

Abstract
Structural maintenance of chromosomes (SMC) proteins play fundamental roles in higher-order chromosome dynamics from bacteria to humans. It has been proposed that the Bacillus subtilis SMC (BsSMC) homodimer is composed of two anti-parallel coiled-coil arms, each having an ATP-binding domain at its distal end. It remains totally unknown, however, how the two-armed structure supports ATP-dependent actions of BsSMC. By constructing a number of mutant derivatives including ‘single-armed’ BsSMC, we show here that the central hinge domain provides a structural flexibility that allows opening and closing of the two arms. This unique structure brings about bimodal regulation of the SMC ATPase cycle. Closing the arm can trigger ATP hydrolysis by allowing an end–end interaction within a dimer (intramolecular mode). When bound to DNA, ATP promotes a dimer–dimer interaction, which in turn activates their DNA-dependent ATPase activity (intermolecular mode). Our results reveal a novel mechanism of ATPase regulation and provide mechanistic insights into how eukaryotic SMC protein complexes could mediate diverse chromosomal functions, such as chromosome condensation and sister chromatid cohesion.
Keywords: Bacillus subtilis/chromosome dynamics/cohesin/condensin/structural maintenance of chromosomes
Introduction
The faithful segregation of genetic information requires highly coordinated structural changes of chromosomes during the cell cycle (reviewed by Koshland and Strunnikov, 1996; Hirano, 2000; Nasmyth et al., 2000). Although the molecular basis underlying these changes remains poorly understood, recent studies have shown that members of the structural maintenance of chromosomes (SMC) family play central roles in higher-order chromosome dynamics from bacteria to humans (reviewed by Hirano, 1999; Strunnikov and Jessberger, 1999; Cobbe and Heck, 2000). Eukaryotic cells have at least four different SMC family members. A specific pair of two different SMC proteins forms a heterodimer, which further associates with non-SMC subunits to produce a large protein complex. For example, SMC2- and SMC4-type proteins act as the core subunits of the 13S condensin complex that is essential for chromosome condensation in Xenopus laevis egg extracts (Hirano and Mitchison, 1994; Hirano et al., 1997) and in yeast (Saka et al., 1994; Strunnikov et al., 1995; Sutani et al., 1999; Freeman et al., 2000). On the other hand, cohesin, a complex containing SMC1- and SMC3-type subunits, is required for establishing the linkage between sister chromatids (Guacci et al., 1997; Michaelis et al., 1997; Losada et al., 1998; Tomonaga et al., 2000). In addition, it has been shown that similar, yet distinct SMC protein complexes participate in dosage compensation in Caenorhabditis elegans (Chuang et al., 1994, 1996) and recombinational repair in mammalian cell extracts (Jessberger et al., 1996).
Unlike eukaryotes, each of the bacterial or archaeal genomes encodes a single smc gene, and its gene product functions as a homodimer (Hirano and Hirano, 1998; Melby et al., 1998). Null mutations of smc genes in Bacillus subtilis and Caulobacter crescentus cause multiple phenotypes, including accumulation of anucleate cells, disruption of nucleoid structure, and misassembly of a protein complex involved in chromosome partitioning (Britton et al., 1998; Jensen and Shapiro, 1999). Thus, the bacterial SMC proteins also play important roles in chromosome segregation, although no clear distinction between the cohesion and condensation processes is observed in the bacterial chromosome cycle. In some bacterial species, such as Escherichia coli and Haemophilus influenzae, SMC functions are executed by a distantly related protein called MukB (reviewed by Hiraga, 2000).
Despite the rich background of their biology, very little is known about how SMC proteins might work at a mechanistic level. SMC proteins are large polypeptides (between 1000 and 1500 amino acids long) that share common structural motifs. The N-terminal domain contains a nucleotide-binding motif (the Walker A motif) and the C-terminal domain contains a conserved sequence termed the DA-box (containing a putative Walker B motif). The central domain consists of two long coiled-coil regions connected by a non-helical hinge sequence. An electron microscopy (EM) study showed that the B.subtilis SMC (BsSMC) dimer has a two-armed symmetrical struc ture, each arm of which consists of a long anti-parallel coiled-coil (Melby et al., 1998). Such an anti-parallel arrangement nicely explains how the two conserved ATP-binding motifs located in the N- and C-terminal domains can make direct contact with each other, possibly constituting an ATP-binding pocket (Saitoh et al., 1994). The EM study also suggested that the central hinge may be structurally flexible, allowing a ‘scissoring’ action of the SMC homodimer. Although this structural model suggests an intriguing potential of SMC actions, no direct biochemical evidence to support this model is currently available.
Biochemical activities associated with SMC proteins have been best characterized using the Xenopus 13S condensin complex. This five-subunit protein complex displays a DNA-stimulated ATPase activity and reconfigures DNA structure in an ATP hydrolysis-dependent manner in vitro. It introduces positive supercoils into relaxed circular DNA in the presence of type I topoisomerases (Kimura and Hirano, 1997) and converts nicked circular DNA into positively knotted forms in the presence of a type II topoisomerase (Kimura et al., 1999). Although the two SMC subunits of 13S condensin are likely to be responsible for binding and hydrolysis of ATP, neither the DNA-stimulated ATPase nor the ATP-dependent supercoiling activity can be supported by a purified SMC heterodimer alone (Kimura and Hirano, 2000). The bacterial SMC protein (BsSMC) exhibits biochemical properties substantially different from those of the eukaryotic condensin complex (Hirano and Hirano, 1998). BsSMC is a simple homodimer that binds preferentially to ssDNA and displays a DNA-stimulated ATPase activity without any associated subunits. It forms large aggregates of ssDNA in an ATP-dependent manner, but this reaction does not require ATP hydrolysis. It remains totally unknown how these biochemical activities are supported by the proposed two-armed structure of SMC proteins or how their mechanical actions are coupled with the cycle of ATP binding and hydrolysis.
In this report, we have used BsSMC as a model system to understand the basic mechanism of SMC actions. By constructing a number of BsSMC mutant derivatives, we show that the flexible hinge connecting the two arms plays a crucial role in modulating intra- and inter-molecular protein–protein interactions of BsSMC, being responsible for bimodal regulation of its ATPase cycle. This novel mechanism of ATPase regulation provides fundamental implications for our understanding of the actions of eukaryotic SMC protein complexes.
Results
Construction of BsSMC mutant derivatives
The central region of BsSMC consists of a putative hinge domain flanked by two long coiled-coil domains (Figure 1A, upper). To understand the role of the hinge domain, we have performed site-directed mutagenesis with a major focus on glycine residues highly conserved in SMC proteins. Since glycines are amino acid residues that potentially break α-helices and contribute to the structural flexibility of polypeptides, we anticipated that they might have important roles in the postulated hinge function. BsSMC has five glycines in the conserved hinge region (G651, G657, G658, G662 and G663), and four of them (except G657) are invariant among bacterial, archaeal and eukaryotic SMC proteins (Figure 1A, lower, left). We constructed seven different mutants in which these glycines were systematically replaced with alanines in different combinations (Figure 1A, lower, right). For simplicity, we call these mutant proteins by their substituted sequences. For example, a double mutant with substitutions in the second and third glycines (G657A/G658A) is referred to as GAAGG. Likewise, the wild-type protein is called GGGGG.

Fig. 1. Design and purification of BsSMC mutant derivatives. (A) (Upper) Coiled-coil prediction of BsSMC (Berger et al., 1995). (Lower, left) Alignment of hinge sequences from different organisms. (Lower, right) The conserved glycine residues (G) were combinatorially replaced with alanines (A) to construct seven different hinge mutants. (B) Expression and purification of BsSMC mutant derivatives. Purified full-length proteins were fractionated by 7.5% SDS–PAGE and visualized by staining with Coomassie Blue (lanes 1–7). The hinge-less mutant, which consists of the N-terminal half (amino acid number 1–476) and C-terminal half (672–1186) fragments, was subjected to 10% SDS–PAGE and stained with Coomassie Blue (lane 8).
We have also introduced a point mutation in the Walker A motif located in the N-terminal domain (K37I; Hirano and Hirano, 1998) or in the putative Walker B motif located in the C-terminal domain (D1117A). SMC proteins share another conserved motif (LSGG; amino acid number 1089–1092), which is present upstream of the Walker B motif. This motif, often referred to as the ‘signature motif’ or the ‘C motif’, is shared with subclasses of ATPases including ATP-binding cassette (ABC) transporters (reviewed by Holland and Blight, 1999) and the double-strand break repair protein Rad50 (Hopfner et al., 2000). Although the function of the C motif remains elusive, it has been implicated in interactions between two ATP-binding domains in these proteins (Jones and George, 1999; Hopfner et al., 2000). Mutations in this motif in the cystic fibrosis transmembrane regulator (CFTR), a member of the ABC transporter family, cause cystic fibrosis (Zielenski and Tsui, 1995). To understand the role of the C motif in SMC functions, we replaced the conserved serine residue (S1090) with arginine, constructing the S1090R mutant.
To simplify purification protocols, each of the wild-type and mutant polypeptides was tagged with a His6 sequence at its C-terminus. The tagged proteins were purified by a combination of nickel-affinity and ion-exchange column chromatography (Figure 1B, lanes 1–7). The His6-tagged wild-type protein purified in this way had the same set of biochemical activities (DNA-binding, ATPase and ssDNA reannealing and aggregation) as the untagged version (Hirano and Hirano, 1998).
Finally, as an attempt to test the anti-parallel dimer model, we designed a strategy for constructing a deletion mutant of BsSMC. The model predicts that each arm of an SMC dimer is composed of the N- and C-terminal halves of a BsSMC polypeptide (Melby et al., 1998). If this is correct, we should be able to make a single arm of BsSMC by co-expressing the two fragments in bacterial cells. When an N-terminal fragment (1–476) or a C-terminal, His6-tagged fragment (672–1186) was expressed separately in E.coli, it formed an insoluble aggregate (data not shown). When the two fragments were co-expressed in the bacterial cells, however, both of them were recovered in a soluble fraction. The two polypeptides tightly associated with each other and co-fractionated by nickel-affinity and ion-exchange column chromatography (Figure 1B, lane 8). Hydrodynamic properties and electron micrographs of this protein were consistent with the prediction that it is composed of a single arm of BsSMC (see below). Since the co-expressed fragments lacked sequences corresponding to the hinge domain, we call this mutant protein ‘hinge-less’.
Hydrodynamic properties of BsSMC mutants
Melby et al. (1998) provided evidence that the central hinge of MukB is flexible, and both MukB and BsSMC showed a range of conformations from the ‘folded rod’, with the hinge completely closed, to the ‘open V’, with the hinge open at angles up to 180°. To test how solution conditions might affect the transition from folded rod to open V, we centrifuged wild-type BsSMC in sucrose gradients under different salt conditions. We found that wild-type BsSMC (GGGGG) undergoes large conformational changes in response to salt titration. Increased concentrations of salt resulted in a progressive decrease in the sedimentation coefficient of BsSMC, from 6.5S at 7.5 mM KCl to 5.0S at 1.0 M KCl (Figure 2). Conversely, the Stokes radius of this protein, as determined by gel filtration, increased from 10.5 nm at 100 mM KCl to 12.5 nm at 1.0 M KCl. The native molecular weight calculated from the two hydrodynamic parameters (Siegel and Monty, 1966) fitted well to the size of a dimer throughout the different salt concentrations tested (Table I). A straightforward interpretation of these results is that the hinge is indeed flexible, allowing a global conformational change of BsSMC from a folded rod at low ionic strength to an open V at high ionic strength. Simulations of bent, rod-like molecules using the Kirkwood (1954) formalism (H.P.Erickson, unpublished) show that the largest decrease in sedimentation coefficient occurs in the initial transition from a fully closed folded rod to an open V molecule with a small angle. For a fully folded rod (0° bend between arms) of 6.5S, the sedimentation coefficient decreases to 5.7S when the hinge opens 20°, then to 5.1S at a 90° bend, and to 4.9S for the fully straightened rod.

Fig. 2. Salt-induced conformational changes of BsSMC and its mutant derivatives. (A) Wild-type BsSMC (GGGGG) or its derivatives (GAAAA and hinge-less) was fractionated on 5–20% sucrose gradients containing different concentrations of KCl and a fixed concentration of MgCl2 (2.5 mM). Fractions were precipitated with trichloroacetic acid (TCA), resolved by 7.5% SDS–PAGE and stained with Coomassie Blue. Five examples of runs at 7.5 mM or 1.0 M KCl are shown here. The positions of three protein standards (BSA [4.6S], open circle; aldolase [7.3S], open triangle; catalase [11.4S], open square) are indicated. (B) Sedimentation coefficients of five different proteins were plotted as a function of KCl concentration.
Table I. Estimation of native molecular weights under different salt conditions.
| Wild type (GGGGG) |
Hinge mutant (GAAAA) |
|||||
|---|---|---|---|---|---|---|
| Sediment (S) | Stokes (nm) | cal MW (kDa) | Sediment (S) | Stokes (nm) | cal MW (kDa) | |
| 100 mM | 6.5 | 10.5 | 281 | 3.6 | 8.6 | 128 |
| 125 mM | 6.5 | 10.8 | 290 | 3.6 | 8.7 | 129 |
| 200 mM | 6.4 | 11.2 | 296 | 3.6 | 9.5 | 141 |
| 500 mM | 5.7 | 11.2 | 263 | 3.6 | 9.7 | 144 |
| 1000 mM | 5.0 | 12.5 | 258 | 3.5 | 10.2 | 147 |
| 2000 mM | 4.9 | 13.3 | 269 | N. T. | N. T. | N. T. |
| Predicted MW | dimer | (276) | monomer | (138) | ||
Sedimentation coefficients (Sediment) and Stokes radii (Stokes) were determined by sucrose gradient centrifugation and gel filtration, respectively. Native molecular weights (cal MW) were calculated from these values (Siegel and Monty, 1966). Predicted molecular weights (predicted MW) are the masses of dimer and monomer predicted from the amino acid sequence of BsSMC. N. T., not tested.
We then studied three hinge mutants (GAAGG, GGGAA and GAAAA) in the sucrose gradient centrifugation assay. We found that these mutations synergistically affected the sedimentation property of BsSMC. The GAAGG mutant displayed consistently lower S values than the wild-type protein at different salt concentrations (Figure 2B). The sedimentation coefficient of GGGAA was even smaller and exhibited a sharp reduction from 5.7S at 7.5 mM KCl to 4.4S at 50 mM KCl. The quadruple mutant GAAAA presented the most severe phenotype (Figure 2): its sedimentation coefficient was constitutively small (∼3.6–4.2S) and showed only a small decrease upon salt titration. We also examined the sedimentation properties of the remaining hinge mutants (AGGGG, AAAGG, AGGAA, AAAAA), each having an additional mutation at G651. We found that the G651A mutation had little, if any, effect on the sedimentation properties of BsSMC in any combinations with other hinge mutations (data not shown).
One explanation for the synergistic decreases in the sedimentation coefficient is that the hinge mutations may increase the angle of the two arms. If this is the case, the quadruple mutant GAAAA may be fixed into a constitutively open configuration with a larger Stokes radius. To look directly for the structural basis for the altered sedimentation property, we performed rotary shadowing EM of the GAAAA molecules. Surprisingly, the EM showed rod-shaped molecules that looked like a single arm of BsSMC (see below), suggesting that the GAAAA mutant was a monomer. Gel filtration of GAAAA under different salt concentrations revealed that the Stokes radius of this mutant protein was much smaller (8.6–10.2 nm) than that of wild-type BsSMC (10.5–12.5 nm; Table I). The molecular weight of GAAAA calculated from the measured sedimentation coefficient and Stokes radius was in excellent agreement with that of a monomer. This conclusion was further supported by the hydrodynamic properties of the hinge-less mutant, which were similar to those of GAAAA (Figure 2; data not shown).
EM of BsSMC mutants
Direct visualization of the mutant proteins by EM provided the final proof that the hinge mutations cause a shift from dimers to monomers. We found that 100% of the wild-type protein (GGGGG) had a two-armed structure, with arm angles from 0 (folded rod) to nearly 180°, as seen previously (Melby et al., 1998; Figure 3A and B). Of the molecules, 53% had the two arms open, 25% had the coils apart but the ends close together (<5 nm apart), and 22% were in the folded rod conformation (Figure 3C, first row). In contrast, 98% of the quadruple mutant (GAAAA) displayed a single-armed structure (Figure 3A and B). Folded rods of the GGGGG dimer were distinguishable from GAAAA monomers by the presence of a globular domain at each end and by their increased thickness (Figure 3C). Single coiled-coil arms were 2.4 ± 0.8 nm (n = 70) wide, whereas folded two-armed molecules were 4.6 ± 1.1 nm (n = 16) wide. The GAAGG and GGGAA mutants were a mixture of two-armed and single-armed structures, and GGGAA had a higher ratio of single-armed rods than GAAGG (Figure 3A and B). In these experiments, proteins were sprayed onto mica in a solution containing 0.2 M ammonium bicarbonate and ∼30% glycerol, and then dried under vacuum before rotary shadowing, making local salt concentrations of the specimen variable during the drying process. Presumably due to this technical problem, no reproducible difference in the arm angles was observed among the two-armed populations of GGGGG, GAAGG and GGGAA. High-magnification images of GAAAA and hinge-less protein are also shown in Figure 3C. As expected, the total length of GAAAA (55.9 ± 2.9 nm, n = 14) was significantly longer than that of the hinge-less mutant (46.2 ± 2.0 nm, n = 13) because the latter lacks the 195 amino acid-long hinge sequence.

Fig. 3. EM of BsSMC and its mutant derivatives. (A) Low magnification of wild type (GGGGG) and three different hinge mutants (GGGAA, GAAGG and GAAAA) visualized by rotary shadowing. Bar, 100 nm. (B) Numbers and percentages of two-armed and single-armed structures in different BsSMC mutants as determined by EM. (C) High magnification of GGGGG [from left to right: ‘open-V’ (two examples), ‘ends-split’, ‘coils-spread’, ‘folded-rod’], GAAAA and hinge-less proteins. Bar, 50 nm.
ATP-binding activity of BsSMC mutants
We then tested the ATP-binding activity of BsSMC by a UV cross-linking method (Figure 4). Wild-type BsSMC bound efficiently to radiolabeled ATP in this assay. The three hinge mutants (GAAGG, GGGAA and GAAAA) and the hinge-less mutant also displayed ATP-binding activities comparable to that of wild type. In the hinge-less mutant, the majority of cross-linked ATP was detected in the N-terminal fragment containing the Walker A motif. Thus, the two-armed structure is not necessarily required for assembling a functional ATP-binding domain within an arm. The ATP-binding activity of BsSMC was abolished in the Walker A (K37I) and putative Walker B (D1117A) mutants, whereas the C motif mutant S1090R exhibited a wild-type level of ATP binding.
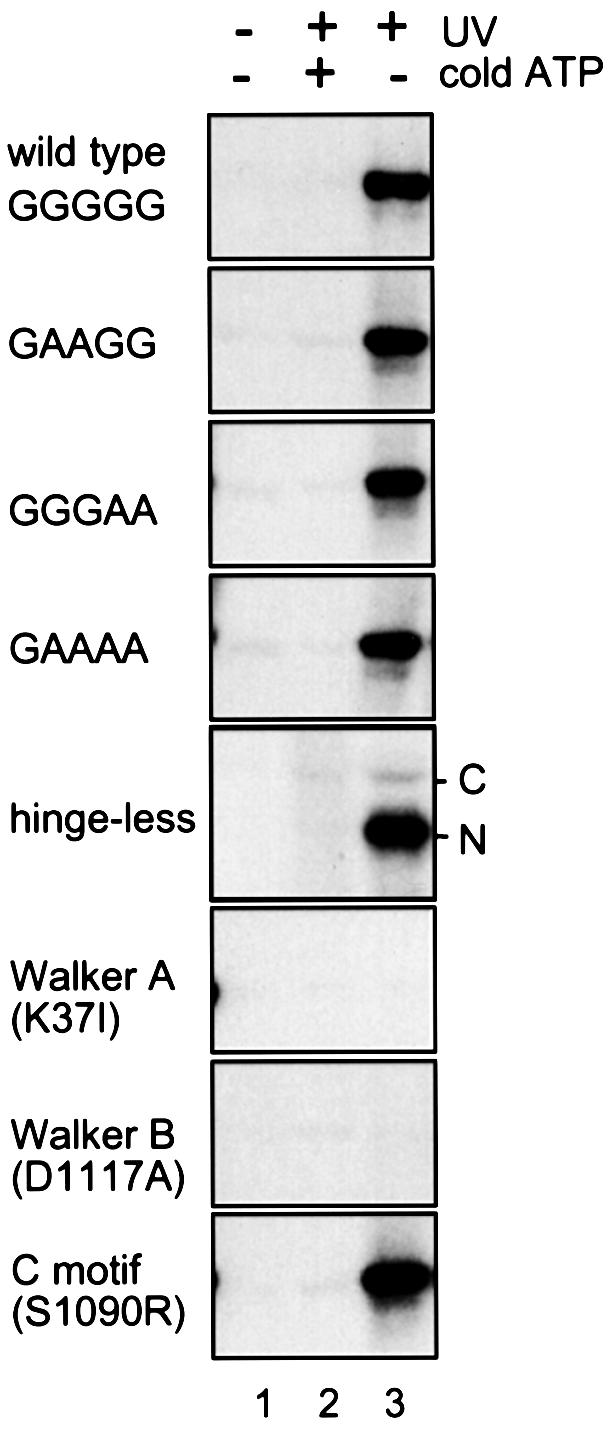
Fig. 4. UV cross-linking of ATP to BsSMC. BsSMC protein was incubated in a buffer containing 7.5 mM KCl and 2.5 mM MgCl2 in the presence of [α-32P]ATP. After incubation at 37°C for 30 min, the sample was irradiated with no UV (lane 1) or UV (lanes 2 and 3) to induce cross-linking in the absence (lanes 1 and 3) or presence (lane 2) of 1 mM non-radioactive ATP. Labeled polypeptides were fractionated by 7.5% SDS–PAGE and visualized by autoradiography.
Two distinct modes of ATPase activation in the absence or presence of DNA
The character of BsSMC ATPase is highly complex: it has DNA-independent and DNA-dependent activities that respond differently to KCl or MgCl2 titration (Hirano and Hirano, 1998). The intrinsic (DNA-independent) ATPase activity of wild-type BsSMC peaked at 50–100 mM KCl (Figure 5A, panel 1) and at 0.5–1.0 mM MgCl2 (Figure 5A, panels 3 and 5). The single-armed hinge mutant (GAAAA) lost its response to KCl, displaying a constitutively low ATPase activity (Figure 5A, panel 1). MgCl2 titration curves of GAAAA were also different from those of the wild-type protein (Figure 5A, panels 3 and 5). The GAAGG and GGGAA mutants had intermediate characters (Figure 5A, panel 3), and the hinge-less ATPase was similar to the GAAAA ATPase (data not shown). These results suggest that the two-armed structure of BsSMC plays an important role in regulating its intrinsic ATPase activity. In contrast, in the presence of DNA, the ATPase activity of GAAAA was comparable to that of wild type at low salt concentrations (5–50 mM KCl) although it declined more sharply at medium salt concentrations (100–200 mM KCl) (Figure 5A, panel 2). MgCl2 titration curves of the wild-type and mutant ATPases were indistinguishable at 5 mM KCl (Figure 5A, panel 4) and very similar at 50 mM KCl (Figure 5A, panel 6). Thus, the DNA-dependent ATPases of the mutant proteins appear to be fully active under these low-salt conditions.

Fig. 5. Two distinct modes of ATPase activation in the absence or presence of DNA. (A) KCl titration at 2 mM MgCl2 (panels 1 and 2), MgCl2 titration at 5 mM KCl (panels 3 and 4), and MgCl2 titration at 50 mM KCl (panels 5 and 6) in the absence (panels 1, 3 and 5) or presence (panels 2, 4 and 6) of φX174 virion DNA (31.2 μM nucleotides). A fixed protein concentration of 300 nM arms (equivalent to 150 nM dimers in the case of wild-type BsSMC) was used for all the proteins. The rate of ATP hydrolysis is expressed as the number of ATP molecules hydrolyzed per second per arm. (B) ATPase-defective mutants. The ATPase rate of four different proteins (wild type, K37I, D1117A and S1090R; 300 nM arms for each) was determined under different KCl/MgCl2 concentrations in the absence (solid bars) or presence (open bars) of φX174 virion DNA (31.2 μM nucleotides). (C) Mixing ATPase assay. A fixed concentration of wild type (300 nM arms) was mixed with increasing concentrations (0–1200 nM arms) of K37I (panel 1), D1117A (panel 2), S1090R (panel 3) or wild type (panel 4). The ATPase activity of the mixtures was determined in the absence (filled circles) or presence (open circles) of φX174 virion DNA (31.2 μM nucleotides). The concentrations of KCl and MgCl2 were 50 and 2 mM, respectively. The activity is shown by percentage of ‘wild-type ratio = 1 (300 nM arms)’. At the highest concentration of wild-type protein [panel 4; total protein ratio = 5 (1500 nM arms)], the rate of ATP hydrolysis is saturated and is no longer proportional to the input amount of protein.
As predicted from the ATP-binding assay, we found that a mutation either in the Walker A (K37I) motif or in the Walker B (D1117A) motif abolishes both the intrinsic and DNA-dependent ATPase activities under a variety of conditions tested (Figure 5B). Interestingly, the C motif mutant (S1090R) was also defective in ATP hydrolysis although its ATP binding was apparently normal (Figure 4). A mixing ATPase assay using these mutants revealed that different molecules of two-armed BsSMC functionally interact with each other on DNA. In this assay, a fixed amount of wild-type BsSMC was mixed with increasing amounts of the K37I, D1117A or S1090R mutants, and the ATPase activity of the mixtures was determined in the absence or presence of DNA. Since ATP hydrolysis by the mutant proteins was at the background level under this condition, the measured hydrolysis of ATP reflected the activity of wild-type protein. In the absence of DNA, the addition of the mutant proteins had little effect on the wild-type ATPase activity (Figure 5C, panels 1–3, –DNA). In contrast, the mutant proteins greatly suppressed wild-type ATPase in the presence of DNA (Figure 5C, panels 1–3, +DNA). The addition of increasing amounts of wild-type protein (instead of the mutant proteins) resulted in a near-proportional increase in the rate of ATP hydrolysis under this condition (Figure 5C, panel 4). This observation ruled out the possibility that the mutant proteins titrate out DNA and thereby reduce the DNA-dependent ATPase activity of wild-type BsSMC. We conclude that the K37I, D1117A and S1090R mutants have dominant-negative effects on the ATPase activity of the wild-type protein only when they interact with each other on DNA.
ATP-stimulated protein–protein interactions of BsSMC on DNA
To further test whether different BsSMC molecules physically interact with each other on DNA, we performed a protein–protein cross-linking assay. In this assay, wild-type or mutant proteins were incubated in the absence or presence of DNA under different nucleotide conditions (no nucleotide, ATP, ATPγS), and cross-linked with bis-maleimidohexane (BMH), a protein cross-linker specific for free sulfhydryl groups (Perukhova et al., 1999). After quenching the cross-linking reaction, protein samples were analyzed by SDS–PAGE followed by immunoblotting. Without DNA, wild-type BsSMC produced a discrete set of bands regardless of the presence of nucleotides (Figure 6A, lanes 1–3, indicated by asterisks). They most likely correspond to the products of intramolecular cross-linking. The two-armed K37I and S1090R mutants produced similar cross-linking patterns under these conditions (Figure 6A, lanes 7–9 and 13–15). In the presence of DNA, however, a new set of larger cross-linked products appeared in wild-type BsSMC. The formation of a ladder of high-molecular-weight bands was greatly enhanced in the presence of ATP (Figure 6A, lanes 4 and 5, indicated by black circles). When ATP was replaced with ATPγS, a unique band of low intensity, instead of the ladder, was observed (Figure 6A, lane 6, indicated by an open triangle). The ATP- or ATPγS-stimulated cross-linking was abolished in K37I, further confirming the crucial role of ATP binding in this reaction (Figure 6A, lanes 10–12). We obtained an indistinguishable result with the Walker B mutant D1117A (data not shown). Interestingly, an opposite effect was observed with the C motif mutant S1090R: it exhibited a tendency to form the ladder even in the absence of ATP or in the presence of ATPγS (Figure 6A, lanes 16–18). This ATP- and DNA-stimulated ladder formation is most likely to represent intermolecular interactions of BsSMC. The single-armed GAAAA mutant also produced a similar ladder (data not shown), suggesting that the two-armed structure is not necessarily required for the ATP-stimulated protein– protein interactions of BsSMC on DNA.

Fig. 6. ATP-dependent protein–protein interactions of BsSMC on DNA. (A) BMH cross-linking. Three different BsSMC proteins [wild type (lanes 1–6), K37I (lanes 7–12), S1090R (lanes 13–18); 450 nM arms] were mixed with the indicated nucleotides in the absence or presence of φX174 virion DNA (15.6 μM nucleotides). After incubation, the proteins were cross-linked with BMH, fractionated in a 2.5–7.5% SDS polyacrylamide gel, and analyzed by immunoblotting with an anti-BsSMC antibody. The positions of the intramolecular cross-linking products (asterisks) and the intermolecular cross-linking products (ATP-stimulated products, filled circles; ATPγS-specific products, open triangles) are indicated. The position of non-cross-linked BsSMC polypeptide (135 kDa) is shown in lane 0. The concentrations of KCl and MgCl2 were 7.5 and 2.5 mM, respectively. (B) Spin-down assay. The same reactions were set up as in (A). After incubation, the mixtures were spun at 16 000 g for 15 min. The supernatants (upper) and pellets (lower) were separated, fractionated by 7.5% SDS–PAGE and stained with silver.
As we showed previously (Hirano and Hirano, 1998), wild-type BsSMC has an ability to form large aggregates of ssDNA in an ATP-dependent manner, as judged by a spin-down assay (Figure 6B, lanes 1–6; see Materials and methods). ATPγS enhanced the aggregation reaction. Such nucleotide dependency was abolished in the Walker A mutant K37I (Figure 6B, lanes 7–12). Conversely, S1090R formed ssDNA aggregates even in the absence of ATP (Figure 6B, lane 16). Thus, there is a correlation between the ability of BsSMC to form the DNA aggregates and its ability to display protein–protein interactions on DNA. Continuous hydrolysis of ATP is likely to affect the dynamics of the protein–protein interactions of BsSMC (see Discussion).
Discussion
Biochemical evaluation of the anti-parallel dimer model
The anti-parallel dimer model predicts two unique characters in the architecture of SMC proteins (Saitoh et al., 1994; Melby et al., 1998). One is the formation of an ATP-binding pocket from two distantly located Walker A and B motifs, and the second is the symmetrical positioning of the two catalytic domains at the end of long arms. The current work offers biochemical evidence to support this structural model and further provides mechanistic implications for the action of this unique class of chromosomal ATPases. Three lines of independent evidence support that BsSMC is composed of two long arms connected by a structurally flexible hinge. First, wild-type BsSMC dimer undergoes a salt-induced large conformational change that is readily detectable by the change in its hydrodynamic properties. Secondly, the successful construction of the hinge-less, single-armed mutant strongly suggests that each arm of BsSMC is composed of its N- and C-terminal halves. Thirdly, site-directed mutagenesis shows that the putative Walker B motif located in the C-terminal domain, together with the N-terminally located Walker A motif, indeed participates in ATP binding and hydrolysis. The two activities are retained in the hinge-less and single-armed GAAAA mutants, providing additional evidence for the anti-parallel arrangement of the N- and C-terminal coiled coils in all these molecules.
Our initial motivation behind the mutagenesis of the hinge domain was to construct a so-called ‘stiff-hinge’ mutant. Since glycines are often found in loop regions of polypeptides and contribute to their structural flexibility, we anticipated that replacement of the conserved glycines with alanines would produce an inflexible hinge. As expected, we have observed that combinations of these mutations synergistically decrease the sedimentation coefficient of BsSMC, being indicative of arm opening. Unexpectedly, however, we have found that one of the severest mutants (GAAAA) disrupts dimerization of BsSMC, resulting in the formation of single-armed monomers. The intermediate mutants (GAAGG and GGGAA) contained a mixed population of monomers and dimers as judged by EM. The progressive decrease in the sedimentation coefficient of these mutants could therefore reflect two related yet distinct effects: an opening of the arms of dimers and a conversion from dimers to monomers. The latter effect was surprising because the original anti-parallel dimer model proposed that dimerization of SMC proteins is mediated by inter-subunit coiled-coil interactions (Melby et al., 1998; Figure 7A, Model I) and because such long coiled-coil interactions are believed to be very stable. We provide two explanations for our current results. First, the hinge region could act as a determinant to specify the overall folding pattern of BsSMC subunits. The functional hinge domain may actively prevent intra-subunit folding and produce dimers by promoting inter-subunit coiled-coil interactions. When the hinge domain is mutated, intra-subunit folding is favored, giving rise to single-armed monomers (Figure 7A, GAAAA). Secondly, there is another possibility for folding of wild-type BsSMC, where each subunit bends to form an intra-subunit, anti-parallel coiled coil, and then the dimer is formed by contacts of the two hinges (Figure 7A, Model II). If this is the case, the hinge mutations may simply weaken the dimerization by altering the hinge–hinge interface. This model, however, would require that the hinge–hinge bond be extremely strong since the wild-type dimer shows no sign of dissociation during sucrose gradient centrifugation even in the presence of 2 M KCl. Clearly, additional evidence will be required to distinguish between the two possibilities.

Fig. 7. Models. (A) Configuration of BsSMC. In wild-type BsSMC, dimerization may be mediated by coiled-coil interactions between two different subunits (Model I). Alternatively, the two subunits are self-folded to form two separate coiled-coil rods, which in turn dimerize by a hinge-mediated interaction (Model II). The GAAAA mutant is likely to be a self-folded monomer (right), and no alternative can be considered for the folding of this mutant. (B) ATP binding and hydrolysis cycle of BsSMC. Putative arrangement of the ATP binding pocket and the C motif is drawn on the basis of the crystal structure of the Rad50 catalytic domain (Hopfner et al., 2000). N and C indicate the N- and C-terminal domains of BsSMC, respectively. (C) Bimodal activation of SMC ATPase. The ATP-bound form of end domains is indicated by T. Each stage of conformational changes of BsSMC on DNA is numbered from 1 to 7. See the text for details.
ATP binding and hydrolysis cycle of BsSMC
The overall structure of SMC proteins is similar to that of the double-strand break repair protein Rad50. A recently reported crystal structure has shown that the globular catalytic domain of Rad50 is composed of N- and C-terminal sequences in which the Walker A and Walker B motifs interact to form an ATP-binding site (Hopfner et al., 2000). Interestingly, ATP binding induces an association of two catalytic domains, with the two ATPs sandwiched in the interface, thereby making a DNA-binding surface. A mutation in the C motif of Rad50 (S793R) abolishes the ATP-induced interaction of the two catalytic domains. By analogy, we infer that the corresponding mutant of BsSMC (S1090R) can not hydrolyze ATP because the mutation prevents a proper interaction between the globular ends, which is required for ATPase activation (Figure 7B). In contrast to Rad50, however, neither the end–end interaction nor ATP binding is essential for the DNA-binding activity of BsSMC (Hirano and Hirano, 1998). The S1090R mutant can bind to DNA and mimics an ATP-bound form of BsSMC although its behavior is not identical to the ATPγS-bound form of wild-type protein, as judged by protein–protein cross-linking. We suggest that ATPγS induces a stable association of two end domains (Figure 7B), thereby restricting the dynamic interaction between different BsSMC molecules (see below).
Bimodal activation of SMC ATPase mediated by the flexible hinge
What is the role of the ATP binding and hydrolysis cycle of BsSMC? As judged by sucrose gradient centrifugation, binding to ATP or ATPγS had little effect on the sedimentation properties of the two-armed and single-armed BsSMC (data not shown). We have no evidence that the ATPase cycle of BsSMC might actively control opening or closing of the two coiled-coil arms. Instead the current results suggest that the ATPase cycle acts as a bimodal switch that regulates the intra- and inter-molecular interactions of BsSMC.
In the absence of DNA, BsSMC displays an intrinsic ATPase activity that is highly dependent on Mg and KCl concentrations. We provide three lines of evidence to suggest that the flexible hinge of BsSMC allows an interaction between the two ATPase domains at the ends, thereby stimulating ATP hydrolysis (Figure 7C, inset). First, the decrease in wild-type ATPase at high-salt concentrations correlates well with its salt-induced change to a more open conformation that may reduce end–end interactions. Secondly, the intrinsic ATPase activity of the single-armed mutant is poor under conditions optimal for two-armed BsSMC. Thirdly, the S1090R mutation, which is expected to prevent the end–end interaction, completely abolishes the ATPase activity of BsSMC although the mutant retains the ability to bind to ATP. This intramolecular mode of ATPase stimulation could also occur on DNA (Figure 7C, stages 1 and 2).
When BsSMC binds to DNA, an additional, independent mode of the ATPase cycle is activated. We speculate that when the SMC molecule binds to DNA the intramolecular association of its catalytic domains is weakened, and the molecule adopts a more open conformation (Figure 7C, stage 3). This idea is supported by our previous observation that wild-type SMC becomes more sensitive to proteolytic cleavage when bound to DNA (Hirano and Hirano, 1998). In this configuration, ATP binding promotes intermolecular interactions of BsSMC on DNA, which can be revealed by protein– protein cross-linking. The ATP-dependent formation of ssDNA aggregates, as judged by the spin-down assay, suggests that such interactions can occur between BsSMC molecules bound to different DNA molecules. Since S1090R is active in both cross-linking and spin-down assays, the intermolecular interactions are likely to be initiated by coiled-coil association (Figure 7C, stage 4), not by end–end association. Subsequent hydrolysis of the bound ATP, however, requires intermolecular end–end interactions (Figure 7C, stage 5) because the ATPase-defective mutants (K37I, D1117A and S1090R) display a dominant-negative effect on the wild-type ATPase. This hydrolysis event is likely to be important for a destabilization of the intermolecular interactions (Figure 7C, stage 6), leading to a turnover of BsSMC (Figure 7C, stage 7).
It is not yet fully understood why protein–protein cross-linking of wild-type BsSMC is apparently poor in the presence of ATPγS. BsSMC has only four cysteines (C119, C437, C826 and C1114) that are cross-linkable with BMH. Since the length of this cross-linking reagent is very short (∼2 nm) compared with that of BsSMC (∼100 nm), BsSMC molecules must be very close together and fortuitously positioned on DNA so that the rare cysteines can be cross-linked intermolecularly. We speculate that intermolecular interactions of BsSMC are highly dynamic in the presence of ATP or when end–end association is prevented by the S1090R mutation (Figure 7C, stages 3 and 4). In this situation, multiple rounds of transient interactions would occur in different angles and positioning, and as a result increase the probability of hitting on cross-linkable states. In the presence of ATPγS, the protein–DNA complex would be trapped into a stable form, in which BsSMC molecules are oriented in such a way that the cysteines are not in close proximity and that intermolecular cross-linking rarely occurs (e.g. Figure 7C, stage 5).
In summary, we propose that the ATPase cycle of BsSMC is regulated by two different modes. Hinge-mediated arm closing can trigger ATP hydrolysis by allowing an end–end interaction within a dimer (intramolecular mode). Upon binding to DNA, the hinge tends to open and the catalytic domains of one dimer can interact with those of other dimers, causing ATP hydrolysis (intermolecular mode). This bimodal model for the activation of SMC ATPase naturally explains the functional importance of the symmetrical two-armed structure connected by the flexible hinge. It remains to be determined whether this model can also be extended to the action of Rad50 since the previous mechanistic study primarily used its isolated catalytic domains (Hopfner et al., 2000).
Implications for the actions of eukaryotic SMC protein complexes
The physiological role for the bimodal activation of BsSMC ATPase is not necessarily apparent because its mechanistic contribution to bacterial chromosome dynamics remains elusive. Nevertheless, we suggest that this model may provide a conceptual framework for our understanding of the actions of eukaryotic SMC protein complexes, condensin and cohesin, whose cellular functions have been better established. It is reasonable to extrapolate that the anti-parallel dimer model also applies to eukaryotic SMC heterodimers. On the basis of this assumption and their physiological roles, we previously hypothesized that condensin and cohesin could function as intramolecular and intermolecular DNA cross-linkers, respectively (Hirano, 1999). Since BsSMC is the prototype of the two eukaryotic SMC protein complexes, it must share common mechanistic characteristics with both complexes. We reason that the intramolecular activation mode of SMC ATPase is exaggerated in condensin’s action (Figure 7C, stages 1 and 2) whereas the intermolecular mode may prevail in cohesin’s action (Figure 7C, stages 4–6). A crucial point of this hypothesis is that the two different modes of the SMC ATPase cycle are mechanistically coupled with the two different modes of DNA interactions supported by condensin and cohesin. Consistently, a recent study by electron spectroscopic imaging revealed that a single condensin complex binds to a single DNA molecule, introducing supercoils into the DNA in an ATP-hydrolysis-dependent manner (D.Bazett-Jones, K.Kimura and T.Hirano, unpublished). Although it remains to be determined how cohesin’s activity is modulated by its ATP binding and hydrolysis cycle (Losada and Hirano, 2001), comparative structural and functional analyses of the two SMC protein complexes in the future should test and refine this model, which is admittedly highly speculative at this moment. We also anticipate that the idea presented here sheds light on how the eukaryotic SMC protein machines might have evolved to acquire their unique activities starting from a versatile prototype of SMC.
When this paper was under review, a crystal structure of the SMC end domain of Thermotoga maritima was published (Lowe et al., 2001). As expected, the fold of N- and C-terminal parts of SMC was very similar to that of Rad50. Since the SMC crystal was in an ATP-unbound form, however, little information was provided regarding the role of ATP binding and hydrolysis in the action of SMC proteins.
Materials and methods
Construction of BsSMC derivatives
A plasmid (pSO133) that expresses wild-type BsSMC with a His6 tag at its C-terminal end was constructed as follows. To modify the 3′-end of the coding sequence of BsSMC, a DNA fragment was first amplified by PCR using pSO104 (Hirano and Hirano, 1998) as a template. The primers used were: BS15 (5′-CGTCAGAGATGTCATCTAGC-3′) and BS28 (5′-GAGGATCCTGAACGAATTCTTTTGTTTC-3′; BamHI site underlined). The amplified fragment was digested with SnaBI and BamHI and inserted back into the SnaBI–BamHI site of pSO104. The full-length BsSMC coding sequence was excised from the resulting plasmid with NdeI and BamHI, and inserted into pET23b(+) (Novagen) to produce pSO133. Point mutations were introduced by using QuikChange site-directed mutagenesis kit (Stratagene) and confirmed by sequencing. The hinge-less BsSMC construct (pSO195) was designed so that a C-terminal fragment (corresponding to amino acid numbers 672–1186) and an N-terminal fragment (1–476) are co-expressed from a single expression vector. The C-terminal coding sequence was amplified by PCR and inserted into the NheI–BamHI site of pRSETA (Invitrogen) to generate pSO166. The primers used were: BS41 (5′-GAGCTAGCCTCCTTG GAAGAAGCCGG-3′; NheI site underlined) and BS36 (5′-GCGGAT CCTTACTGAACGAATTCTTTTGTTTC-3′; BamHI site underlined; an artificial termination codon is shown in bold). The N-terminal coding sequence was inserted into pSO166 in the following two steps. It was first amplified by PCR and inserted into the NdeI–NcoI site of pRSETA. The primers used were: BS43R (5′-GCCATATGTTCCTCAAAC GTTTAGAC-3′; NdeI site underlined; an initiation codon shown in bold) and BS42 (5′-GCCCATGGTTACAGAGCGGATTCATTTTTTTC-3′; NcoI site underlined; an artificial termination codon shown in bold). Then a different pair of primers was used to amplify the whole transcription unit containing the insert and its flanking regulatory elements. The primers used were: BS48 (5′-GTCCCCGGGGATCCC GCGAAATTAATACG-3′; SmaI site underlined) and BS49 (5′-GTC CCCGGGATATAGTTCCTCCTTTCAGC-3′; SmaI site underlined). The amplified fragment was digested with SmaI and inserted into the NaeI site of pSO166, giving rise to pSO195. In this construct, the C-terminal fragment has a His6 tag at its N-terminal end.
Purification of BsSMC derivatives
A 1-l culture of BL21(DE3)pLysS containing an expression plasmid was grown at 37°C in Luria–Bertani (LB) broth with antibiotics, and expression of BsSMC was induced by the addition of 50 µM isopropyl-β-d-thiogalactopyranoside (IPTG) at 30°C for 2 h. Cells were harvested, resuspended in 40 ml of lysis buffer (50 mM Na-phosphate pH 8.0, 300 mM NaCl, 10% glycerol), and subjected to three rounds of freeze–thaw. The cell suspension was supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF) and 0.5 mg/ml lysozyme, incubated on ice for 30 min, and sonicated. 2-mercaptoethanol was then added at a final concentration of 5 mM. After a spin at 23 000 g for 30 min, the supernatant was loaded onto a 7-ml Ni–NTA metal affinity column (Qiagen; column size, 1.0 × 8.8 cm). The column was washed with 70 ml of wash-1 buffer (50 mM Na-phosphate pH 6.0, 300 mM NaCl, 50 mM imidazole, 10% glycerol, 5 mM 2-mercaptoethanol) and then with 14 ml of wash-2 buffer (50 mM Na-phosphate pH 7.5, 300 mM NaCl, 50 mM imidazole, 10% glycerol, 5 mM 2-mercaptoethanol). Proteins were eluted with a 30-ml imidazole gradient (50–500 mM) containing 50 mM Na-phosphate pH 7.5, 300 mM NaCl, 10% glycerol and 5 mM 2-mercaptoethanol. The peak fractions were pooled (∼12 ml) and dialyzed against buffer M (20 mM K–HEPES pH 7.7, 1 mM EDTA, 10% glycerol, 0.1 mM PMSF) containing 50 mM KCl and 5 mM 2-mercaptoethanol. The dialysate was applied to a 5-ml Q–Sepharose FF column (Amersham Pharmacia Biotech; 1.0 × 6.3 cm), and fractionated with a 30-ml KCl gradient (100–600 mM) in buffer M containing 5 mM 2-mercaptoethanol. The peak fractions were pooled (∼8 ml), dialyzed against buffer M containing 50 mM KCl and 1 mM 2-mercaptoethanol, aliquotted, and stored at –70°C. The typical yield of BsSMC from a 1-l culture was ∼1.5 mg. In some cases, the Q–Sepharose fraction was applied to a 1-ml denatured DNA cellulose column (Amersham Pharmacia Biotech; 1.0 × 1.2 cm), and fractionated with a 6-ml KCl gradient (0.05–0.6 M) in buffer M containing 5 mM 2-mercaptoethanol. The peak fractions were pooled (∼2 ml), dialyzed and stored as described above. Inclusion or omission of the final purification step did not affect the functional properties of the BsSMC proteins obtained. Purification of the single-armed protein (expressed from pSO195) was performed in the same way except that Q–Sepharose FF was replaced with SP–Sepharose FF (Amersham Pharmacia Biotech). The concentrations of purified BsSMC were determined by SDS–PAGE followed by Coomassie Blue stain using bovine serum albumin (BSA) as a standard. To directly compare the activities of the two-armed and single-armed proteins, all protein concentrations are expressed in moles of BsSMC arms rather than moles of dimers or monomers.
Sucrose gradient centrifugation and gel filtration
Sucrose gradient centrifugation was carried out as described previously (Hirano and Hirano, 1998). For salt titration experiments, indicated concentrations of KCl were added into both protein solutions and sucrose gradients. Three protein standards (BSA, 4.6S; aldolase, 7.3S; catalase, 11.4S) were run with BsSMC proteins in the same gradients. Gel filtration was performed using a Superose 6 column (Amersham Pharmacia Biotech) that had been pre-equilibrated with buffers containing the indicated concentrations of KCl. Three protein standards (aldolase, 4.8 nm; ferritin, 6.1 nm; thyroglobulin, 8.5 nm) were used to calibrate the gel filtration column under different salt conditions. The Stokes radius of BsSMC was determined by extrapolating calibration curves made with Kaleidagraph (Synergy Software). The native molecular weight was calculated as described by Siegel and Monty (1966) using a partial specific volume of 0.725.
Electron microscopy
EM was performed as described previously (Melby et al., 1998). In brief, purified BsSMC proteins were spun through 5-ml glycerol gradients (15–40%) in 0.2 M ammonium bicarbonate at 135 000 g for 16 h at 4°C. Peak fractions were recovered, rotary shadowed (Fowler and Erickson, 1979), and photographed at 50 000× in an electron microscope (model 301; Philips Electron Optics).
UV cross-linking of ATP
UV cross-linking of ATP to BsSMC was performed by a procedure as described by Ishimi (1997). Reaction mixtures (10 µl) contained 20 mM Tris–HCl pH 7.5, 7.5 mM KCl, 2.5 mM MgCl2, 1 mM dithiothreitol, 165 nM [α-32P]ATP (3000 Ci/mmol) and 450 nM BsSMC arms. For competition, unlabeled MgATP was added at a final concentration of 1 mM. After incubation at 37°C for 30 min, the mixtures were transferred to a microwell plate on ice, and UV (254 nm) was irradiated to induce cross-linking. After 30 min, the reactions were stopped by the addition of 8 mM unlabeled ATP and 1 mg/ml BSA. Protein samples were precipitated with TCA, fractionated by SDS–PAGE and analyzed by autoradiography.
Protein–protein cross-linking
Protein–protein cross-linking with BMH (Pierce) was performed as described by Perukhova et al. (1999) with minor modifications. Reaction mixtures (10 ml) contained 20 mM K–HEPES pH 7.7, 7.5 mM KCl, 2.5 mM MgCl2 and 450 nM BsSMC arms. When necessary, MgATP or MgATPγS was included at a final concentration of 1 mM. The mixtures were spun at 16 000 g for 15 min, and the supernatants were taken. After a pre-incubation at 37°C for 5 min, φX174 virion DNA (15.6 μM nucleotides) or buffer alone was added and incubated at 37°C for 30 min. BMH was then added at a final concentration of 2 mM, and incubated at 37°C for another 30 min. After quenching the cross-linking reaction by the addition of 1 μl of 1 M 2-mercaptoethanol, the protein samples were fractionated by 2.5–7.5% gradient SDS–PAGE and analyzed by immunoblotting with an antibody raised against the C-terminal peptide of BsSMC (Hirano and Hirano, 1998).
Other assays
ATPase assays were performed as described previously (Hirano and Hirano, 1998) with minor modifications. The concentration of unlabeled ATP in the reaction mixtures was 500 µM (instead of 300 µM) in Figure 5A and B, and was further increased to 1000 µM in Figure 5C in order to maintain the linearity of the rate of ATP hydrolysis in the high protein concentrations tested. Spin-down assays were performed as described by Hirano and Hirano (1998).
Acknowledgments
Acknowledgements
We thank Keiji Kimura for instruction in gel filtration and members of the Hirano laboratory for critically reading the manuscript. This work was supported in part by grants from the Pew Scholars Program in the Biomedical Sciences (to T.H.) and the NIH (to T.H. and H.P.E.).
References
- Berger B., Wilson,D.B., Wolf,E., Tonchev,T., Milla,M. and Kim,P.S. (1995) Predicting coiled-coils by use of pairwise residue correlations. Proc. Natl Acad. Sci. USA, 92, 8259–8263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton R.A., Lin,D.C.-H. and Grossman,A.D. (1998) Characterization of a prokaryotic SMC protein involved in chromosome partitioning. Genes Dev., 12, 1254–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang P.-T., Albertson,D.G. and Meyer,B.J. (1994) DPY-27: a chromosome condensation protein homolog that regulates C.elegans dosage compensation through association with the X chromosome. Cell, 79, 459–474. [DOI] [PubMed] [Google Scholar]
- Chuang P.-T., Lieb,J.D. and Meyer,B.J. (1996) Sex-specific assembly of a dosage compensation complex on the nematode X chromosome. Science, 274, 1736–1739. [DOI] [PubMed] [Google Scholar]
- Cobbe N. and Heck,M.M. (2000) SMCs in the world of chromosome biology: from prokaryotes to higher eukaryotes. J. Struct. Biol., 129, 123–143. [DOI] [PubMed] [Google Scholar]
- Fowler W.E. and Erickson,H.P. (1979) Trinodular structure of fibrinogen. Confirmation by both shadowing and negative stain electron microscopy. J. Mol. Biol., 134, 241–249. [DOI] [PubMed] [Google Scholar]
- Freeman L., Aragon-Alcaide,L. and Strunnikov,A.V. (2000) The condensin complex governs chromosome condensation and mitotic transmission of rDNA. J. Cell Biol., 149, 811–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guacci V., Koshland,D. and Strunnikov,A. (1997) A direct link between sister chromatid cohesion and chromosome condensation revealed through the analysis of MCD1 in S.cerevisiae. Cell, 91, 47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraga S. (2000) Dynamic localization of bacterial and plasmid chromosomes. Annu. Rev. Genet., 34, 21–59. [DOI] [PubMed] [Google Scholar]
- Hirano M. and Hirano,T. (1998) ATP-dependent aggregation of single-stranded DNA by a bacterial SMC homodimer. EMBO J., 17, 7139–7148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano T. (1999) SMC-mediated chromosome mechanics: a conserved scheme from bacteria to vertebrates? Genes Dev., 13, 11–19. [DOI] [PubMed] [Google Scholar]
- Hirano T. (2000) Chromosome cohesion, condensation and separation. Annu. Rev. Biochem., 69, 115–144. [DOI] [PubMed] [Google Scholar]
- Hirano T. and Mitchison,T.J. (1994) A heterodimeric coiled-coil protein required for mitotic chromosome condensation in vitro. Cell, 79, 449–458. [DOI] [PubMed] [Google Scholar]
- Hirano T., Kobayashi,R. and Hirano,M. (1997) Condensins, chromosome condensation protein complexes containing XCAP-C, XCAP-E and a Xenopus homolog of the Drosophila Barren protein. Cell, 89, 511–521. [DOI] [PubMed] [Google Scholar]
- Holland I.B. and Blight,M.A. (1999) ABC ATPases, adaptable energy generators fuelling transmembrane movement of a variety of molecules in organisms from bacteria to humans. J. Mol. Biol., 293, 381–399. [DOI] [PubMed] [Google Scholar]
- Hopfner K.-P., Karcher,A., Shin,D.S., Craig,L., Arthur,L.M., Carney,J.P. and Tainer,J.A. (2000) Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC–ATPase superfamily. Cell, 101, 789–800. [DOI] [PubMed] [Google Scholar]
- Ishimi Y. (1997) A DNA helicase activity is associated with an MCM4, -6 and -7 protein complex. J. Biol. Chem., 272, 24508–24513. [DOI] [PubMed] [Google Scholar]
- Jensen R.B. and Shapiro,L. (1999) The Caulobacter crescentus smc gene is required for cell cycle progression and chromosome segregation. Proc. Natl Acad. Sci. USA, 96, 10661–10666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessberger R., Riwar,B., Baechtold,H. and Akhmedov,A.T. (1996) SMC proteins constitute two subunits of the mammalian recombination complex RC-1. EMBO J., 15, 4061–4068. [PMC free article] [PubMed] [Google Scholar]
- Jones P.M. and George,A.M. (1999) Subunit interactions in ABC transporters: towards a functional architecture. FEMS Microbiol. Lett., 179, 187–202. [DOI] [PubMed] [Google Scholar]
- Kimura K. and Hirano,T. (1997) ATP-dependent positive supercoiling of DNA by 13S condensin: a biochemical implication for chromosome condensation. Cell, 90, 625–634. [DOI] [PubMed] [Google Scholar]
- Kimura K. and Hirano,T. (2000) Dual roles of the 11S regulatory subcomplex in condensin functions. Proc. Natl Acad. Sci. USA, 97, 11972–11977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K., Rybenkov,V.V., Crisona,N.J., Hirano,T. and Cozzarelli,N.R. (1999) 13S condensin actively reconfigures DNA by introducing global positive writhe: implications for chromosome condensation. Cell, 98, 239–248. [DOI] [PubMed] [Google Scholar]
- Kirkwood J.G. (1954) The general theory of irreversible processes in solutions of macromolecules. J. Polym. Sci., 12, 1–14. [Google Scholar]
- Koshland D. and Strunnikov,A. (1996) Mitotic chromosome condensation. Annu. Rev. Cell Dev. Biol., 12, 305–333. [DOI] [PubMed] [Google Scholar]
- Losada A. and Hirano,T. (2001) Intermolecular DNA interactions stimulated by the cohesin complex in vitro: implications for sister chromatid cohesion. Curr. Biol., 11, 268–272. [DOI] [PubMed] [Google Scholar]
- Losada A., Hirano,M. and Hirano,T. (1998) Identification of Xenopus SMC protein complexes required for sister chromatid cohesion. Genes Dev., 12, 1986–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe J., Cordell,S.C. and van den Ent,F. (2001) Crystal structure of the SMC head domain: an ABC ATPase with 900 residues antiparallel coiled coil inserted. J. Mol. Biol., 306, 25–35. [DOI] [PubMed] [Google Scholar]
- Melby T.E.G., Ciampaglio,C.N., Briscoe,G. and Erickson,H.P. (1998) The symmetrical structure of structural maintenance of chromosomes (SMC) and MukB proteins: long, antiparallel coiled coils, folded at a flexible hinge. J. Cell Biol., 142, 1595–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis C., Ciosk,R. and Nasmyth,K. (1997) Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell, 91, 35–45. [DOI] [PubMed] [Google Scholar]
- Nasmyth K., Peters,J.-M. and Uhlmann,F. (2000) Splitting the chromosome: cutting the ties that bind sister chromatids. Science, 288, 1379–1385. [DOI] [PubMed] [Google Scholar]
- Perukhova G., Van Komen,S., Vergano,S., Klein,H. and Sung,P. (1999) Yeast Rad54 promotes Rad51-dependent homologous DNA pairing via ATP hydrolysis-driven change in DNA double helix conformation. J. Biol. Chem., 274, 29453–29462. [DOI] [PubMed] [Google Scholar]
- Saitoh N., Goldberg,I., Wood,E.R. and Earnshaw,W.C. (1994) ScII: an abundant chromosome scaffold protein is a member of a family of putative ATPases with an unusual predicted tertiary structure. J. Cell Biol., 127, 303–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saka Y., Sutani,T., Yamashita,Y., Saitoh,S., Takeuchi,M., Nakaseko,Y. and Yanagida,M. (1994) Fission yeast cut3 and cut14, members of a ubiquitous protein family, are required for chromosome condensation and segregation in mitosis. EMBO J., 13, 4938–4952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel L.M. and Monty,K.J. (1966) Determination of molecular weights and friction ratios of proteins in impure systems by use of gel filtration and density gradient centrifugation: application to crude preparations of sulfite and hydroxylamine reductases. Biochim. Biophys. Acta, 112, 346–362. [DOI] [PubMed] [Google Scholar]
- Strunnikov A.V. and Jessberger,R. (1999) Structural maintenance of chromosomes (SMC) proteins: conserved molecular properties for multiple biological functions. Eur. J. Biochem., 263, 6–13. [DOI] [PubMed] [Google Scholar]
- Strunnikov A.V., Hogan,E. and Koshland,D. (1995) SMC2, a Saccharomyces cerevisiae gene essential for chromosome segregation and condensation, defines a subgroup within the SMC family. Genes Dev., 9, 587–599. [DOI] [PubMed] [Google Scholar]
- Sutani T., Yuasa,T., Tomonaga,T., Dohmae,N., Takio,K. and Yanagida,M. (1999) Fission yeast condensin complex: essential roles of non-SMC subunits for condensation and cdc2 phosphorylation of Cut3/SMC4. Genes Dev., 13, 2271–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomonaga T. et al. (2000) Characterization of fission yeast cohesin: essential anaphase proteolysis of Rad21 phosphorylated in the S phase. Genes Dev., 14, 2757–2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielenski J. and Tsui,L.-C. (1995) Cystic fibrosis: genotypic and phenotypic variations. Annu. Rev. Genet., 29, 777–807. [DOI] [PubMed] [Google Scholar]