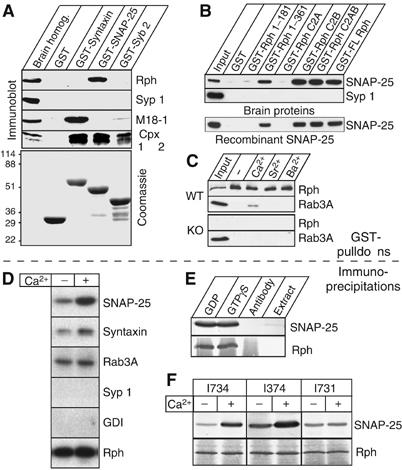
Binding of rabphilin to SNAP-25. (A) Pulldowns of rat brain proteins with immobilized GST, GST-syntaxin, GST-SNAP-25, and GST-synaptobrevin (in 50 mM HEPES pH 7.2, 0.1 M NaCl, 4 mM EGTA, 2 mM MgCl2, 1 mM DTT, protease inhibitor cocktail (Roche), and 0.5% Triton X-100). Bound proteins were analyzed by immunoblotting (top panels) for rabphilin (Rph), synaptophysin 1 (Syp 1), Munc18-1 (M18-1) and complexin 1 and 2 (Cpx 1 & 2). Bottom panel shows a Coomassie-blue stained gel of the GST-proteins to illustrate that similar amounts of protein were employed. (B) Pulldowns of rat brain proteins (upper panel) or recombinant SNAP-25 (lower panel) with immobilized GST-fusion proteins containing full-length rabphilin (GST-FL Rph) or rabphilin fragments (GST-Rph1-181 or -Rph1-361=residues 1–181 or 1–361 of rabphilin; GST-Rph C2A, C2B, or C2AB=C2A-, C2B-, or double C2A/B-domain fragment of rabphilin; GST=GST only control). Bound proteins were analyzed by immunoblotting for SNAP-25 and for synaptophysin 1 (Syp 1; used as a negative control). (C) Effects of divalent cations on the interaction of rabphilin with SNAP-25. Solubilized synaptic vesicle proteins from wild type (WT) and rabphilin KO mice (KO) were bound to GST-SNAP-25 in the presence of 1 mM of the indicated divalent cations; bound proteins were analyzed by immunoblotting. (D) Immunoprecipitations of rabphilin from detergent-solubilized synaptosomes with a polyclonal antibody to the N-terminus of rabphilin (I734) in the presence or absence of 1 mM Ca2+. Bound proteins were analyzed by immunoblotting with monoclonal antibodies to SNAP-25, syntaxin 1, Rab3A, synaptophysin 1 (Syp 1), GDI, or rabphilin (Rph). (E, F) Analysis of SNAP-25 co-immunoprecipitations with rabphilin as a function of GDP versus GTPγS (E; antibody=‘antibody only' control, extract=control with the detergent-solubilized synaptosome extract and protein G-Sepharose only), or as a function of independent rabphilin antibodies (I734 and I374=antibodies to the rabphilin N-terminal half; I731=antibody to the rabphilin C-terminal half) in the presence or absence of Ca2+ (F). In (E) and (F), the smear below the rabphilin band is caused by the IgG from the immunoprecipitations.
Does SNAP-25 bind to rabphilin directly or indirectly? To investigate this, we compared the binding of native brain SNAP-25 and of recombinant SNAP-25 to immobilized GST-fusion proteins of full-length rabphilin and of fragments of rabphilin, thereby reversing the orientation of the initial GST-pulldown experiments. We found that brain and recombinant SNAP-25 were equally efficiently retained by GST-rabphilins (Figure 1B), indicating a direct interaction. Both the N-terminal half of rabphilin that includes its Zn2+-finger domain and phosphorylation sites (Fykse et al, 1995; Stahl et al, 1996), and its C-terminal C2B domain captured SNAP-25 (Figure 1B). The SNAP-25 binding of the N-terminal half of rabphilin was not investigated further because this region in our hands is often subject to nonspecific interactions, possibly because part of it is natively unfolded. The binding of the rabphilin C2B domain was specific because the synaptotagmin 1 C2B domain, which is structurally similar to the rabphilin C2B domain, was unable to pull down SNAP-25 (data not shown).
Since the rabphilin C2B domain is a Ca2+-binding domain (Ubach et al, 1999), we next tested the effects of various divalent cations on the rabphilin/SNAP-25 interaction (Figure 1C). Divalent cations had no effect on the ability of GST-SNAP-25 to pull down rabphilin from wild-type mouse brain homogenates (Figure 1C). As a negative control, we tested brain homogenates from rabphilin KO mice (Schlüter et al, 1999), but found no binding (Figure 1C). Moreover, we observed no significant retention of Rab3A, also used as a negative control because these experiments were carried out in the absence of GTP.
We next examined whether endogenous brain rabphilin and SNAP-25 interact with each other. We immunoprecipitated rabphilin from rat brain homogenates under conditions that favor SNARE complex assembly, and probed for co-immunoprecipitated proteins by immunoblotting. We found that SNAP-25 and syntaxin 1 were co-immunoprecipitated with rabphilin, whereas synaptophysin and GDI were not (Figure 1D). The co-immunoprecipitation of SNAP-25 with rabphilin was independent of GTP (Figure 1E). Since the GST-pulldowns indicated that the C2B domain of rabphilin is, at least in part, responsible for the binding of SNAP-25, we examined the relative ability of antibodies directed to the N-terminal half of rabphilin (I734 and I374) or to its C-terminal C2 domains (I731) to co-immunoprecipitate SNAP-25 with rabphilin. We found that although the three antibodies used immunoprecipitated similar amounts of rabphilin, the C-terminal antibody was less potent than the N-terminal antibodies in co-immunoprecipitating SNAP-25 (Figure 1F), consistent with a binding of SNAP-25 to the C-terminal C2B domain of rabphilin. A modest increase of SNAP-25 binding in the presence of Ca2+ was observed, possibly because Ca2+ stabilizes the C2 domains of rabphilin.
Most C2 domains form Ca2+-dependent phospholipid complexes (reviewed in Nalefski and Falke, 1996; Rizo and Südhof, 1998). Previous studies showed that the rabphilin C2B domain is an effective Ca2+-binding domain (Ubach et al, 1999), but no general Ca2+-dependent phospholipid binding was detected (Li et al, 1994; Chung et al, 1998). To determine whether the rabphilin C2B domain forms Ca2+-dependent phospholipid complexes, we employed a solution binding assay (Fernandez et al, 2001). This assay was required because the standard GST-pulldown assay for phospholipid binding does not reliably detect phospholipid binding to all C2 domains (Fernandez et al, 2001). We observed no apparent Ca2+-dependent binding of the C2A domain to phospholipids. The isolated C2B domain, however, bound to the liposomes with a high apparent Ca2+ affinity (1–2 μM); the same Ca2+-dependent binding was observed for the double C2A/B domain fragment (Supplementary Figure 2). The high apparent Ca2+ affinity of the rabphilin C2B domain/phospholipid complex corresponds well to the high intrinsic Ca2+ affinity of the C2B domain (Ubach et al, 1999), suggesting that the C2B domain of rabphilin, similar to the C2A domain of synaptotagmin 1, can form both Ca2+-dependent phospholipid complexes and Ca2+-independent complexes with a SNARE protein.
The bottom surface of the rabphilin C2B domain with the α-helix binds to SNAP-25
To investigate the nature and stoichiometry of the rabphilin/SNAP-25 complex, we used NMR spectroscopy as previously employed in determining the structure of the rabphilin C2B domain (Ubach et al, 1999). Using recombinant, 15N-labeled protein, we recorded 1H–15N heteronuclear single quantum coherence (HSQC) spectra from the C2B domain in the presence and absence of unlabeled SNAP-25, with or without a saturating concentration of Ca2+ (Figure 2). In 1H–15N HSQC spectra of 15N-labeled proteins, each nonproline residue is represented by a single cross-peak, whose position reflects the microenvironment of the corresponding amide group. Thus, changes in 1H–15N spectra of an 15N-labeled protein provide a sensitive method to monitor binding of an unlabeled protein and to map binding sites.
Figure 2.
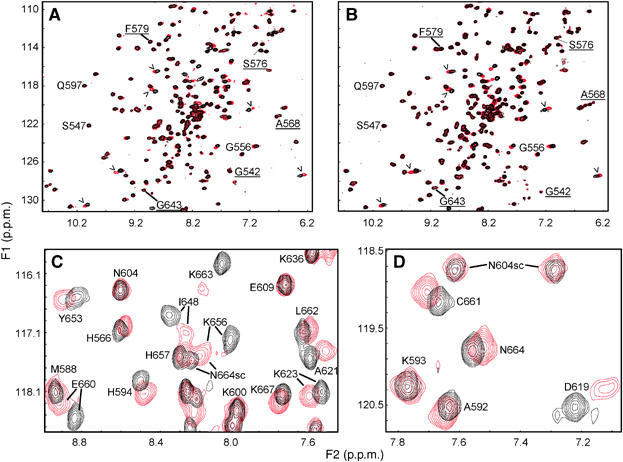
Characterization of the rabphilin C2B domain binding to SNAP-25 by NMR spectroscopy. (A, B) 1H-15N HSQC spectra of the 15N-labeled C2B domain from rabphilin (75 μM) in the absence (black) or presence (red) of unlabeled SNAP-25 (75 μM). Spectra were acquired in 0.2 mM EDTA (A) or 10 mM Ca2+ (B). To facilitate comparison, a few cross-peaks that do not change with SNAP-25 are identified (underlined if the peaks are not altered by Ca2+, and not underlined if they shift with Ca2+). The subset of peaks that move after addition of SNAP-25 are marked by arrowheads; these peaks exhibit analogous changes in the presence and absence of Ca2+. (C, D) Expansions of the 1H-15N HSQC spectra from the rabphilin C2B domain (75 μM) recorded in 10 mM Ca2+ without SNAP-25 (black) or with 150 μM SNAP-25 (red). Note that some peaks shift substantially with SNAP-25 while others do not. Residue assignments are indicated for some of the cross-peaks.
1H–15N HSQC spectra of the 15N-labeled rabphilin C2B domain (75 μM) were acquired in the absence (Figure 2A) and presence (Figure 2B) of Ca2+, without (black contours) or with (red contours) an equimolar concentration of SNAP-25 (75 μM). SNAP-25 caused general line broadening of cross-peaks in the HSQC spectra of the C2B domain, arising from the larger size of the SNAP-25/C2B-domain complex. In addition, SNAP-25 caused shifts in a small subset of the 1H–15N HSQC cross-peaks, induced by binding of SNAP-25 to the corresponding residues. Line broadening of C2B-domain cross-peaks occurred independently of the presence or absence of Ca2+. The broadening was less severe than would be expected for a 25 kDa protein, probably because free SNAP-25 is largely unfolded, and SNAP-25 bound to the rabphilin C2B domain is only partially folded at the site of interaction.
The shifts in a subset of 1H–15N HSQC cross-peaks of the rabphilin C2B domain upon SNAP-25 binding can be better observed in the expansions shown in Figures 2C and D (acquired at a higher concentration of SNAP-25 (150 μM) to ensure saturation of the interaction). The assignment of the 1H–15N HSQC cross-peaks of the rabphilin C2B domain (Ubach et al, 1999) made it possible to identify the residues corresponding to the shifted cross-peaks; their positions in the structure of the C2B domain are shown in Figure 3. C2 domains are composed of stable β-sandwiches with flexible loops emerging at the ‘top' (the side that binds Ca2+) and the ‘bottom' (the side that does not bind Ca2+; Rizo and Südhof, 1998). Strikingly, SNAP-25 binding exclusively shifted cross-peaks corresponding to residues from the ‘bottom' surface of the rabphilin C2B domain, which does not bind Ca2+. The cross-peak shifts induced by SNAP-25 were concentrated in the long bottom α-helix that is unique to C2B domains of rabphilins and synaptotagmins (Ubach et al, 1999; Fernandez et al, 2001), and were identical in the presence and absence of Ca2+ (Figures 2A and B).
Figure 3.
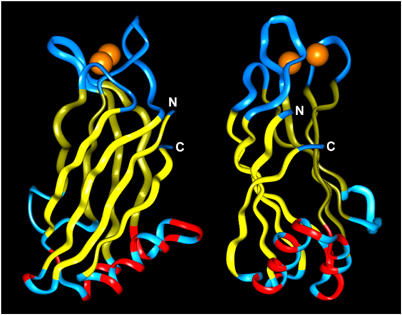
Ribbon diagram of the C2B domain of rabphilin: identification of the SNAP-25 binding site defined by chemical shift changes. The figure displays views of the rabphilin C2B domain with a 90° rotation around the vertical axis. The Ca2+-binding loops are shown on top with two Ca2+ ions bound (orange; Ubach et al, 1999). β-Strands are displayed in yellow. Residues that changed upon SNAP-25 binding are shown in red. Note that these changes are restricted to the bottom α-helix and the adjacent bottom loop. N- and C-termini are indicated by white ‘N' and ‘C'.
Several conclusions can be drawn from the NMR results. First, SNAP-25 appears to bind to the rabphilin C2B domain in a stoichiometric 1:1 complex. This conclusion is based on the significant cross-peak shifts observed when the two proteins were mixed in a 1:1 ratio, and on the fact that the magnitude of these shifts increased only slightly when SNAP-25 was doubled (Figure 2 and data not shown). Second, analogous cross-peak shifts were observed in the presence and absence of Ca2+, confirming that the interaction of the C2B domain with SNAP-25 does not require Ca2+. Third, all of the shifted cross-peaks are from residues residing on the bottom surface of the rabphilin C2B domain, mostly in the unique α-helix that is characteristic of C2B domains (Figure 3).
Deletion of rabphilin increases recovery from use-dependent depression
The binding of rabphilin to SNAP-25 suggests a function for rabphilin in exocytosis, but extensive previous analyses of rabphilin KO mice failed to detect a phenotype (Schlüter et al, 1999). These analyses, however, might have missed a phenotype that manifests only when synaptic transmission becomes dependent on SNARE complex assembly. One model of docking and priming of vesicles at the synapse suggests that vesicles dock in a SNARE-independent manner, and are subsequently primed to become Ca2+-responsive by a SNARE-dependent mechanism, allowing Ca2+ to trigger fusion pore opening by binding to synaptotagmin (Südhof, 1995). According to this concept, SNARE-complex assembly becomes rate limiting for the recovery of the RRP of primed vesicles at a synapse after this pool has been depleted by a high-frequency stimulus train. Therefore, we tested whether deletion of rabphilin alters the rate of recovery of the EPSC after use-dependent depression.
We cultured embryonic hippocampal neurons from littermate wild type and rabphilin KO mice at a high density at which these cultures form extensive synaptic networks (Supplementary Figure 3), and monitored postsynaptic responses to field stimulation by whole-cell recordings. We first measured steady-state EPSCs during low-frequency stimulation (60 stimuli at 0.4 Hz), then applied a high-frequency stimulus train that induced massive synaptic depression (1200 stimuli at 20 Hz), and finally determined the rate of recovery of the EPSCs during low-frequency stimulation (60 stimuli at 0.4 Hz; Figure 4A). As described before (Schlüter et al, 1999), we observed no difference between rabphilin-deficient and control neurons in the size of the initial EPSCs (Figure 4B) or the extent of use-dependent depression (Figure 4D). However, deletion of rabphilin dramatically accelerated the recovery of EPSCs after termination of the high-frequency stimulus train (Figure 4E).
Figure 4.
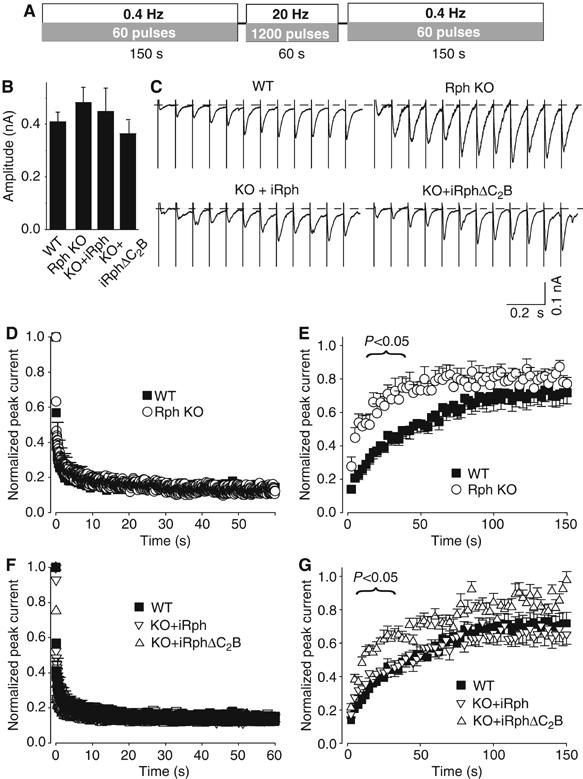
Effect of the deletion of rabphilin on the recovery of synaptic responses from use-dependent depression. (A) Experimental design. Cultured neurons from wild type or rabphilin KO mice were either analyzed without further manipulations (WT and Rph KO), or rabphilin-deficient neurons were infected with lentivirus expressing full-length rabphilin (iRph) or rabphilin lacking the C2B domain (iRΔC2B). Neurons were stimulated at 0.4 Hz for 150 s to determine the initial EPSC amplitude, then at 20 Hz for 60 s to cause use-dependent depression, and finally again at 0.4 Hz for 150 s to monitor recovery of synaptic responses. (B) Bar graph of initial EPSC amplitudes. (C) Representative traces during synaptic recovery (only the first 100 ms of the first 12 pulses are shown for clarity). (D, E) Depression of synaptic responses in WT and rabphilin KO neurons during 20 Hz stimulation (D; peak currents were normalized to the first response), and recovery of synaptic responses from synaptic depression during 0.4 Hz stimulation (E; normalized to the average amplitude of each cell during the initial 0.4 Hz stimulation). Rabphilin-deficient synapses recovered significantly faster up to the 21st pulse (P<0.05). (F, G) Depression of synaptic responses during 20 Hz stimulation in WT neurons and in rabphilin KO neurons expressing full-length iRph or C-terminally truncated iRph (iRΔC2B) (F), and subsequent recovery of synaptic responses in these neurons (G). For (B) and (D–G), data shown are means±s.e.m. (number of neurons analyzed in four independent cultures: WT, n=14; Rph KO, n=11; iRph, n=12; iRphΔC2B, n=13).
The recovery time course was fitted with a two-exponential function 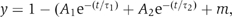 where A1 and A2 and τ1 and τ2 are the amplitudes and time constants of the first and second component, respectively, and m is an offset value to correct for incomplete recovery of the EPSC at the end of the monitoring period). Deletion of rabphilin produced a >2-fold increase in the amplitude of the first component, and a corresponding decrease in the second component of recovery (WT: A1, 0.219±0.038; A2, 0.547±0.066 (n=14 cells/3 cultures); KO: A1, 0.587±0.068; A2, 0.331±0.059 (n=11 cells/3 cultures); P<0.0001; also resulting in different offset values; WT: m=0.241±0.051; KO: m=0.135±0.041). The time constants of the two recovery components, however, were not significantly altered (WT: τ1, 4.5±0.6 s; τ2, 57.0±4.1 s; KO: τ1, 3.5±0.6 s; τ2, 55.4±5.3 s (n's as above); P>0.25). Thus, deletion of rabphilin shifts vesicles from a slowly recovering into a swiftly recovering mode, suggesting that rabphilin normally functions to control re-priming of vesicles after extensive synaptic activity.
where A1 and A2 and τ1 and τ2 are the amplitudes and time constants of the first and second component, respectively, and m is an offset value to correct for incomplete recovery of the EPSC at the end of the monitoring period). Deletion of rabphilin produced a >2-fold increase in the amplitude of the first component, and a corresponding decrease in the second component of recovery (WT: A1, 0.219±0.038; A2, 0.547±0.066 (n=14 cells/3 cultures); KO: A1, 0.587±0.068; A2, 0.331±0.059 (n=11 cells/3 cultures); P<0.0001; also resulting in different offset values; WT: m=0.241±0.051; KO: m=0.135±0.041). The time constants of the two recovery components, however, were not significantly altered (WT: τ1, 4.5±0.6 s; τ2, 57.0±4.1 s; KO: τ1, 3.5±0.6 s; τ2, 55.4±5.3 s (n's as above); P>0.25). Thus, deletion of rabphilin shifts vesicles from a slowly recovering into a swiftly recovering mode, suggesting that rabphilin normally functions to control re-priming of vesicles after extensive synaptic activity.
Is the effect of the rabphilin deletion on recovery due to its interaction with SNAP-25? As a first test of this question, we examined whether the rabphilin KO phenotype can be rescued by expression of wild-type rabphilin, or of truncated rabphilin that lacks the C2B domain and thus does not bind to SNAP-25 via its C2B domain. Both proteins were efficiently expressed in the cultured neurons with recombinant lentiviruses (Supplementary Figure 4). We found that wild-type rabphilin, expressed with a lentivirus, reversed the rabphilin KO phenotype (Figures 4B, C, F and G; numerical values: KO with WT lentivirus, τ1, 3.8±0.6 s; τ2, 65.7±5.8 s; A1, 0.294±0.035; A2, 0.466±0.059; m=0.241±0.051 (n=12 cells/3 cultures)). Among others, this result shows that the increase in recovery kinetics in the rabphilin KO is not a developmental abnormality since it can be rescued by expression of rabphilin in postmitotic neurons. In contrast to full-length rabphilin, truncated rabphilin was unable to rescue the rabphilin KO phenotype (Figure 4G; KO with lentivirus expressing truncated rabphilin, τ1, 4.0±0.5 s; τ2, 62.5±4.6 s; A1, 0.495±0.046; A2, 0.441±0.037 (n=13 cells/3 cultures); P=0.002; m=0.091±0.025; P=0.011 (P-values are for the comparison of KO cultures infected with wild type and C-terminally truncated rabphilin expressing lentivirus)).
Effect of rabphilin on spontaneous and sucrose-induced synaptic activity in synaptobrevin-deficient synapses
To test by a different approach whether rabphilin functionally interacts with SNARE proteins, we made use of previously generated synaptobrevin KO mice (Schoch et al, 2001; Deák et al, 2004). We performed electrophysiological analyses of embryonic hippocampal neurons cultured from littermate mice that were generated in two breeding schemes: wild type and synaptobrevin KO mice obtained in crosses of heterozygous synaptobrevin 2 KO mice, and rabphilin KO mice that either lack or contain synaptobrevin 2 obtained in crosses of mice that were homozygous for the rabphilin KO and heterozygous for the synaptobrevin KO. The overall idea behind these experiments was that deletion of synaptobrevin should create a state analogous to that of use-dependent depression. In both cases, the RRP is depleted and assembled SNARE complexes are largely absent; thus, if rabphilin normally inhibits Ca2+-dependent exocytosis arising from such a state, its deletion may also amplify the remaining exocytosis present in synaptobrevin-deficient neurons.
We first examined the properties of spontaneous synaptic events (‘minis'; Figure 5A). As reported previously, synaptobrevin-deficient neurons exhibited a ∼10 fold decrease in the frequency of spontaneous events (Schoch et al, 2001), whereas rabphilin-deficient neurons displayed no significant change (Schlüter et al, 1999). In the rabphilin/synaptobrevin double KO neurons, mini frequency was decreased even further than in synaptobrevin KO neurons (Figure 5B). The amplitude of minis was slightly larger in synaptobrevin-deficient neurons than in wild-type neurons (Figure 5C), consistent with the increase in vesicle size in synaptobrevin-deficient neurons (Deák et al, 2004). Rise times tended to be longer for synaptobrevin-deficient synapses, although the difference only reached significance for the comparison between synaptobrevin/rabphilin double KO mice versus rabphilin single KO mice (Figure 5D).
Figure 5.
Spontaneous synaptic responses (‘minis') and hypertonic sucrose-evoked synaptic responses in cultured hippocampal neurons. (A) Representative traces of spontaneous synaptic events monitored in 1 μM tetrodotoxin in neurons from wild type (WT), synaptobrevin KO (Syb2 KO), rabphilin KO (Rph KO), and synaptobrevin/rabphilin double KO mice (SR DKO). (B) Frequency of spontaneous synaptic events. Only the statistically significant difference between synaptobrevin-deficient and synaptobrevin/rabphilin-double deficient synapses is marked. (C) Amplitudes of spontaneous synaptic events. Synaptobrevin-deficient neurons exhibit larger amplitudes than synaptobrevin-containing neurons, but this difference is statistically significant only for the indicated comparison. (D) Rise times (measured as time required for reaching the half-maximum) of spontaneous synaptic events. Double KO neurons (SR DKO) exhibit significantly slower rise times than rabphilin single KO neurons. In (B–D), all data shown are means±s.e.m. (n=19 SR DKO; 10 Syb2 KO; 8 WT; 8 Rph KO). (E) Representative traces of synaptic responses evoked with hypertonic sucrose (0.5 Osm). (F) Summary graph of synaptic responses to sucrose monitored in WT and various single- and double-KO neurons. Responses are calculated as the cumulative charge transfer integrated over a 2 s interval at the peak of the response (means±s.e.m.; n=16 for SR DKO, n=12 for rph KO, n=6 for syb2 KO and n=4 for WT).
We next assessed the size of the RRP by measuring synaptic responses to hypertonic sucrose (Rosenmund and Stevens, 1996; Figure 5E). KO of synaptobrevin caused a ∼10-fold decrease in sucrose responses. Additional deletion of rabphilin did not change the average size of sucrose-induced responses; similarly, the single KO of rabphilin also did not alter sucrose responses (Figure 5F). The same result was obtained when the entire time period of the sucrose response was analyzed instead of the acute phase (data not shown). Thus, based on spontaneous and on sucrose-induced release, additional deletion of rabphilin on top of the synaptobrevin KO does not generally increase the releasability of synaptic vesicles.
Evoked synaptic responses in rabphilin/synaptobrevin double KO neurons
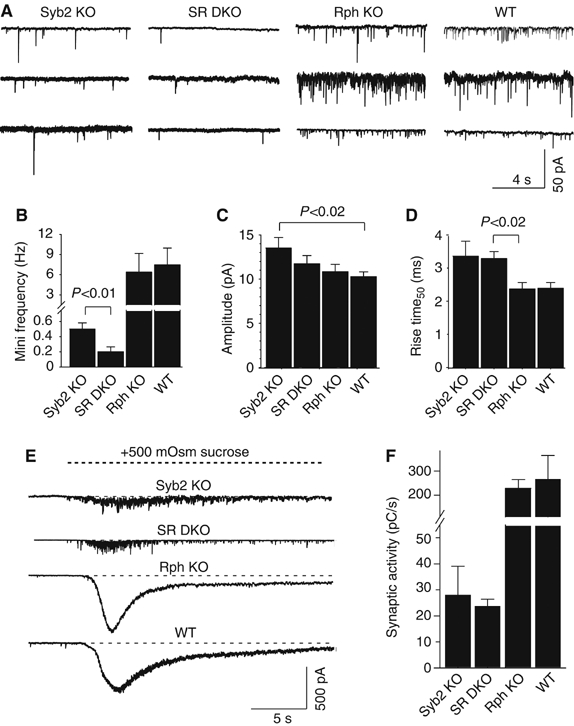
We next examined synaptic responses to action potentials induced by 1 Hz field stimulation (Figure 6A). Synaptobrevin-deficient neurons responded only to 34.2±7.8% of action potentials (n=11), whereas wild type and rabphilin KO neurons responded to 100% of action potentials (Figures 6B and C). Upon deletion of rabphilin, response rates increased >2-fold in synaptobrevin-deficient neurons (to 78.5±7.8%; n=12), making synaptic events almost completely reliable. In addition, the rabphilin deletion enhanced the average amplitude of synaptic responses ∼2-fold in synaptobrevin-deficient but not wild-type neurons (synaptobrevin KO=32.7±7.9 pA (n=11); double KO=76.9±22.6 pA (n=12); wild type=757.8±145.0 pA (n=4); rabphilin KO=709.8±140.9 pA (n=7); Figure 6D). In these experiments, neurons likely receive hundreds of synaptic inputs, with each stimulus eliciting release at a subset of inputs depending on their release probability. Thus, the observed low response rate in synaptobrevin-deficient neurons combined with the small amplitude of responses means that in these synapses, the actual release probability is very low, and that the additional deletion of rabphilin in synaptobrevin-deficient neurons boosts this release probability much more than the ∼2-fold increase observed for total responses.
Figure 6.
Synaptic responses to 1 Hz stimulation. (A) Representative traces from whole-cell recordings during 1 Hz field stimulations. Only the first 200 ms of each response is shown for clarity. (B) Diary plots of responses recorded from synaptobrevin KO neurons (Syb2 KO) and double synaptobrevin/ rabphilin KO neurons (SR DKO). (C) Fraction of successful stimuli during 60 pulses at 1 Hz. (D) Amplitudes of evoked responses. Data in (C, D) are means±s.e.m. (n=11 Syb2 KO cells; 7 SR DKO cells; 7 Rph KO cells; 4 WT cells).
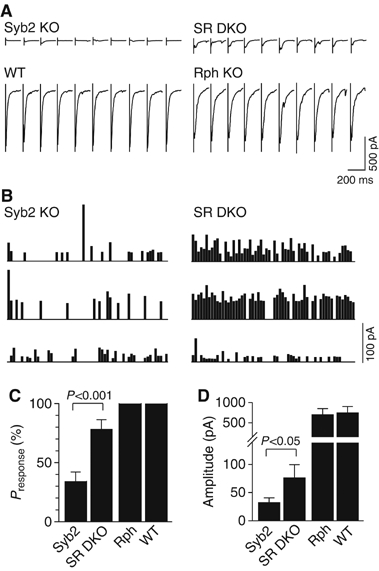
Finally, we investigated the responses of mutant synapses to 10 Hz stimulation. At this stimulation frequency, synaptobrevin-deficient synapses exhibit strong facilitation (Deák et al, 2004; see Figures 7A and B). Deletion of both rabphilin and synaptobrevin converted this facilitation into depression (Figures 7A–C), while deletion of rabphilin alone had no effect on the synaptic depression (Figure 4). These results provide independent confirmation of the conclusion (Figure 6) that deletion of rabphilin on the background of the synaptobrevin KO significantly increases the release probability. In parallel, we also measured the size of the recycling pool of synaptic vesicles in mutant synapses using fluorescent FM-dye staining and destaining (Deák et al, 2004). As described before, the pool size labeled by a single depolarization with 90 mM K+ was significantly decreased in synaptobrevin KO neurons compared to wild-type controls (∼3-fold; Schoch et al, 2001; Deák et al, 2004). Additional deletion of rabphilin significantly increased the pool size in synaptobrevin-deficient neurons (∼2 fold; Figure 7E), consistent with the increase in Ca2+-triggered release.
Figure 7.
Synaptic responses to 10 Hz stimulation. (A) Representative traces from whole-cell recordings obtained during 10 Hz stimulations of synaptobrevin KO neurons (Syb2 KO) and synaptobrevin/rabphilin double KO neurons (SR DKO). Responses to pulse # 1–10 and 191–200 are shown. (B, C) Diary plots of synaptic responses in three cells from synaptobrevin KO mice and synaptobrevin/rabphilin double KO mice during 10 Hz stimulation. (D) Normalized amplitudes of synaptic responses during 10 Hz stimulation from synaptobrevin KO neurons (Syb2 KO; n=9) and synaptobrevin/rabphilin double KO neurons (SR DKO; n=9). Synaptic responses were normalized to the average of the first five pulses in the train, and are shown as means±s.e.m. Only the difference between the synaptobrevin KO neurons and the three other genotypes is statistically significant (P<0.05). (E) Size of the recycling pools of synaptic vesicles measured by FM2-10 staining and destaining. FM2-10-loaded synaptic boutons were destained with 5 × 90 mM KCl/2 mM CaCl2 (n=12 (610 synapses) for SR DKO, n=10 (632 synapses) for Rph KO, n=10 (486 synapses) for syb2 KO, and n=9 (533 synapses) for WT in four independent cultures). Data shown are histograms of the distribution of fluorescent loss (in arbitrary units); the difference between synaptobrevin-deficient synapses containing or lacking rabphilin is statistically significant (P<0.01).
Discussion
Rabphilin is an abundant, evolutionarily conserved protein that interacts with Rab3 as a function of GTP (Shirataki et al, 1993; Li et al, 1994), binds Ca2+ directly via its C2 domains (Ubach et al, 1999), and together with Rab3 cycles on and off synaptic vesicles during exocytosis (Geppert et al, 1994; Stahl et al, 1996). Deletions of rabphilin in mice (Schlüter et al, 1999) or C. elegans (Staunton et al, 2001) failed to produce a major phenotype, although a genetic interaction with SNARE proteins was observed in C. elegans (Staunton et al, 2001). In the present study, we describe a physiological function for rabphilin in regulating neurotransmitter release, and offer a possible mechanistic explanation for this function. Our study reports three principal findings: (1) In vitro, the C2B domain of rabphilin binds to the SNARE protein SNAP-25 in a Ca2+-independent interaction that is mediated by the ‘bottom', Ca2+-independent surface of the C2B domain. (2) Deletion of rabphilin causes a dramatic increase in the rate at which synapses recover from use-dependent synaptic depression. (3) Deletion of rabphilin also enhances release in synapses lacking the SNARE protein synaptobrevin; in these synapses, a small amount of residual Ca2+-triggered release remains that is increased >2-fold upon the additional deletion of rabphilin. Besides providing physiological evidence for a function of rabphilin in regulating neurotransmitter release, these findings have—as described below—implications for our thinking about the mechanism of action of C2 domains and the control of catalysis of membrane fusion by SNARE proteins.
Interaction of rabphilin with SNAP-25
We observed a stoichiometric interaction of the rabphilin C2B domain with the SNARE protein SNAP-25 (Figures 1, 2 and 3). Our data expand a GST-pulldown study that was published while this paper was under submission (Tsuboi and Fukuda, 2005) by demonstrating with immunoprecipitations that endogenous rabphilin and SNAP-25 interact with each other, and by using NMR spectroscopy to identify the ‘bottom' surface of the rabphilin C2B domain as the binding site. The validity of the rabphilin/SNAP-25 complex is supported by three lines of evidence: (1) Pulldowns of rat brain proteins with immobilized GST-fusion proteins showed that SNAP-25 captures rabphilin, and rabphilin captures SNAP-25 (Figure 1). No other SNARE protein bound rabphilin. (2) A rabphilin/SNAP-25 complex was immunoprecipitated from rat brain homogenates with multiple antibodies, demonstrating that the endogenous proteins interact with each other (Figure 1). (3) The NMR experiments revealed that the complex of the C2B domain of rabphilin with SNAP-25 is stoichiometric (Figure 2), and involves a defined sequence in rabphilin (Figure 3).
The selective interaction of SNAP-25 with the conserved α-helix at the bottom of the rabphilin C2B domain represents the first known protein interaction mediated by the Ca2+-independent surface of a C2 domain, and provides a first binding activity for the unique α-helix found at the bottom of C2B domains. This finding suggests a general mechanism by which C2 domains could simultaneously perform multiple, Ca2+-dependent and Ca2+-independent binding reactions via binding sites at the top and bottom loops of the domain. The interaction of SNAP-25 with rabphilin suggests an explanation for the changes in exocytosis induced by introduction of high concentrations of rabphilin or rabphilin fragments into bovine chromaffin cells, PC12 cells, or squid synapses (Chung et al, 1995, 1998; Burns et al, 1998; Tsuboi and Fukuda, 2005). It seems likely that the large excess of rabphilin in these experiments interferes with normal SNARE function beyond the physiological role of rabphilin, as is often observed in dominant-negative interference experiments, providing an explanation for why these experiments lead to phenotypes that bear no resemblance to the physiological KO phenotype.
Phenotype of rabphilin-deficient synapses
We reanalyzed the rabphilin KO phenotype in cultured neurons, and confirmed previous results obtained by slice physiology that standard synaptic parameters were not impaired by deletion of rabphilin (Schlüter et al, 1999). Guided by the observed rabphilin/SNAP-25 interaction, we then tested whether deletion of rabphilin alters recovery of synaptic responses after the RRP has been depleted by high-frequency stimulation. We found that synaptic responses recovered much faster in the absence than in the presence of rabphilin. Curve fitting uncovered two recovery phases of synaptic responses in these experiments; deletion of rabphilin did not alter the kinetics of these phases, but produced a large shift (∼3-fold) from the slower to the faster phase. The effect of the rabphilin deletion was rescued by acute viral expression of full-length rabphilin, demonstrating that the phenotype was not due to a developmental abnormality or a homeostatic compensatory reaction. Rescue was not achieved with a C-terminally truncated rabphilin lacking the C2B domain, consistent with the notion that the action of rabphilin may depend, at least in part, on the interaction of the C2B domain with SNAP-25.
One interpretation of these experiments is that vesicles recover into the RRP by two separate reactions, and that rabphilin normally channels vesicles from the faster into the slower reaction. This premise is consistent with our finding that rabphilin binds to SNAP-25/syntaxin heterodimers, but not fully assembled SNARE complexes and may thus, at least transiently, interfere with cognate SNARE interactions (Figure 1 and Supplementary Figure 1). The efficacy and frequency dependence of neurotransmission during natural spike trains depends on the kinetics of depression as well as recovery (Zucker and Regehr, 2002). A possible network role for an inhibitory function of rabphilin could be to prevent rebound hyperexcitability after bursts of activity; that is, rabphilin may allow regulation of the kinetics of recovery without altering the frequency dependence of depression. For instance, when synapses are in a depressed state, a single action potential that follows a silent period may result in full excitability, thus causing network imbalance. Naturally, a role for rabphilin in controlling such excitability would be most powerful if this function itself could be regulated. Future experiments will have to test whether such regulation of rabphilin occurs, for example by Ca2+-binding to its C2 domains or by phosphorylation.
Mechanism of fusion in synaptobrevin-deficient synapses
At the synapse, the SNARE proteins synaptobrevin/VAMP, syntaxin 1, and SNAP-25 form the core fusion machinery that mediates neurotransmitter release (Jahn et al, 2003). It was thus surprising that deletions of synaptobrevin (Schoch et al, 2001) or SNAP-25 (Washbourne et al, 2002) did not abolish release. Specifically, in synaptobrevin-deficient neurons, hypertonic sucrose and action potentials still elicit ∼10 and ∼2–3% of wild-type release, respectively (Schoch et al, 2001), and high-frequency stimulation facilitates release (Deák et al, 2004). Moreover, although the size of the recycling vesicle pool labeled with FM-dyes by a single round of K+-depolarization is smaller in synaptobrevin-deficient than in wild-type synapses, the total size of the pool that is labeled with FM-dyes by repeated rounds of K+-depolarization is identical in synaptobrevin-deficient and wild-type synapses (Deák et al, 2004). Thus, synaptobrevin-deficient synapses contain the same total number of fusion-competent vesicles as wild-type synapses, but the fraction of fusion-competent vesicles that are primed is decreased ∼10-fold, and a smaller subset of the primed vesicles in mutant synapses than in wild-type synapses undergoes exocytosis in response to Ca2+.
We now find that deleting rabphilin strongly potentiates the residual Ca2+-triggered release in synaptobrevin-deficient synapses, but does not increase sucrose-induced release. The observed effect is not small: release is increased >2-fold (Figure 6), and synaptic facilitation during high-frequency stimulus trains is converted into depression (Figure 7). Our findings thus suggest that in synaptobrevin-deficient synapses, rabphilin normally suppresses the Ca2+-triggering of vesicles; reversal of this suppression by accumulating residual Ca2+ during repetitive stimulation may explain, at least in part, the facilitation of release observed in synaptobrevin-deficient synapses during repetitive stimulation (Deák et al, 2004). The overall effect agrees well with the acceleration of recovery from use-dependent depression by the deletion of rabphilin (Figure 4), because synapses after use-dependent depression or after deletion of synaptobrevin both lack an RRP produced by assembled SNARE complexes. This also explains why deletion of rabphilin produces no increase in release in standard rabphilin KO mice. Finally, an important implication of our observations is that the residual release in synaptobrevin-deficient synapses likely involves SNAP-25, in agreement with the promiscuous nature of SNARE protein interactions.
In contrast to the above observations on evoked neurotransmission, deleting rabphilin further decreased the rate of spontaneous fusion in the synaptobrevin-deficient synapses. The recent finding that spontaneous fusion events in part originate from a distinct set of vesicles (Sara et al, 2005) may help reconcile these contradictory results. For instance, if these two sets of vesicles normally compete for fusion due to a limited set of release sites, relieving inhibition imposed by rabphilin on evoked fusion may bring a competitive advantage and reduce the propensity of spontaneous fusion. This scenario is consistent with the normal rate of spontaneous fusion seen in rabphilin KOs where in the presence of synaptobrevin evoked fusion already possesses a significant advantage.
In summary, we suggest that rabphilin physiologically interacts with SNAP-25 in docked and primed vesicles, and that this interaction may inhibit the remaining Ca2+-triggered release in synaptobrevin-deficient synapses. These findings are consistent with the genetic interaction of rabphilin with SNARE proteins in C. elegans (Staunton et al, 2001), and the functional interaction of SNARE proteins with Rab3 in Aplysia (Johannes et al, 1996). An implication is that consistent with the rab3 KO phenotype (Schlüter et al, 2004), the rab3/rabphilin complex normally fine-tunes the transition of primed vesicles containing partially or fully assembled SNARE complexes to Ca2+-responsive vesicles. Regulation of this transition step likely is a set-point resulting in short-term synaptic plasticity, and rabphilin may contribute to this regulation during increased synaptic activity. Our data suggest a mechanism that may explain these observations, and provide molecular evidence for a physiological function of rabphilin as a regulator of the SNARE complex.
Materials and methods
Mouse breeding and hippocampal cultures
Synaptobrevin 2/rabphilin double KO mice were obtained from timed matings of mice that were heterozygous for the synaptobrevin KO (Schoch et al, 2001) and homozygous for the rabphilin KO (Schlüter et al, 1999), while synaptobrevin 2 and rabphilin single KO mice were generated from standard heterozygous matings. High-density cultures of hippocampal neurons were prepared on Matrigel coated 12 mm coverslips (∼3 coverslips/hippocampus) as described (Schoch et al, 2001), and used at 12–24 days in vitro.
Electrophysiology
Synaptic responses were monitored in pyramidal cells by whole-cell patch-clamp recordings using an Axopatch 200B amplifier and Clampex 8.0 software (Axon Instruments). Recordings were filtered at 2 kHz and sampled at 200 μs. The pipette internal solution included (in mM): 115 Cs-MeSO3, 10 CsCl, 5 NaCl, 0.1 CaCl2, 10 HEPES, 4 Cs-BAPTA, 20 TEA-Cl, 4 Mg-ATP, 0.3 mM Na2-guanosine-triphosphate, and 10 lidocaine N-ethyl-bromide, pH 7.35 (300 mOsm). A hypertonic solution, prepared by addition of 500 mM sucrose to the Tyrode solution, was applied to proximal dendrites. Field stimulation was achieved through parallel platinum electrodes immersed into the perfusion chamber delivering 24 mA pulses of 1 ms.
Plasmid constructions and protein expression
Recombinant GST-fusion proteins were purified on glutathione agarose as unlabeled or uniformly 15N-labeled (the recombinant C2B domain of rabphilin; residues 524–684; Ubach et al, 1999). Proteins were used as affinity matrices immobilized on glutathione agarose, or cleaved from the GST-moiety with thrombin and purified by size exclusion chromatography. See the Supplementary Materials for a complete list of plasmids used.
NMR spectroscopy was performed with 15N-labeled proteins as described (Ubach et al, 1999).
Miscellaneous procedures
All antibodies used were described previously (see Supplementary Data). Phospholipid binding assays using liposomes, immunofluorescence labeling experiments, and FM fluorescent dye imaging were carried out as described (Schoch et al, 2001; Shin et al, 2002; Deák et al, 2004). SDS–PAGE and immunoblotting were performed using standard procedures. Immunoprecipitations and GST-pulldown experiments were performed essentially as described (Li et al, 1994; McMahon et al, 1995; see Supplementary Materials). Paired Student's t-test or variance analysis was used to determine statistical significance (P<0.05).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Information
Acknowledgments
This study was supported by grants from the NIH (MH 066198 to ETK and NS40944 to JR) and by investigatorships from the Howard Hughes Medical Institute (to RJ and TCS). We thank Ms I Kornblum, A Roth, N Hamlin and E Borowicz for technical assistance.
References
- Burns ME, Sasaki T, Takai Y, Augustine GJ (1998) Rabphilin-3A: a multifunctional regulator of synaptic vesicle traffic. J Gen Physiol 111: 243–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE, Janz R, Tzounopoulos T, Südhof TC, Malenka RC, Nicoll RA (1997) The synaptic vesicle protein Rab3A is essential for mossy fiber long term potentiation in the hippocampus. Nature 388: 590–593 [DOI] [PubMed] [Google Scholar]
- Castillo PE, Schoch S, Schmitz F, Südhof TC, Malenka RC (2002) RIM1α is required for presynaptic long-term potentiation. Nature 415: 327–330 [DOI] [PubMed] [Google Scholar]
- Chung SH, Song WJ, Kim K, Bednarski JJ, Chen J, Prestwich GD, Holz RW (1998) The C2 domains of Rabphilin3A specifically bind phosphatidylinositol 4,5-bisphosphate containing vesicles in a Ca2+-dependent manner. In vitro characteristics and possible significance. J Biol Chem 273: 10240–10248 [DOI] [PubMed] [Google Scholar]
- Chung SH, Takai Y, Holz RW (1995) Evidence that the Rab3a-binding protein, rabphilin3a, enhances regulated secretion. Studies in adrenal chromaffin cells. J Biol Chem 270: 16714–16718 [DOI] [PubMed] [Google Scholar]
- Dai H, Tomchick DR, Garcia J, Südhof TC, Machius M, Rizo J (2005) Crystal structure of the RIM2 C2A-domain at 1.4 A resolution. Biochemistry 44: 13533–13542 [DOI] [PubMed] [Google Scholar]
- Darchen F, Goud B (2000) Multiple aspects of Rab protein action in the secretory pathway: focus on Rab3 and Rab6. Biochimie 82: 375–384 [DOI] [PubMed] [Google Scholar]
- Deák F, Schoch S, Liu X, Südhof TC, Kavalali ET (2004) Synaptobrevin is essential for fast synaptic-vesicle endocytosis. Nat Cell Biol 11: 1102–1108 [DOI] [PubMed] [Google Scholar]
- Fernandez I, Arac D, Ubach J, Gerber SH, Shin O, Gao Y, Anderson RGW, Südhof TC, Rizo J (2001) Three-dimensional structure of the synaptotagmin 1 C2B-domain: Synaptotagmin 1 as a phospholipid binding machine. Neuron 32: 1057–1069 [DOI] [PubMed] [Google Scholar]
- Fukuda M (2005) Versatile role of Rab27 in membrane trafficking: focus on the Rab27 effector families. J Biochem (Tokyo) 137: 9–16 [DOI] [PubMed] [Google Scholar]
- Fykse EM, Li C, Südhof TC (1995) Phosphorylation of rabphilin-3A by Ca2+/Calmodulin- and cAMP-dependent protein kinases in vitro. J Neurosci 15: 2385–2395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geppert M, Bolshakov VY, Siegelbaum SA, Takei K, De Camilli P, Hammer RE, Südhof TC (1994) The role of Rab3A in neurotransmitter release. Nature 369: 493–497 [DOI] [PubMed] [Google Scholar]
- Geppert M, Goda Y, Stevens C, Südhof TC (1997) Rab3A regulates a late step in synaptic vesicle fusion. Nature 387: 810–814 [DOI] [PubMed] [Google Scholar]
- Hata Y, Slaughter CA, Südhof TC (1993) Synaptic vesicle fusion complex contains unc-18 homologue bound to syntaxin. Nature 366: 347–351 [DOI] [PubMed] [Google Scholar]
- Izumi T, Gomi H, Kasai K, Mizutani S, Torii S (2003) The roles of Rab27 and its effectors in the regulated secretory pathways. Cell Struct Funct 28: 465–474 [DOI] [PubMed] [Google Scholar]
- Jahn R, Lang T, Südhof TC (2003) Membrane fusion. Cell 112: 519–533 [DOI] [PubMed] [Google Scholar]
- Johannes L, Doussau F, Clabecq A, Henry JP, Darchen F, Poulain B (1996) Evidence for a functional link between Rab3 and the SNARE complex. J Cell Sci 109: 2875–2884 [DOI] [PubMed] [Google Scholar]
- Koushika SP, Richmond JE, Hadwiger G, Weimer RM, Jorgensen EM, Nonet ML (2001) A post-docking role for active zone protein Rim. Nat Neurosci 4: 997–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Takei K, Geppert M, Daniell L, Stenius K, Chapman ER, Jahn R, De Camilli P, Südhof TC (1994) Synaptic targeting of rabphilin-3A, a synaptic vesicle Ca2+/phospholipid-binding protein, depends on Rab3A/3C. Neuron 13: 885–898 [DOI] [PubMed] [Google Scholar]
- Lin RC, Scheller RH (2000) Mechanisms of synaptic vesicle exocytosis. Annu Rev Cell Dev Biol 16: 19–49 [DOI] [PubMed] [Google Scholar]
- McMahon HT, Missler M, Li C, Südhof TC (1995) Complexins: cytosolic proteins that regulate SNAP receptor function. Cell 83: 111–119 [DOI] [PubMed] [Google Scholar]
- Nalefski EA, Falke JJ (1996) The C2 domain Ca2+-binding motif: structural and functional diversity. Protein Sci 5: 2375–2390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonet ML, Staunton JE, Kilgard MP, Fergestad T, Hartwieg E, Horvitz HR, Jorgensen EM, Meyer BJ (1997) Caenorhabditis elegans rab-3 mutant synapses exhibit impaired function and are partially depleted of vesicles. J Neurosci 17: 8061–8073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizo J, Südhof TC (1998) C2-domains, structure and function of a universal Ca2+-binding domain. J Biol Chem 273: 15879–15882 [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF (1996) Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron 16: 1197–1207 [DOI] [PubMed] [Google Scholar]
- Sara Y, Virmani T, Deak F, Liu X, Kavalali ET (2005) An isolated pool of vesicles recycles at rest and drives spontaneous neurotransmission. Neuron 17: 563–573 [DOI] [PubMed] [Google Scholar]
- Schlüter OM, Schmitz F, Jahn R, Rosenmund C, Südhof TC (2004) A complete genetic analysis of neuronal Rab3 function. J Neurosci 24: 6629–6637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlüter OM, Schnell E, Verhage M, Tzonopoulos T, Nicoll RA, Janz R, Malenka RC, Geppert M, Südhof TC (1999) Rabphilin knock-out mice reveal rat rabphilin is not required for Rab3 function in regulating neurotransmitter release. J Neurosci 19: 5834–5846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoch S, Castillo PE, Jo T, Mukherjee K, Geppert M, Wang Y, Schmitz F, Malenka RC, Südhof TC (2002) RIM1α forms a protein scaffold for regulating neurotransmitter release at the active zone. Nature 415: 321–326 [DOI] [PubMed] [Google Scholar]
- Schoch S, Deák F, Königstorfer A, Mozhayeva M, Sara Y, Südhof TC, Kavalali ET (2001) SNARE function analysed in synaptobrevin/VAMP knockout mice. Science 294: 1117–1122 [DOI] [PubMed] [Google Scholar]
- Shin O-H, Rizo J, Südhof TC (2002) Synaptotagmin function in dense core vesicle exocytosis studied in cracked PC12 cells. Nat Neurosci 5: 649–656 [DOI] [PubMed] [Google Scholar]
- Shirataki H, Kaibuchi K, Sakoda T, Kishida S, Yamaguchi T, Wada K, Miyazaki M, Takai Y (1993) Rabphilin-3A, a putative target protein for smg p25A/Rab3A p25 small GTP-binding protein related to synaptotagmin. Mol Cell Biol 13: 2061–2068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl B, Chou JH, Li C, Südhof TC, Jahn R (1996) Rab3 reversibly recruits rabphilin to synaptic vesicles by a mechanism analogous to raf recruitment by ras. EMBO J 15: 1799–1809 [PMC free article] [PubMed] [Google Scholar]
- Staunton J, Ganetzky B, Nonet ML (2001) Rabphilin potentiates SNARE function independently of Rab3. J Neurosci 21: 9255–9264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC (1995) The synaptic vesicle cycle: a cascade of protein-protein interactions. Nature 375: 645–653 [DOI] [PubMed] [Google Scholar]
- Tsuboi T, Fukuda M (2005) The C2B domain of rabphilin directly interacts with SNAP-25 and regulates the docking step of dense core vesicle exocytosis in PC12 cells. J Biol Chem 280: 39253–39259 [DOI] [PubMed] [Google Scholar]
- Ubach J, Garcia J, Nittler MP, Südhof TC, Rizo J (1999) The C2B-domain of rabphilin: structural variations in a janus-faced domain. Nat Cell Biol 1: 106–112 [DOI] [PubMed] [Google Scholar]
- Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg T, Missler M, Geuze H, Südhof TC (2000) Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science 287: 864–869 [DOI] [PubMed] [Google Scholar]
- Wang Y, Okamoto M, Schmitz F, Hofmann K, Südhof TC (1997) RIM: A putative Rab3-effector in regulating synaptic vesicle fusion. Nature 388: 593–598 [DOI] [PubMed] [Google Scholar]
- Wang Y, Sugita S, Südhof TC (2000) The RIM/NIM family of neuronal C2-domain proteins. Interactions with Rab3 and a new class of Src homology 3 domain proteins. J Biol Chem 275: 20033–20044 [DOI] [PubMed] [Google Scholar]
- Wang Y, Südhof TC (2003) Genomic definition of RIM proteins: evolutionary amplification of a family of synaptic regulatory proteins. Genomics 81: 126–137 [DOI] [PubMed] [Google Scholar]
- Washbourne P, Thompson PM, Carta M, Costa ET, Mathews JR, Lopez-Bendito G, Molnar Z, Becher MW, Valenzuela CF, Partridge LD, Wilson MC (2002) Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat Neurosci 5: 19–26 [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG (2002) Short-term synaptic plasticity. Annu Rev Physiol 64: 355–405 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Information