

Abstract
Classic mechanisms of tumor response to chemotherapy include apoptosis and mitotic catastrophe. Recent studies have suggested that cellular senescence, a terminal proliferationarrest seen in vitro, may be invoked during the exposure of cancer cells to chemotherapeutic agents. To identify markers associated specifically with the cellular senescence phenotype, we utilized expression data from cDNA microarray experiments identifying transcripts whose expression levels increased as human prostate epithelial cells progressed to senescence. When screened against other growth-inhibitory conditions, including quiescence and apoptosis, many of these transcripts were also upregulated, indicating that similar pathways occur between apoptosis and senescence. A senescent-like phenotype was then induced in several prostate cancer cell lines using 5-aza-2′-deoxycytidine, doxorubicin, or Docetaxel. Treatment with these agents resulted in a significant increase in the induction of senescence-specific genes when compared to nonsenescent conditions. The performance of the panel was improved with fluorescence-activated cell sorting using PKH26 to isolate nonproliferating, viable, drug-treated populations, indicating that a heterogeneous response occurs with chemotherapy. We have defined an RNA-based gene panel that characterizes the senescent phenotype induced in cancer cells by drug treatment. These data also indicate that a panel of genes, rather than one marker, needs to be utilized to identify senescence.
Keywords: Senescence, cancer, differentiation, cancer therapy, prostate
Introduction
Cellular senescence has been traditionally defined as a terminal phenotype that is manifest as primary cells undergo prolonged passage in culture [1]. Senescent cells are terminally arrested, often express senescence-associated β-galactosidase (SA-β-gal), and assume a characteristic enlarged, granular, flattened appearance in culture. A similar phenotype can be achieved in both normal and transformed cells in response to various stimuli including oxidative stress, radiation, activated oncoproteins, and agents that cause DNA or other cellular damage [2,3]. This terminal growth arrest is a molecular and cellular phenotype distinct from apoptotic or catastrophic cell death. The growth arrest that characterizes cellular senescence may be induced by lower doses of drugs or differentiating agents that result in cytostatic growth arrest, which persists even following removal of the agent [3].
The onset of senescence in primary cells is mediated by numerous factors including telomere shortening and alterations in DNA methylation [1]. Discrete changes in genes involved in growth regulation, differentiation, and cell signaling have been characterized in human prostate epithelial cells (HPECs) and other cell types during senescence [4–6]. Several of these genes, such as the cyclin-dependent kinase (Cdk) inhibitor p21WAF1/Cip1/Sdi1 and genes downstream from p53, have been shown to mediate the growth arrest observed at senescence [7,8]. Other genes, including the Cdk inhibitor p16INK4a, are involved in the maintenance of senescence [7]. These pathways are frequently targeted for inactivation in the development of epithelial cancers, including prostate cancer [9,10]. Studies also suggest that members of the insulin-like growth factor (IGF) axis, including IGF2 and its IGF-binding proteins (e.g., IGFBP5), constitute another “senescence” pathway [4,5]. Reconstitution of senescence-associated genes in immortal cancer cells by cell fusion, or by selected chromosomal or gene transfection leads to reestablishment of the senescent phenotype, indicating that a finite number of senescence genes and pathways exist [11–15].
In immortalized cells, several stimuli, including DNA damage, oxidative stress, DNA methylation alterations, and histone deacetylase inhibition, may induce a senescent phenotype that has been termed “cellular” senescence to distinguish it from replicative or passage-dependent senescence [16]. Many chemotherapeutics function through these and other mechanisms, and it has been suggested that the latent senescent phenotype can be reactivated in cancer cells [3]. Recently, it was demonstrated that primary murine lymphomas respond to chemotherapy by engaging an apoptosis or a senescence program controlled by p53 and p16INK4a [17]. Blocking apoptosis by expressing bcl2 results in senescence, indicating that senescence may represent an alternate mechanism for cell arrest in some circumstances. These studies have relied on the identification of senescence-associated-β-galactosidase (SA-β-gal), an enzyme activity induced during replicative senescence in primary cells. However, SA-β-gal can be induced in other stress-associated states, and is expressed at detectable levels in some proliferating immortalized cancer cells, leading to concern regarding its specificity as a marker of senescence [18–20]. Therefore, the development of a panel of senescence-specific molecular markers would be useful in the identification of cells displaying the terminal arrest and morphology associated with senescence.
Strategies to inhibit malignant growth by disabling proliferation, rather than killing cancer cells, have been understudied and represent an area of intense interest. In this manuscript, we identify and confirm a series of molecular markers that distinguish senescence in primary and tumor cells of epithelial origin. These markers are upregulated in senescence, in contrast to apoptosis and quiescence. We find that these markers are induced after the treatment of prostate cancer cells in vitro with chemotherapeutics that induce a senescent phenotype. Reexpression of two of these markers, CXCL14/BRAK and P311, has been found to inhibit cellular and tumor growth [21,22]. These data indicate that a subset of genes upregulated during senescence in primary cells are also induced in cancer cells undergoing cellular senescence. Given that drug-induced senescence may occur with chemotherapy, these markers may assist in identifying and exploiting the senescent phenotype in human cancer treatment.
Materials and Methods
Cell Culture and Drug Treatment
Primary HPECs were established and maintained as described previously [7]. HPECs were forced to undergo quiescence by contact inhibition for 96 hours in serumdepleted media. HPECs were treated with staurosporine and thapsigargin from 4 to 48 hours to induce apoptosis. The selected concentration to maximize apoptosis was 1 µg/ml staurosporine and 1 µM thapsigargin. The degree of apoptosis was determined through quantitation of fragmented nuclei—a hallmark of the apoptotic process. Cells were fixed for 30 minutes on ice in 1% phosphate-buffered saline (PBS)-buffered paraformaldehyde. Cells were washed in PBS and resuspended in PBS containing 0.1% NP-40 and 1 µg/ml Hoechst 33342. Fragmented nuclei were visualized by fluorescence microscopy. The percent fragmented nuclei compared to intact nuclei was determined in three independent experiments.
The prostate cancer cell lines, DU145 and LNCaP, were plated the day before the drug treatment so that they would be 50% confluent on the day of treatment. The topoisomerase II inhibitor, doxorubicin (Sigma, St. Louis, MO), or microtubule-stabilizing agent, Docetaxel (Aventis Pharmaceuticals Products Inc., Bridgewater, NJ), was added to the cells at days 1 and 3. Regular growth medium DMEM with 5% FBS was then resumed from days 4 to 7 when the cells were harvested for downstream analyses. 5-Aza-2′-deoxycytidine (DAC; Sigma), a methyltransferase inhibitor, dosed continuously for 7 days before harvest. A series of dose-ranging studies was performed, as previously described [15]: 5 to 200 nM for doxorubicin, 0.1 to 10 nM for Docetaxel, and 0.25 to 250 µM for DAC. The final drug doses were chosen based on induction of the senescence phenotype as assessed by phenotype and fluorescence-activated cell sorting (FACS) analysis 7 days after initial drug treatment. All cells were incubated at 37°C with 5% CO2 during the drug treatment. SA-β-gal staining was performed as previously described and five random high-power fields were counted for staining in each culture [23].
Quantitative Reverse Transcriptase Polymerase Chain Reaction (RT-PCR) Gene Quantitation
Total RNA was isolated from drug-treated cells using the RNeasy RNA isolation kit (Qiagen). On column DNase treatment was used to eliminate DNA contamination. One microgram of total RNA was used to make cDNA. Quantitative PCR was performed by monitoring in real time the increase in fluorescence of the SYBR Green dye using an iCycler detection system (Bio-Rad) as described [4]. For comparison of transcript levels between samples, a standard curve of cycle thresholds for several serial dilutions of a cDNA sample was established. This value was then used to calculate the relative abundance of each gene. These values were then normalized to the relative amounts of 18S cDNA. All PCR reactions were performed in duplicate. Sequences of primers used for PCR analysis are available on request. We compared the proportion of the nine genes that were observed to have at least a two-fold increase between the senescent conditions and the nonsenescent conditions using an unpaired t-test. Prior to testing, the proportions were transformed using an arcsine square-root transformation in order to better meet the assumptions of the analysis. A P-value less than .05 was considered significant.
Cell Cycle Analysis, Viability, and Apoptosis
Cells were trypsinized in 0.05% trypsin, 0.53 mM EDTA-4Na in Hank's Balanced Salt Solution (Cellgro) and resuspended in growth media with 10 µg/ml of propidium iodide (Sigma) and analyzed for cell viability by flow cytometry. Another aliquot of the cells that were harvested at the same time point was washed with cold 1x PBS followed by fixation in 90% ethanol overnight at -20°C. Cells were centrifuged, resuspended in PBS, then 0.5% NP-40 solution was applied to the cells with RNase A to a final concentration of 250 µg/ml. Propidium iodide stock solution was added to the cells to 10 µg/ml and incubated at 37°C for 5 min. The stained cells were analyzed for DNA content and cell cycle distribution. Data was acquired on a FACScan benchtop flow cytometer (Becton Dickinson, San Jose, CA). Ten thousand events were acquired with CellQuest software (Becton Dickinson). Cell cycle position was determined with MODFiT LT (Verity, Topsham, Maine).
For PKH26 analysis of cell division, 1 x 107 cells were trypsinized and stained with PKH26-GL (Sigma) according to manufacturer's protocol. Cells were replated, treated with the individual drugs and PKH26 fluorescence was examined seven days after initial treatment by FACS analysis. Cells with high fluorescence represent nondividing cells whereas cells with low fluorescence represent cells that have undergone cell divisions. FlowJo software (Tree Star, Inc.) was used to distinguish the proliferating and growth arrest cell populations. Total RNA was isolated from half of the sorted cells and the remainder were plated and stained for SA-β-gal.
Results
Identification of Genes with Increased mRNA Expression in Senescent Cells
To identify genes specifically upregulated with cellular senescence, we initially focused on genes induced in HPECs when passaged to senescence in vitro. Previously, we had generated gene expression profiles from primary prostate epithelial cells and identified 93 independent image clones consisting of known genes, ESTs, and hypothetical proteins that were significantly induced in senescence [24]. To confirm gene induction, three independent primary HPECs cultures were grown to senescence, RNA isolated and converted to cDNA. Real-time quantitative RT-PCR (QPCR) was performed to monitor gene expression by comparing proliferating and senescent samples. The induction of 46 genes was confirmed, of which 42 were characterized and 4 were hypothetical proteins (Supplemental Table 1). Subsequent analyses were performed on robustly induced genes (change of greater than four-fold), refining the group to 17 genes (Table 1).
Table 1.
Gene Expression in HPEC Senescence.
| Gene | Accession Number | Fold Change |
| CSPG2 | NM_004385 | 25 |
| PLXDC2 | AY358486 | 21 |
| EST | BG391777 | 19 |
| Wnt5a | NM_003392 | 18 |
| DOC1 | NM_182909 | 17 |
| FBOX32 | AK125888 | 12 |
| OLFML2A | BC054001 | 12 |
| IGF2 | AK074614 | 11 |
| Adlican | AF245505 | 10 |
| COL1A1 | Z74615 | 9.0 |
| MMP2 | AL832088 | 8.6 |
| RKHD3 | AK131424 | 8.4 |
| CKTSF1B1 | AF110137 | 5.5 |
| MFAP2 | BC028033 | 5.2 |
| CXCL14 | NM_004887 | 4.6 |
| P311 | BC011050 | 4.6 |
| MDK | BM473044 | 4.1 |
The expression of 93 genes previously identified to be induced in HPEC senescence by microarray [4] were assayed by QPCR in three additional HPEC cultures undergoing senescence.
Those genes induced greater than four-fold are shown above.
Gene expression was standardized to 18S rRNA expression, and the ratio of expression in senescent cells to the expression in proliferating cultures was determined.
Identification of Senescence-Specific Genes
Other cell cycle arrest and stress states may share gene expression changes in common with senescence. To define a senescence-specific gene panel, we initiated growth arrest and apoptosis by several means. First, growth arrest was achieved by propagating HPECs and fibroblasts to confluence. Cells were then maintained in a 100% confluent and arrested state for 4 days in serum-depleted media. HPECs did not stain for SA-β-gal at confluence, in contrast to fibroblasts, which were positive as previously documented [25]. Arrested cells were harvested, and the 17 genes robustly induced at senescence (above) were analyzed by QPCR. This experiment revealed that FBXO32, PLXDC2, AK024270, OLFML2A, and Wnt5a were also induced at confluence arrest (Table 2). These five genes did not undergo further analysis.
Table 2.
The Effect of Apoptosis and Confluence Arrest on Genes Induced in HPEC Senescence.
| Gene | Confluence Arrest | Staurosporine (1 µg/ml) | Thapsigargin (1 µM) | |||||
| HPEC | Fibroblast | 4 hr | 12 hr | 48 hr | 24 hr | 48 hr | ||
| CSPG2 | 1.1 | 1.0 | 1.0 | 1.1 | 1.0 | 0.5 | 0.2 | |
| PLXDC2 | 1.1 | 2.3 | 0.8 | 1.9 | 2.5 | 1.1 | 3.1 | |
| EST (BG39177) | 0.6 | 0.8 | 1.1 | 0.8 | 0.5 | 1.6 | 4.3 | |
| Wnt5a | 1.2 | 3.9 | 1.1 | 0.4 | 0.2 | 1.4 | 17.2 | |
| DOC1 | 0.8 | 1.5 | 1.2 | 0.1 | 0.2 | 0.5 | 0.4 | |
| FBOX32 | 0.9 | 5.1 | 1.1 | 0.6 | 1.1 | 2.4 | 4.6 | |
| OLFML2A | 1.0 | 23.0 | 1.0 | 0.8 | 1.1 | 0.6 | 0.6 | |
| IGF2 | 0.8 | 0.8 | 1.0 | 1.4 | 2.4 | 0.6 | 0.3 | |
| Adlican | 1.5 | 0.8 | 1.0 | 0.7 | 1.2 | 1.0 | 0.9 | |
| COL1A1 | 0.5 | 0.6 | 1.2 | 0.5 | 0.4 | 0.6 | 0.9 | |
| MMP2 | 0.8 | 1.4 | 1.0 | 1.2 | 1.1 | 0.5 | 0.7 | |
| RKHD3 | 0.8 | 0.9 | 1.0 | 1.1 | 2.0 | 0.8 | 1.1 | |
| CKTSF1B1 | 1.1 | 1.0 | 1.0 | 1.8 | 13.0 | 1.5 | 0.9 | |
| MFAP2 | 0.8 | 1.0 | 0.9 | 0.3 | 0.2 | 1.7 | 0.4 | |
| CXCL14 | 1.9 | 1.0 | 0.9 | 0.9 | 0.9 | 0.2 | 0.3 | |
| P311 | 0.9 | 1.0 | 0.9 | 0.6 | 0.6 | 0.3 | 0.5 | |
| MDK | 1.1 | 1.8 | 1.0 | 2.6 | 3.9 | 1.2 | 1.0 | |
Cells were induced to undergo growth arrest in response to confluence, or apoptosis by drug treatment.
The relative ratio of gene expression, determined by QPCR, on drug treatment/confluence to that of the untreated cells is listed.
Highlighted genes did not increase expression under these conditions.
Shared signaling pathways exist between senescence and apoptosis (i.e., p53-related) [1], yet we hypothesized that specific genes are induced with senescence. We induced apoptosis using two non-DNA-damaging drugs, staurosporine and thapsigargin. Staurosporine is a known kinase inhibitor [26], whereas thapsigargin induces apoptosis by directing an intracellular calcium flux [27]. DNA-damaging agents have been documented to induce a senescent phenotype and were avoided [3]. HPECs were treated with 1 µg/ml staurosporine or 1 µM thapsigargin for up to 48 hours, and nuclei were visualized under fluorescent microscopy after Hoechst 33342 staining for fragmentation and condensation. Both drugs effectively lead to reduced viability and initiated apoptosis (Figure 1). Gene expression analysis from cells treated with apoptosis-inducing drugs revealed that CKTSF1B1, PLXDC2, MDK, and IGF2 were induced by greater than two-fold with staurosporine treatment (Table 2). BC071787, FBOX32, PLXDC2, and Wnt5a were induced with thapsigargin treatment. Eight of 17 genes induced with senescence at high levels were also induced with apoptosis. The remaining genes define a nine-gene “senescence-specific” panel that is induced in senescent epithelial cells, but not with growth arrest or apoptosis (Table 2).
Figure 1.

HPEC apoptotic induction. HPECs were treated with 1 µg/ml staurosporine (A) or 1 µM thapsigargin (B). At the indicated time points, cells were trypan blue-stained for viability and fragmented nuclei were detected with Hoechst 33342 and fluorescent microscopy. The percentages of viable and apoptotic cells are shown ± SD.
Cellular Senescence Induction in Prostate Cancer Cells
This senescence panel was then applied to several models of drug-induced cellular senescence using prostate cancer cell lines. Analyses were performed on whole cultures and sorted populations. We used chemical agents previously reported to induce a senescent-like phenotype including DAC, a methyltransferase inhibitor [28], and doxorubicin, which acts primarily to inhibit the topoisomerase II complex [29]. In addition, Docetaxel, a microtubule-stabilizing agent active in prostate cancer, was tested [30]. Dose-ranging studies were performed and cells were assessed at day 7 after initial drug exposure. By this time point, initial cell death processes had been completed and the viable senescent phenotype, as defined below and by trypan blue exclusion staining, becomes predominant at selected doses.
Senescence was determined through the use of several criteria. The first was the development of a flattened, enlarged morphology (Figure 2A). Second, flow analysis was used to confirm this increase in size and cellular complexity by documenting an increase in side scatter (SSC). Third, growth arrest was recognized by a proliferative failure of treated cultures. Finally, polyploidy, indicating multinucleation, a feature of senescent cells [31,32], was evident in treated cells (Figure 2B). Drug concentrations were optimized for DAC, doxorubicin, and Docetaxel to reproducibly induce a senescent-like phenotype in DU145 and LNCaP cells (Table 3). DU145 cells treated with Docetaxel did not induce a senescent phenotype based on these criteria at any dose.
Figure 2.

Treatment of prostate cancer cell lines with senescence-inducing drugs. (A) DU145 treated with doxorubicin (25 nM) after 7 days reveals that cancer cells are growth-arrested and have a viable phenotype consistent with senescence. Untreated cells (left, x400) show the standard cuboidal morphology. In contrast, doxorubicin-treated cells (right, x400) are greatly enlarged, flattened, and contain well-defined, occasionally multiple nuclei. (B) FACS analysis of DNA content distribution of a typical exponentially growing control cell population is composed of two peaks (G1/G0 and G2/M phase) and a valley of S phase. Little change is seen in DU145 cells treated with 5 nM doxorubicin. A significant shift toward a higher FL2 area and additional peaks (higher DNA content area) are seen in senescent cultures (25 nM), indicating the development of polyploid nuclei, a feature of senescent cells. (C) FACS analysis of cell division after PKH26 staining of drug-treated (Table 3) and untreated LNCaP prostate cancer cells. Cells with high fluorescence represent nondividing cells (right shift) and cells with low fluorescence represent cells that have undergone cell divisions. The population of cells with increased fluorescence after drug treatment (decreased cell proliferation) was analyzed for senescent panel gene expression.
Table 3.
Induction of Senescence Characteristics in DU145, PPC-1, and LNCaP Cells after Chemotherapy Treatment for 7 Days.
| Cell Line | Drug | Dose | Viability (%) | Complexity Change*,† (%) | Ploidy Change* (%) | Apoptosis‡ (%) |
| DU145 | Control | - | 95 | - | - | 0.3 |
| DAC | 0.4 µM | 57 | 66 | 129 | 2.0 | |
| Docetaxel | 1 nM§ | 87 | -32 | 100 | 0.3 | |
| Doxorubicin | 5 nM§ | 94 | -29 | -35 | 0 | |
| 25 nM | 85 | 65 | 335 | 0.9 | ||
| LNCaP | Control | - | 79 | - | - | 2 |
| DAC | 75 µM | 51 | 63 | 60 | 0.8 | |
| Docetaxel | 5 nM | 48 | 83 | 20 | 0.5 | |
| Doxorubicin | 5 nM§ | 83 | -6 | 10 | 0.8 | |
| 25 nM | 65 | 22 | 120 | 0.9 | ||
Cells were harvested and analyzed by flow cytometry for viability, complexity (SSC), ploidy, and apoptosis.
Represents percentage change from untreated cells.
Complexity refers to SSC.
Apoptosis is the absolute percentage calculated from flow analysis and confirmed by Annexin V staining.
Selected drug concentrations and doses that did not induce senescence are also shown.
To further refine the senescent population in these drug-treated cultures, DU145- and LNCaP-treated cells were stained prior to treatment with PKH26, a lipophilic fluorescent compound that incorporates into the plasma membrane. PKH26 fluorescence decreases with each cell division. With the exception of Docetaxel-treated DU145 cells, all drug doses at day 7 demonstrated a significant increase (over 50%) in the high-fluorescence fraction compared to control cultures (Figure 2C). A defined single peak was also found, indicating that the majority of cells had decreased proliferation. A comparison of SA-β-gal staining of high-fraction fluorescence cells revealed significant increases (three-fold to five-fold) in staining for drug-treated DU145 cells compared to control (data not shown). LNCaP cells were unable to be replated after sorting and were not analyzed for SA-β-gal. These data demonstrate that drug-treated cultures were generated with the phenotypic and cell cycle characteristics of induced cellular senescence.
Induction of the “Senescence Panel” during Drug-Induced Cellular Senescence
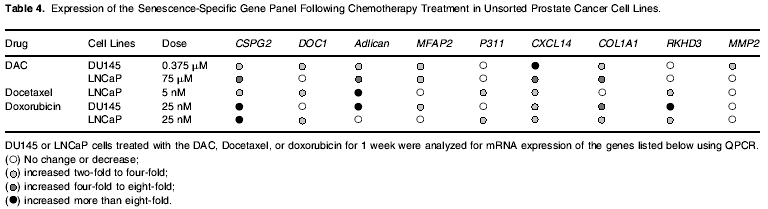
The nine-gene senescence panel was then assessed using QPCR in the whole (Table 4) and PKH26-sorted cultures after drug treatment. Analysis of the expression of the nine genes in five unsorted senescent cultures (45 data points) resulted in the more than two-fold induction of the senescence markers in 30 cases. CSPG2 and CXCL14 expression was consistently increased in senescence, over 20-fold in doxorubicin-treated cells, when compared to untreated cells. Adlican, COL1A1, DOC1, MFAP2, and RKHD3 were upregulated in the majority (60–80%) of senescent conditions. Both DAC and doxorubicin were equally proficient in inducing the nine-gene senescent gene panel. A comparison of gene expression under conditions that did not induce the senescent criteria to those that did (Table 3) revealed that only two (22%) of the nine genes showed at least a two-fold increase under the nonsenescent conditions, in contrast to six (67%) under senescent conditions (P = .0008). Thus, a panel of genes specifically upregulated in primary cell senescence is also found to be significantly increased with induced cellular senescence in immortalized cancer cells.
For PKH26-stained cells, the high-staining (i.e., growthinhibited) fraction was compared to proliferating, FACS-sorted control cultures. A greater than two-fold increase in expression was seen in 35 of 45 genes (five sorted cultures), and 31 of these increases were over four-fold. All markers were increased in DAC- and doxorubicin-treated DU145 cells. This demonstrates that enrichment of the viable, growth-arrested population improves the precision of the senescence panel.
Discussion
Chemotherapy induces a senescence-like terminal proliferation arrest that is distinct from apoptosis and mitotic catastrophe [3]. SA-β-gal staining has been utilized to putatively identify this phenotype in aging human tissues and human cancers; however, this marker lacks specificity [19,20]. In contrast to apoptosis, confirmatory markers have not been developed for senescent human cells. We previously identified a number of genes whose expression was increased or decreased in normal prostate epithelial cells when developing a senescent phenotype after limited passage in culture [4]. In the current manuscript, we identified a subset of genes that are highly upregulated at senescence in prostate epithelial cells, and screened these genes against cells undergoing apoptosis or confluence arrest. Our initial finding is that many genes were found to demonstrate increased expression in both apoptosis and senescence, consistent with pathways in common between these two processes. However, senescence-specific genes were identified. We applied this panel to a series of cancer cells that had a senescent phenotype induced using various chemotherapeutic agents, and find consistent increases in expression in these models. During immortalization, genes in senescence pathways may be inactivated, either through genetic or epigenetic means, and bypassing senescence requires each of these pathways to be altered [11]. The reexpression of the majority, but not all, of our gene markers suggests that reactivation of one or more of these pathways occurs with establishment of the senescent phenotype in cancer cells. This indicates the need for utilizing a panel of genes, rather than one gene, to evaluate for senescence.
Chemotherapy type and dose, as well as genetic context, are variables that contribute to determining whether a cell becomes apoptotic or senescent [3]. We optimized a series of drugs with different therapeutic mechanisms to induce a senescent phenotype. Senescence is a differentiated phenotype that is defined in primary cells based on morphology, viability, cell cycle arrest, aneuploidy, and the expression of SA-β-gal [1,31,33]. Similar criteria were utilized for treated cancer cells. We find that 8 of 17 genes induced at senescence were also increased with apoptosis, suggesting common pathways between the two processes. None of these genes is known to be directly regulated by p53, an important gene in signaling both these events [34]. Importantly, genes specifically upregulated in senescence were identified, confirming that distinct senescence pathways exist [11]. Chemotherapeutic drugs that did not induce a senescent phenotype in certain cell types despite variations in dosing and schedule (i.e., DU145 with Docetaxel) were encountered. Recently, it was demonstrated that primary murine lymphomas respond to chemotherapy by engaging an apoptosis or a senescence program controlled by p53 and p16INK4a [17]. Blocking apoptosis by expressing bcl2 results in senescence, indicating that similar signaling pathways may trigger these phenotypes, but the fate of a cancer cell is dependent on the genetic context.
The majority of genes in the senescence-specific panel were induced with onset of the senescent phenotype in the cancer cell lines studied. Our data show that CSPG2 and CXCL14 mRNA upregulations are highly and consistently associated with prostate cells that have undergone senescence through either passage or chemotherapeutic treatment regimens. These genes are also upregulated with human fibroblasts grown to senescence (data not shown). The other genes induced in our senescence-specific panel (DOC1, MFAP2, Adlican, COL1A1, MMP2, and P311) demonstrate more of a dependence on the drug and cell types examined in unsorted cultures. However, these genes were uniformly upregulated by enriching the treated cells for the viable, low-proliferation senescent fraction (high PKH26). These findings indicate that the senescence response to chemotherapy is heterogeneous even with an optimization of dose and timing. Although CSPG2 and CXCL14 appear to be the most reliable senescence-specific molecular markers in this model, we would advocate using multiple markers because senescence genes and pathways are often deleted or mutated.
The genes identified in the senescence panel represent diverse biologic functions. Putative structural roles have been assigned to DOC1 (deleted in ovarian cancer) and MFAP2, a microfibril-associated glycoprotein [35,36]. Alterations in these genes may reflect the enlarged, flattened morphology that characterizes senescence. Others are secreted (e.g., Adlican, COL1A1, CSPG2, CXCL14, and MMP2). Adlican has been found to be overexpressed in skin fibroblasts from centurians, but its function is unknown [37]. A debate exists whether senescent cells may provide an environment that stimulates or inhibits tumor growth [38]. Several secreted genes, MMP2 and CSPG2, are proteases found to have increased levels in the peritumor extracellular matrix of cancers, suggesting a role in promoting tumor invasion [39,40]. Alternately, senescence genes may impede cancer progression. The overexpression of the chemokine CXCL14 inhibits the growth of LAPC4 prostate cancer xenografts [21] and prevents angiogenesis [41]. The expression of P311, a gene involved in smooth muscle myogenesis, leads to neuron differentiation and the induction of p21 expression, a gene important in signaling senescence [22]. The diverse biologic functions of these genes should, with further study, lend insights into common pathways involved in modulating senescence in cancer cells.
This study identifies and confirms a molecular definition of senescence, based on mRNA expression, in human cancer cells after treatment with chemotherapy. Senescence is a distinct form of cell arrest activated with cancer therapy, including chemotherapy and possibly radiation. Even though senescent tumor cells are not destroyed but arrested, this may be a feasible approach to controlling cancer. However, some genes upregulated at senescence may have protumorigenic effects so the definitive effects of this phenotype remain to be determined [38]. Studies have detected SA-β-gal expression in human and mouse tumors after chemotherapy, suggesting that senescence occurs in vivo [17,42]. The current study defines a series of genes useful in identifying senescent cells, and further analyses will confirm their application to senescence in vivo.
Acknowledgements
We thank Igor Roninson for his advice and review of this manuscript, and Glen Leverson for statistical support. Special thanks also to John P. Livesey for his support of this cancer research.
Abbreviations
- SA-β-gal
senescence-associated β-galactosidase
- HPEC
human prostate epithelial cell
- QPCR
quantitative reverse transcriptase polymerase chain reaction
- FACS
fluorescence-activated cell sorting
- DAC
5-aza-2′-deoxycytidine
Footnotes
This work was supported by The Prostate Foundation (formerly CapCURE) and the National Institutes of Health (CA76184 and CA97131).
Steven R. Schwarze and Vivian X. Fu have contributed equally to this work.
References
- 1.Campisi J. Cancer, aging and cellular senescence. [Review; 60 references] In Vivo. 2000:183–188. [PubMed] [Google Scholar]
- 2.Torres K, Horwitz SB. Mechanisms of Taxol-induced cell death are concentration dependent. Cancer Res. 1998:3620–3626. [PubMed] [Google Scholar]
- 3.Chang BD, Broude EV, Dokmanovic M, Zhu H, Ruth A, Xuan Y, Kandel ES, Lausch E, Christov K, Roninson IB. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res. 1999:3761–3767. [PubMed] [Google Scholar]
- 4.Schwarze SR, DePrimo SE, Grabert LM, Fu VX, Brooks JD, Jarrard DF. Novel pathways associated with bypassing cellular senescence in human prostate epithelial cells. J Biol Chem. 2002:14877–14883. doi: 10.1074/jbc.M200373200. [DOI] [PubMed] [Google Scholar]
- 5.Untergasser G, Koch HB, Menssen A, Hermeking H. Characterization of epithelial senescence by serial analysis of gene expression: identification of genes potentially involved in prostate cancer. Cancer Res. 2002:6255–6262. [PubMed] [Google Scholar]
- 6.Zhang H, Pan KH, Cohen SN. Senescence-specific gene expression fingerprints reveal cell-type-dependent physical clustering of up-regulated chromosomal loci. Proc Natl Acad Sci USA. 2003:3251–3256. doi: 10.1073/pnas.2627983100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schwarze SR, Shi Y, Fu VX, Watson PA, Jarrard DF. Role of cyclin-dependent kinase inhibitors in the growth arrest at senescence in human prostate epithelial and uroepithelial cells. Oncogene. 2001:8184–8192. doi: 10.1038/sj.onc.1205049. [DOI] [PubMed] [Google Scholar]
- 8.Chang BD, Watanabe K, Broude EV, Fang J, Poole JC, Kalinichenko TV, Roninson IB. Effects of p21WAF1/Cip1/Sdi1 on cellular gene expression: implications for carcinogenesis, senescence, and age-related diseases. Proc Natl Acad Sci USA. 2000:4291–4296. doi: 10.1073/pnas.97.8.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jarrard DF, Bova GS, Ewing CM, Pin SS, Nguyen SH, Baylin SB, Cairns P, Sidransky D, Herman JG, Isaacs WB. Deletional, mutational, and methylation analyses of CDKN2 (p16/MTS1) in primary and metastatic prostate cancer. Genes Chromosomes Cancer. 1997:90–96. [PubMed] [Google Scholar]
- 10.Gonzalgo ML, Isaacs WB. Molecular pathways to prostate cancer. J Urol. 2003:2444–2452. doi: 10.1097/01.ju.0000085381.20139.b6. [DOI] [PubMed] [Google Scholar]
- 11.Sasaki M, Honda T, Yamada H, Wake N, Barrett JC, Oshimura M. Evidence for multiple pathways to cellular senescence. Cancer Res. 1994:6090–6093. [PubMed] [Google Scholar]
- 12.Pereira-Smith OM, Smith JR. Genetic analysis of indefinite division in human cells: identification of four complementation groups. Proc Natl Acad Sci USA. 1988:6042–6046. doi: 10.1073/pnas.85.16.6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Itahana K, Dimri G, Campisi J. Regulation of cellular senescence by p53. [Review; 78 references] Eur J Biochem. 2001:2784–2791. doi: 10.1046/j.1432-1327.2001.02228.x. [DOI] [PubMed] [Google Scholar]
- 14.Jarrard DF, Sarkar S, Shi Y, Yeager TR, Magrane G, Kinoshita H, Nassif N, Meisner L, Newton MA, Waldman FM, et al. p16/pRb pathway alterations are required for bypassing senescence in human prostate epithelial cells. Cancer Res. 1999:2957–2964. [PubMed] [Google Scholar]
- 15.Chang BD, Xuan Y, Broude EV, Zhu H, Schott B, Fang J, Roninson IB. Role of p53 and p21waf1/cip1 in senescence-like terminal proliferation arrest induced in human tumor cells by chemotherapeutic drugs. Oncogene. 1999:4808–4818. doi: 10.1038/sj.onc.1203078. [DOI] [PubMed] [Google Scholar]
- 16.Chang BD, Swift ME, Shen M, Fang J, Broude EV, Roninson IB. Molecular determinants of terminal growth arrest induced in tumor cells by a chemotherapeutic agent. Proc Natl Acad Sci USA. 2002:389–394. doi: 10.1073/pnas.012602599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beausejour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, Campisi J. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. 2003:4212–4222. doi: 10.1093/emboj/cdg417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang BD, Broude EV, Dokmanovic M, Zhu H, Ruth A, Xuan Y, Kandel ES, Lausch E, Christov K, Roninson IB. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res. 1999:3761–3767. [PubMed] [Google Scholar]
- 19.Robles SJ, Adami GR. Agents that cause DNA double strand breaks lead to p16INK4a enrichment and the premature senescence of normal fibroblasts. Oncogene. 1998:1113–1123. doi: 10.1038/sj.onc.1201862. [DOI] [PubMed] [Google Scholar]
- 20.Ramirez RD, Morales CP, Herbert BS, Rohde JM, Passons C, Shay JW, Wright WE. Putative telomere-independent mechanisms of replicative aging reflect inadequate growth conditions. Genes Dev. 2001:398–403. doi: 10.1101/gad.859201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwarze SR, Luo J, Isaacs WB, Jarrard DF. Modulation of CXCL14 (BRAK) expression in prostate cancer. Prostate. 2005:67–74. doi: 10.1002/pros.20215. [DOI] [PubMed] [Google Scholar]
- 22.Fujitani M, Yamagishi S, Che YH, Hata K, Kubo T, Ino H, Tohyama M, Yamashita T. P311 accelerates nerve regeneration of the axotomized facial nerve. J Neurochem. 2004:737–744. doi: 10.1111/j.1471-4159.2004.02738.x. [DOI] [PubMed] [Google Scholar]
- 23.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwarze SR, DePrimo SE, Grabert LM, Fu VX, Brooks JD, Jarrard DF. Novel pathways associated with bypassing cellular senescence in human prostate epithelial cells. J Biol Chem. 2002:14877–14883. doi: 10.1074/jbc.M200373200. [DOI] [PubMed] [Google Scholar]
- 25.Severino J, Allen RG, Balin S, Balin A, Cristofalo VJ. Is beta-galactosidase staining a marker of senescence in vitro and in vivo? Exp Cell Res. 2000:162–171. doi: 10.1006/excr.2000.4875. [DOI] [PubMed] [Google Scholar]
- 26.Xu C, Meikrantz W, Schlegel R, Sager R. The human papilloma virus 16E6 gene sensitizes human mammary epithelial cells to apoptosis induced by DNA damage. Proc Natl Acad Sci USA. 1995:7829–7833. doi: 10.1073/pnas.92.17.7829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tombal B, Weeraratna AT, Denmeade SR, Isaacs JT. Thapsigargin induces a calmodulin/calcineurin-dependent apoptotic cascade responsible for the death of prostatic cancer cells. Prostate. 2000:303–317. doi: 10.1002/1097-0045(20000601)43:4<303::aid-pros10>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 28.Weller EM, Poot M, Hoehn H. Induction of replicative senescence by 5-azacytidine: fundamental cell kinetic differences between human diploid fibroblasts and NIH-3T3 cells. Cell Prolif. 1993:45–54. doi: 10.1111/j.1365-2184.1993.tb00005.x. [DOI] [PubMed] [Google Scholar]
- 29.Chang BD, Broude EV, Dokmanovic M, Zhu H, Ruth A, Xuan Y, Kandel ES, Lausch E, Christov K, Roninson IB. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res. 1999:3761–3767. [PubMed] [Google Scholar]
- 30.Hussain M, Smith DC, El Rayes BF, Du W, Vaishampayan U, Fontana J, Sakr W, Wood D. Neoadjuvant docetaxel and estramustine chemotherapy in high-risk/locally advanced prostate cancer. Urology. 2003:774–780. doi: 10.1016/s0090-4295(02)02519-0. [DOI] [PubMed] [Google Scholar]
- 31.Nichols WW, Buynak EB, Bradt C, Hill R, Aronson M, Jarrell BE, Mueller SN, Levine EM. Cytogenetic evaluation of human endothelial cell cultures. J Cell Physiol. 1987:453–462. doi: 10.1002/jcp.1041320307. [DOI] [PubMed] [Google Scholar]
- 32.Dimri GP, Campisi J. Molecular and cell biology of replicative senescence. Cold Spring Harbor Symp Quant Biol. 1994:67–73. doi: 10.1101/sqb.1994.059.01.010. [Review; 28 references] [DOI] [PubMed] [Google Scholar]
- 33.Chang BD, Broude EV, Dokmanovic M, Zhu H, Ruth A, Xuan Y, Kandel ES, Lausch E, Christov K, Roninson IB. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res. 1999:3761–3767. [PubMed] [Google Scholar]
- 34.Lowe SW, Ruley HE, Jacks T, Housman DE. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell. 1993:957–967. doi: 10.1016/0092-8674(93)90719-7. [DOI] [PubMed] [Google Scholar]
- 35.Faraco J, Bashir M, Rosenbloom J, Francke U. Characterization of the human gene for microfibril-associated glycoprotein (MFAP2), assignment to chromosome 1p36.-p35, and linkage to D1S170. Genomics. 1995:630–637. doi: 10.1016/0888-7543(95)80004-6. [DOI] [PubMed] [Google Scholar]
- 36.Mok SC, Wong KK, Chan RK, Lau CC, Tsao SW, Knapp RC, Berkowitz RS. Molecular cloning of differentially expressed genes in human epithelial ovarian cancer. Gynecol Oncol. 1994:247–252. doi: 10.1006/gyno.1994.1040. [DOI] [PubMed] [Google Scholar]
- 37.Chondrogianni N, de CMS, Franceschi C, Gonos ES. Cloning of differentially expressed genes in skin fibroblasts from centenarians. Biogerontology. 2004:401–409. doi: 10.1007/s10522-004-3188-1. [DOI] [PubMed] [Google Scholar]
- 38.Roninson IB. Tumor cell senescence in cancer treatment. Cancer Res. 2003:2705–2715. [PubMed] [Google Scholar]
- 39.Kawamata H, Uchida D, Hamano H, Kimura-Yanagawa T, Nakashiro KI, Hino S, Omotehara F, Yoshida H, Sato M. Active MMP2 in cancer cell nests of oral cancer patients: correlation with lymph node metastasis. Int J Oncol. 1998:699–704. [PubMed] [Google Scholar]
- 40.Suwiwat S, Ricciardelli C, Tammi R, Tammi M, Auvinen P, Kosma VM, LeBaron RG, Raymond WA, Tilley WD, Horsfall DJ. Expression of extracellular matrix components versican, chondroitin sulfate, tenascin, and hyaluronan, and their association with disease outcome in node-negative breast cancer. Clin Cancer Res. 2004:2491–2498. doi: 10.1158/1078-0432.ccr-03-0146. [DOI] [PubMed] [Google Scholar]
- 41.Shellenberger TD, Wang M, Gujrati M, Jayakumar A, Strieter RM, Burdick MD, Ioannides CG, Efferson CL, El Naggar AK, Roberts D, et al. BRAK/CXCL14 is a potent inhibitor of angiogenesis and a chemotactic factor for immature dendritic cells. Cancer Res. 2004:8262–8270. doi: 10.1158/0008-5472.CAN-04-2056. [DOI] [PubMed] [Google Scholar]
- 42.te Poele RH, Okorokov AL, Jardine L, Cummings J, Joel SP. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res. 2002:1876–1883. [PubMed] [Google Scholar]