

Abstract
A major link between inflammation and cancer is provided by NF-κB transcription factors. IkkβΔhep mice, which specifically lack IκB kinase β (IKKβ), an activator of NF-κB, in hepatocytes, are unable to activate NF-κB in response to proinflammatory stimuli, such as TNF-α. Surprisingly, IkkβΔhep mice are hypersusceptible to diethylnitrosamine (DEN)-induced hepatocarcinogenesis. Because defective NF-κB activation promotes sustained c-Jun N-terminal kinase (JNK) activation in cells exposed to TNF-α, whose expression is induced by DEN, and JNK activity is required for normal hepatocyte proliferation, we examined whether increased susceptibility to DEN-induced hepatocarcinogenesis in IkkβΔhep mice requires JNK activation. Hepatocytes express both JNK1 and JNK2, but previous studies indicate that JNK1 is more important for hepatocyte proliferation. We therefore investigated this hypothesis using mice homozygous for a JNK1 deficiency either in wild-type or IkkβΔhep backgrounds. In both cases, mice lacking JNK1 were much less susceptible to DEN-induced hepatocarcinogenesis. This impaired tumorigenesis correlated with decreased expression of cyclin D and vascular endothelial growth factor, diminished cell proliferation, and reduced tumor neovascularization. Whereas hepatocyte-specific deletion of IKKβ augmented DEN-induced hepatocyte death and cytokine-driven compensatory proliferation, disruption of JNK1 abrogated this response. In addition to underscoring the importance of JNK1-mediated hepatocyte death and compensatory proliferation, these results strongly suggest that the control of tissue renewal through the IKK and JNK pathways plays a key role in liver carcinogenesis.
Keywords: hepatocellular carcinoma
Hepatocellular carcinoma (HCC), the most common type of liver cancer, is the third leading cause of cancer deaths worldwide (1). Epidemiological studies suggest that the major risk factors for HCC are persistent infection with hepatitis B and C virus (HBV and HCV) and exposure to genotoxic and cytotoxic chemicals, including high levels of ethanol, all of which cause chronic liver injury and inflammation (2). Although relatively uncommon in the U.S., the incidence of HCC has been increasing rapidly in the past decade because of the current HCV epidemic, affecting nearly 2% of the U.S. population. In humans, HCC almost inevitably develops in the setting of chronic hepatitis or cirrhosis, conditions in which hepatocytes are killed and resident inflammatory cells (Kupffer cells), as well as newly recruited inflammatory cells (macrophages, neutrophils, NK and NKT cells), are activated to produce cytokines that drive the compensatory proliferation of surviving hepatocytes. Although the precise molecular role of chronic liver inflammation in the pathogenesis of HCC remains to be fully elucidated, it is likely to promote HCC development through cycles of hepatocyte death and compensatory proliferation (3). For instance, infection with woodchuck hepatitis virus, which eventually culminates in HCC, greatly enhances the proliferative activity of hepatocytes, most of which are quiescent in adult liver (4).
The pathogenesis of HCC has been studied extensively in mice, a species in which it can be induced by exposure to chemicals such as diethylnitrosamine (DEN), transgenic expression of protooncogenes and growth factors, or other genetic manipulations, including expression of hepatitis B virus surface antigen, which results in chronic liver inflammation (5–7). In an attempt to understand the molecular mechanisms by which inflammation promotes cancer development, we (8) and others (9) have targeted transcription factor NF-κB, a major molecular coordinator of inflammatory responses (10, 11). In both colitis-associated cancer and in inflammation-promoted cholestatic carcinoma caused by inactivation of the Mdr2 P-glycoprotein transporter (12), inhibition of NF-κB activation markedly reduced tumorigenesis (8, 9). The mechanism proposed to explain these results was increased apoptotic death of NF-κB-deficient preneoplastic cells and decreased production of NF-κB-dependent growth factors and cytokines by inflammatory cells (11).
Surprisingly, however, a hepatocyte-specific knockout of IκB kinase β (IKKβ), the predominant catalytic subunit of the IKK complex that is required for activation of NF-κB by TNF-α and other proinflammatory stimuli (13), was found to enhance rather than attenuate the development of DEN-induced HCC (3). By contrast, inactivation of IKKβ in hematopoietic-derived cells, most likely in resident liver macrophages known as Kupffer cells, inhibited development of HCC in DEN-treated mice (3). As in other cells, NF-κB provides an important survival function in hepatocytes (13, 14). Thus its absence enhances hepatocyte killing by DEN, but because of the regenerative capacity of the liver, increased hepatocyte death results in a more extensive compensatory proliferative response (3). We therefore proposed that a deficiency in hepatocyte NF-κB enhances the formation of HCC in DEN-exposed IkkβΔhep mice by stimulating compensatory proliferation (3).
One of the mechanisms through which NF-κB provides its survival function in TNF-α-exposed cells is inhibition of prolonged JNK activation (15, 16). Indeed, TNF-α or DEN administration (which induces TNF-α production) to IkkβΔhep mice resulted in longer-lasting JNK activation relative to wild-type mice (3, 17). In addition to promoting the death of TNF-α-exposed NF-κB- (or IKKβ-) deficient cells (17), JNK activation is also required for hepatocyte proliferation after partial hepatectomy (18). Thus, prolonged JNK activation may be a critical contributor to the increased susceptibility of IkkβΔhep mice to DEN-induced HCC.
Here we present a critical evaluation of the role played by JNK1 in DEN-induced hepatocellular carcinogenesis. JNK1, one of the two major JNK isozymes expressed in hepatocytes (19), was previously shown to be the dominant JNK isoform in stimulation of cell proliferation and death (20, 21). We now show that mice that are homozygous for a JNK1 deficiency either in a wild-type or an IkkβΔhep background are much less susceptible to DEN-induced HCC development. Furthermore, JNK1-deficient tumors from either wild-type or IkkβΔhep mice exhibit lower proliferative rates and a substantial decrease in expression of cyclin D polypeptides.
Results
Loss of JNK1 Attenuates Liver Cancer Development.
A single injection of DEN to 2-week-old male mice results in efficient HCC induction (22). Previously, we found that DEN administration rapidly stimulates IKK and JNK signaling (3). A specific deletion of IKKβ in hepatocytes (IkkβΔhep mice) potentiated DEN-induced JNK activation and hepatocarcinogenesis. We suspected that increased JNK activity may contribute to the elevated susceptibility of IkkβΔhep mice to DEN-mediated carcinogenesis. Of the two JNK loci expressed in the liver, Jnk1 and Jnk2, Jnk1-encoded isoforms are mainly responsible for cell proliferation (20) and cell death (21). To determine the role of JNK1 in DEN-induced hepatocarcinogenesis, wild-type and Jnk1−/− male mice, both of the C57BL/6 background, were injected with DEN (5 or 25 mg/kg) on postnatal day 15. All mice given DEN developed typical HCCs within 10 months, but the number of detectable HCCs was reduced by ≈5-fold by the JNK1 deficiency, and so were maximal tumor diameters (Fig. 1A and B). Notably, many HCCs in wild-type mice, but none in Jnk1−/− mice, were large with obvious signs of neovascularization (Fig. 1A). A low dose (5 mg/kg) of DEN also resulted in a substantially reduced HCC load in Jnk1−/− mice compared with wild-type counterparts (Fig. 1C). Thus, JNK1 is required for efficient HCC induction in response to DEN administration.
Fig. 1.

Loss of JNK1 decreases DEN-induced tumor development. (A) Livers of 10-month-old male wild-type (Wt) and Jnk1−/− mice that were given a high dose of DEN (25 mg/kg) at 15 days of age. Arrowheads, neovascularization. (B) Numbers of tumors (≥0.5 mm) and maximal tumor sizes (diameters) in livers of male wild-type (Wt, n = 10) and Jnk1−/− (n = 6) mice 10 months after DEN (25 mg/kg) injection. Horizontal bars indicate averages. (C) Numbers of tumors (≥0.5 mm) and maximal tumor sizes (diameters) in livers of male wild-type (Wt, n = 16) and Jnk1−/− (n = 15) mice 8 months after injection of a low dose of DEN (5 mg/kg). Results are means ± SE. ∗, P < 0.05 vs. wild-type mice.
JNK1-Deficient Tumors Exhibit Decreased Cell Proliferation and Neovascularization.
To investigate the mechanisms by which disruption of JNK1 attenuated hepatocarcinogenesis, we examined the extent of cell proliferation in HCCs isolated 8 months after DEN administration. All wild-type mice given a low dose of DEN developed HCCs (Fig. 2A). Immunohistochemistry revealed many proliferating cell nuclear antigen (PCNA)-positive cells at the border of tumors and around blood vessels (Fig. 2A). Cells positive for phosphorylated c-Jun and ATF2, both of which are JNK1 targets (19), were also found around blood vessels (Fig. 2A). JNK1-deficient HCCs contained fewer PCNA-positive cells than wild-type HCCs, but there were no significant differences in the numbers of apoptotic cells or cells positive for phospho-ATF2 (Fig. 2B). Although the JNK1-deficient tumors contained fewer cells positive for phosphorylated c-Jun than wild-type tumors, the differences were of borderline significance. We also examined expression of known cell-cycle regulators and tumor suppressors in tumor and nontumor liver tissue. Levels of cyclin D, c-Jun, and CDK1 were substantially higher in HCCs compared with nontumor tissues (Fig. 2C). Both cyclin D and VEGF were considerably reduced in Jnk1−/− tumors relative to wild-type tumors (Fig. 2C). A more modest reduction was seen at the level of c-Jun expression. Previously, a complete c-Jun deficiency in hepatocytes was found to fully prevent the development of HCC in DEN-injected mice (23). In addition, the JNK1 deficiency attenuated expression of cyclin D1 and, to a lesser extent, VEGF mRNAs (Fig. 2D and data not shown).
Fig. 2.

Cyclin D, VEGF, cell proliferation, and neoangiogenesis are reduced in Jnk1−/− tumors. (A) Immunohistological analysis of wild-type and Jnk1−/− HCCs. N, noncancerous liver tissues; T, tumors. Original magnification: hematoxylin/eosin (H&E), ×50; others, ×200. (B) Frequencies of proliferating (PCNA-positive), apoptotic (TUNEL-positive), and phospho-c-Jun- or phospho-ATF2-positive cells in wild-type (Wt, n = 10) and Jnk1−/− (n = 10) HCCs. Results are means ± SE. ∗, P < 0.05 vs. wild-type mice. (C) Expression of cell cycle proteins, p53, and VEGF. Lysates of microdissected HCCs (HCC; two separate samples) or nontumor liver tissue (Liver) from DEN-treated mice were gel-separated and immunoblotted with antibodies to the indicated proteins. (D) Effects of JNK1 on cyclin gene expression. RNA from tumors (T) and nontumor liver tissues (NT) were analyzed by real-time quantitative PCR, and the tumor (T)/nontumor tissue (NT) ratio of expression of the different cyclin genes was determined. Results are means ± SE (n = 4). ∗, P < 0.05 vs. wild-type mice (Wt). (E) Expression of VEGF in HCCs. Cryosections were immunostained with polyclonal VEGF antibody. N, noncancerous liver tissues; T, tumors. Original magnification: ×200. Distinction between tumor and noncancerous liver tissue was made by hematoxylin/eosin staining. (F) Intratumoral microvessel density in wild-type (Wt, n = 3) and Jnk1−/− (n = 3) HCCs. Cryosections were immunostained with anti-CD31 antibody, and microvessels were counted per high-power fields (HPF; original magnification: ×400). Data are presented as means ± SE. ∗, P < 0.05 vs. wild-type mice. Corresponding representative photomicrographs (original magnification: ×200) are shown.
As shown in Fig. 2E, cancer cells represent the major source of VEGF in these tumors, and JNK1 disruption showed a marked reduction of intratumoral microvascular density by ≈60% based on immunostaining for CD31, a marker for endothelial cells (Fig. 2F). No differences in p53 levels were detected between tumors and normal liver tissues of the two genotypes (Fig. 2C), and c-Myc expression levels were not affected either (data not shown). Thus, JNK1 mostly affects cell proliferation and neovascularization within developed tumors, possibly through a partial reduction in c-Jun expression, which may influence the regulation of cyclin D and/or VEGF expression. As discussed below, the effect on VEGF may be indirect and could simply be a reflection of the different growth rates of JNK1-positive and -negative tumors.
Loss of JNK1 Attenuates DEN-Induced Death and Compensatory Proliferation.
The substantial decrease in tumor multiplicity in Jnk1−/− mice suggests that JNK1 may act not only during tumor progression but also during initiation or early tumor promotion. To reveal the basis for the decreased susceptibility of Jnk1−/− mice to chemical carcinogenesis, we examined the immediate (acute) effects of DEN on signal transduction, cell behavior, and liver integrity in precancerous tissue. Measurements of circulating liver enzymes and TUNEL assays revealed a modest decrease in hepatocyte death in livers of DEN-treated Jnk1−/− mice relative to similarly treated wild-type counterparts (Fig. 3A). Histological analysis confirmed the presence of fewer necrotic cells after DEN administration in Jnk1−/− livers than in wild-type counterparts (Fig. 3A). Prolonged and robust JNK activation promotes cell death (16, 24). DEN administration activated JNK in livers of both wild-type and Jnk1−/− mice, and loss of JNK1 diminished the magnitude and duration of JNK activation (Fig. 3B). Activator protein 1 (AP-1) DNA-binding activity was also induced by DEN, and overall it was lower, especially at the 4-h time point, in Jnk1−/− mice than in wild-type counterparts (Fig. 3C). These data suggest that JNK1 contributes to acute DEN-induced hepatocyte death as well as to compensatory proliferation. The involvement of JNK1 in the acute cell death induced by DEN stands in contrast to the unaltered rate of apoptosis seen in JNK1-deficient tumors.
Fig. 3.

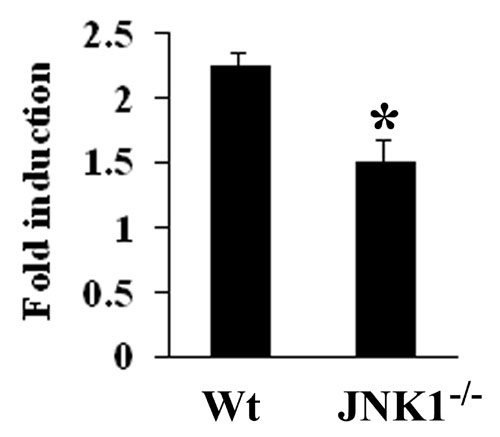
Loss of JNK1 attenuates both cell death and compensatory hepatocyte proliferation after DEN treatment. (A) Effects of JNK1 on cell death and proliferation. Mice were injected with DEN at t = 0, and alanine transaminase (ALT) levels in serum were determined at the indicated time points. The extent of liver cell apoptosis and proliferation was determined by TUNEL staining or BrdU labeling, respectively. The extent of necrosis was determined as described (3). Results are means ± SE. ∗, P < 0.05 vs. wild-type mice (Wt). (B) JNK1 contributes to DEN-induced JNK activity. Mice were treated as above, and their livers isolated when indicated and homogenized. JNK activity was determined by solid-state kinase assay with c-Jun as the substrate. Protein recovery was determined by immunoblotting with anti-JNK1 and -JNK1/2 antibodies. Ratio, relative JNK activity. (C) AP-1 DNA-binding activity in nuclear extracts of DEN-treated livers. Livers were isolated when indicated, nuclear extracts were prepared, and AP-1 activity was assessed by a mobility-shift assay. Ratio, relative AP-1 activity. (D) Effects of JNK1 on cytokine gene expression. Mice were treated as above, and liver RNA was extracted at the indicated times. Levels of cytokine mRNAs were determined by real-time quantitative PCR. Results are means ± SE (n = 4). ∗, P < 0.05 vs. wild-type mice (Wt).
It should be noted, however, that only a small fraction of all hepatocytes undergoes cell death in response to a carcinogenic dose of DEN, but because of the high regenerative capacity of the liver, this amount of cell death should be sufficient to trigger compensatory proliferation of surviving cells. Indeed, BrdU labeling showed the presence of proliferating hepatocytes in DEN-exposed mice (Fig. 3A). Proliferating cells are most common around clusters of apoptotic cells in centrilobular lesions, confirming that DEN-induced cell loss triggers compensatory hepatocyte proliferation (3). This proliferative response was modestly, but significantly, reduced in Jnk1−/− mice (Fig. 3A).
Hepatic mitogens such as TNF-α and IL-6 that are produced by Kupffer cells, and HGF, which is produced by hepatic stellate cells (HSC), are required for compensatory hepatocyte proliferation in various experimental models (25). The JNK1 deficiency attenuated expression of TNF-α, IL-6, and HGF mRNAs at the 24-h time point, most likely by decreasing the extent of liver injury (Fig. 3D). The proinflammatory cytokine IL-1 is involved in tumor progression, possibly through induction of VEGF, which in turn stimulates neoangiogenesis (26). IL-1β can also induce TNF-α and IL-6 (27), as well as cause NF-κB-dependent induction of cyclooxygenase-2 (COX2) and hypoxia-inducible factor 1α (28). IL-1β also induces expression of monocyte chemoattractant protein-1 (MCP-1), a chemokine that plays an important role in angiogenesis and tumor growth (29). The mRNA levels of IL-1β were markedly lower 4 h after DEN administration in Jnk1−/− relative to wild-type mice, but not at 24 h (Fig. 3D). The absence of JNK1 had no significant effect on COX2 or MCP-1 mRNA expression (Fig. 3D and data not shown).
Growth factor production by macrophages and Kupffer cells is thought to involve an inflammatory signal transduction cascade triggered by substances released by necrotic cells (11). To examine the role of JNK1 in monocytes in this inflammatory response, we challenged wild-type and Jnk1−/− bone marrow cells with dead hepatocytes. Necrotic hepatocytes triggered production of the proinflammatory cytokines IL-6, TNF-α, and IL-1β, but the JNK1 deficiency had only a marginal effect on cytokine production in this system (Fig. 6, which is published as supporting information on the PNAS web site).
Loss of JNK1 Attenuates Caspase-Dependent Apoptosis in the Liver.
In previous work, we found that the main proapoptotic function of prolonged JNK1 activation in the context of TNF-receptor 1 signaling is activation of the E3 ubiquitin ligase Itch, which targets the NF-κB-inducible antiapoptotic protein c-FLIPL to proteasomal degradation (17). In wild-type mice, injection of DEN led to almost complete disappearance of c-FLIPL by 4 h, but c-FLIPL expression had recovered by 24 h (Fig. 4A). Loss of JNK1 largely prevented the transient decrease in c-FLIPL levels, and this was accompanied by inhibition of caspase 3 cleavage (Fig. 4A). JNK1 was activated at 4 h after DEN, but not at 24 h (Fig. 3B). Thus, DEN-induced c-FLIPL degradation also depends on JNK1 activation. DEN injection also led to induction of p53, and this response was partially attenuated in Jnk1−/− mice (Fig. 4A). Puma and Noxa are important apoptotic mediators induced by p53 (30). The mRNA levels of Puma and Noxa were significantly lower in DEN-injected Jnk1−/− mice than in similarly treated wild-type counterparts (Fig. 4B). These results suggest that, in addition to c-FLIPL degradation, JNK1 contributes to DEN-induced apoptosis by promoting the activation of p53. The mechanism of p53 modulation by JNK1 remains to be elucidated.
Fig. 4.

JNK1 potentiates DEN-induced apoptotic cell death. (A) Effects of JNK1 on apoptotic signaling. Mice were injected with DEN, and their livers were isolated at the indicated times and homogenized. Protein extracts were gel-separated and immunoblotted with the indicated antibodies. (B) Effects of JNK1 on p53 target gene expression. Mice were treated as above, and total liver RNA was extracted at 24 h after DEN injection. Expression of p53 target genes was measured by real-time quantitative PCR. Results are means ± SE (n = 4). ∗, P < 0.05 vs. wild-type mice (Wt).
Ablation of JNK1 Reverses the Increased Susceptibility to Hepatocarcinogenesis Caused by Hepatocyte-Specific Deletion of IKKβ.
NF-κB activation attenuates prolonged JNK activation in response to TNF-receptor 1 engagement and, when NF-κB activation is inhibited, JNK activation makes an important contribution to TNF-α-induced cell death (13, 16, 17, 31, 32). To assess the role of sustained JNK activation in the elevated susceptibility of IkkβΔhep mice to DEN-induced hepatocarcinogenesis, IkkβΔhep and Jnk1−/− mice were intercrossed to generate IkkβΔhep/Jnk1−/− mice. When treated with DEN (5 mg/kg) at 15 days of age, IkkβΔhep/Jnk1−/− mice developed 3-fold fewer tumors than IkkβΔhep/Jnk1+/+ (IkkβΔhep) mice at 8 months of age, and a similar decrease was found in maximal tumor diameters (Fig. 5A). To understand how loss of JNK1 affects tumor growth, the expression of cell-cycle regulators and tumor suppressors in tumor and nontumor samples was analyzed. As seen before, PCNA, cyclin D, CDK1, CDK2, and VEGF were elevated in HCCs compared with nontumor tissues (Fig. 5B). PCNA, cyclin D, and VEGF levels were substantially lower in IkkβΔhep/Jnk1−/− tumors than in IkkβΔhep tumors (Fig. 5B).
Fig. 5.

JNK1 disruption reverses increased hepatocyte death and susceptibility to HCC formation caused by the absence of hepatocyte IKKβ. (A) Numbers of tumors (≥0.5 mm) and maximal tumor sizes (diameters) in livers of male IkkβΔhep/Jnk1+/+ (Δhep, n = 10) and IkkβΔhep/Jnk1−/− (Δhep/JNK1−/−, n = 12) mice 8 months after DEN (5 mg/kg) injection. (B) Expression of cell-cycle-related proteins. Lysates of microdissected HCCs (HCC; two separate samples) or nontumor liver tissue (Liver) from DEN-treated mice were gel-separated and analyzed by immunoblotting as above. IkkβF/F mice, F/F; IkkβΔhep mice, Δhep; IkkβΔhep/Jnk1−/− mice, Δhep/JNK1−/−. (C) Effects of JNK1 on cell death and proliferation in IkkβΔhep mice. Mice of the indicated genotypes were injected with DEN, and ALT levels in serum were determined at the indicated times. The extents of liver cell apoptosis and proliferation were determined by TUNEL staining or BrdU labeling, respectively. Results are means ± SE. ∗, P < 0.05 vs. IkkβΔhep mice.
As was done with single mutant mice, we examined the early effects of DEN on signal transduction and cell behavior in IkkβΔhep/Jnk1−/− and IkkβΔhep livers. Appearance of circulating liver enzymes and TUNEL assays indicated less hepatocyte death in DEN-treated IkkβΔhep/Jnk1−/− mice relative to similarly treated IkkβΔhep mice (Fig. 5C). In fact, the extent of DEN-induced hepatocyte death in IkkβΔhep/Jnk1−/− mice was not significantly different from what was seen in IKKβ-containing IkkβF/F mice. Histological analysis also revealed fewer necrotic cells after DEN administration in IkkβΔhep/Jnk1−/− and IkkβF/F mice than in IkkβΔhep mice (data not shown). In addition, BrdU labeling showed lower numbers of proliferating hepatocytes in IkkβΔhep/Jnk1−/− and IkkβF/F mice than in IkkβΔhep mice after DEN exposure (Fig. 5C). JNK1 deficiency does not affect NF-κB activity in DEN-treated livers (data not shown). Thus, the ablation of JNK1 reversed all of the effects of IKKβ deletion on acute DEN-induced hepatocyte death and compensatory proliferation, as the IkkβΔhep/Jnk1−/− double mutants responded to DEN-exposure just like the IkkβF/F controls. Taken together, the inability to properly terminate JNK1 activation caused by the absence of hepatocyte IKKβ and NF-κB seems to be the major contributor to the increased susceptibility of IkkβΔhep mice to DEN-induced hepatocarcinogenesis.
Discussion
In previous studies of the role played by transcription factor NF-κB in chemically induced hepatocarcinogenesis, we made the surprising observation that IkkβΔhep mice, which specifically lack the IKKβ catalytic subunit of the IKK complex in hepatocytes and therefore are defective in NF-κB activation in these cells (13), are hypersusceptible to DEN-induced hepatocarcinogenesis (3). In this study, we examined the molecular underpinnings of the mechanism that account for the elevated susceptibility of IkkβΔhep mice to chemically induced hepatocarcinogenesis. We focused our attention on JNK1, because previous investigations have revealed that in the absence of NF-κB (or IKKβ), both TNF-α and DEN, which induces TNF-α release from Kupffer and endothelial cells (Fig. 7, which is published as supporting information on the PNAS web site), cause sustained rather than transient JNK activation in hepatocytes (3, 13, 16, 17). Furthermore, of the two JNK loci expressed in hepatocytes, it is the Jnk1 locus that is the main source of proapoptotic JNK isoforms responsible for TNF-α-induced death of NF-κB-deficient cells (17, 21). We therefore generated IkkβΔhep/Jnk1−/− compound mutants and compared their susceptibility to DEN-induced hepatocyte death, compensatory proliferation, and hepatocarcinogenesis to that of IkkβΔhep/Jnk1+/+ and the IKKβ-expressing IkkβF/F parental strain, from which the IkkβΔhep mutant is derived. Consistent with our expectation, the genetic loss of JNK1 isoforms completely reversed the effect of IKKβ deletion in hepatocytes on DEN-induced hepatocyte death, compensatory proliferation, and hepatocarcinogenesis. Thus, the prolonged activation of JNK1 is a principal mechanism by which the absence of hepatocyte IKKβ results in increased susceptibility to chemically induced hepatocarcinogenesis.
The JNK subgroup of mitogen-activated protein kinases is activated primarily by cytokines and exposure to environmental stress (33). An important target for the JNK signaling pathway is the AP-1 transcription factor composed of Jun, Fos, and related bZIP subunits (19). JNK-mediated phosphorylation increases the transcriptional activity of c-Jun and other AP-1 proteins (19). Importantly, DEN-induced hepatocarcinogenesis was shown to require c-Jun (23), which is also needed for fibroblast and hepatocyte proliferation (18, 19). Surprisingly, however, JNK-mediated N-terminal phosphorylation of c-Jun, which is needed for Ras-induced fibroblast transformation (34), is not required for DEN-induced hepatocarcinogenesis (23). Yet, disruption of JNK1 decreased chemically induced hepatocarcinogenesis and DEN-induced AP-1 activity and led to a modest decrease in c-Jun protein levels. At this point, it is not clear whether the small decrease in c-Jun expression levels or AP-1 activity fully accounts for the reduction in DEN-induced tumorigenesis caused by the absence of JNK1. Nevertheless, the absence of JNK1 resulted in decreased expression of cyclin D proteins, which are important for hepatocyte (35) and hepatoma (36) cell proliferation. These effects are consistent with previous findings showing that cyclin D1 and D2 are AP-1 target genes (19, 37) and suggest that the reduction in AP-1 activity and c-Jun expression may be of importance after all. After DEN administration, JNK activity is rapidly increased (3), and our results now show that the JNK1 isoforms are the ones responsible for much of the ensuing increase in cyclin D expression and hepatocyte proliferation. Correspondingly, Jnk1−/− mice are much less susceptible to DEN-induced hepatocarcinogenesis, and the ablation of JNK1 reverses all of the excess hepatocyte death, compensatory proliferation, and HCC load seen in IkkβΔhep mice.
The carcinogen used in this study, DEN, is metabolized into an alkylating agent that induces DNA damage and mutations (38) as well as hepatocyte death (39). However, mutations alone are probably insufficient for hepatocarcinogenesis. In adult mice, because of its reduced toxicity, DEN is a weak carcinogen that requires assistance that can be provided by a plethora of tumor promoters that somehow induce hepatocyte proliferation (22). Many of these tumor promoters, such as CCl4, enhance hepatocyte proliferation through indirect mechanisms, and their primary effect is cytotoxicity that eventually triggers compensatory proliferation. Compensatory proliferation is critical for tumor promotion, causing initiated hepatocytes to enter the cell cycle and transmit oncogenic mutations to their progeny (5). In addition to cell death, DEN-induced hepatocyte proliferation depends on the production of hepatic mitogens, including TNF-α, IL-6, and HGF (3). Kupffer cells produce TNF-α and IL-6, and endothelial cells produce TNF-α, whereas HGF is mainly produced by HSC, which are related to myofibroblasts (40). The role of these mitogens in hepatocyte proliferation after partial hepatectomy is well established (41, 42), and HGF is required for DEN-induced hepatocarcinogenesis (43). Disruption of JNK1 modestly reduces the expression of intrahepatic TNF-α and IL-6 mRNAs but has a more dramatic effect on the expression of intrahepatic HGF mRNA. Thus JNK1 also contributes to liver cell proliferation by enhancing growth factor production. Nonetheless, because sustained activation of JNK1 caused by the absence of NF-κB is intrinsic to the hepatocyte and does not apply to other cell types in which IKKβ is still expressed, the primary site of JNK1 action in the present case appears to be the hepatocyte.
During hepatocarcinogenesis, the proliferative advantage of genetically altered hepatocytes is associated with enhanced apoptosis (44, 45), and both apoptotic and necrotic cell death may trigger compensatory hepatocyte proliferation. Disruption of JNK1 inhibited caspase-dependent apoptosis in the liver as well as frank necrosis (refs. 13 and 16 and this study). Thus the mitogenic and procarcinogenic function of JNK1 is at least partially linked to its diametrically opposed roles in induction of hepatocyte death and proliferation. In this context, an important target for JNK1 is the ubiquitin ligase Itch (46), which, upon JNK1-mediated activation (47), ubiquitinates the NF-κB-dependent antiapoptotic protein c-FLIPL and targets it to proteasomal degradation (17). Here, we show that JNK1 is also required for DEN-induced c-FLIPL degradation and subsequent apoptosis. The tumor suppressor p53 is another potential target for proapoptotic signaling by JNK1, which may regulate p53 activity and expression. Loss of JNK1 reduced the expression of p53 mRNA and protein, and its downstream effectors, Puma and Noxa, in DEN-treated livers (Fig. 4 and Fig. 8, which is published as supporting information on the PNAS web site). In contrast, loss of Itch reduced the expression of Noxa but not Puma mRNAs (Fig. 9, which is published as supporting information on the PNAS web site) and remarkably inhibited DEN-induced liver injury (Fig. 10, which is published as supporting information on the PNAS web site). Although the potential role of p53 as a target for JNK1 remains to be elucidated, Itch might be involved in JNK1-mediated hepatocyte death via Noxa induction as well as c-FLIPL degradation. Thus, the role of Itch in hepatocarcinogenesis merits further investigation.
It is well established that apoptotic cell death is antiinflammatory, whereas necrotic cell death provides a proinflammatory impetus (48). Yet, excessive apoptosis can lead to secondary necrosis, and JNK1 ablation reduces both apoptosis and necrosis (refs. 16 and 17 and this study). Necrosis-elicited inflammatory signal transduction can activate macrophage-like Kupffer cells to produce hepatic mitogens. Thus the effect of JNK1 on expression of mitogens in the liver could be indirect and may simply be due to the lower level of necrotic injury in Jnk1−/− mice. Correspondingly, JNK1-deficient and wild-type macrophages produced similar levels of hepatic mitogens when incubated with the same amount of necrotic hepatocytes prepared by freeze thawing. The same applies for the expression of cyclin D polypeptides, which may be affected by JNK1 both directly through AP-1-binding sites in the cyclin D1 (19) and D2 (37) promoters and indirectly through induction of compensatory proliferation.
Although reduced hepatocyte injury and compensatory proliferation were seen up to several days after DEN exposure in Jnk1−/− mice, deletion of the Jnk1 locus also decreased tumor growth 10 months later. Although the cell type and mechanism(s) underlying this effect remain to be identified, we suggest that disruption of JNK1 might attenuate the necrotic death of stressed hepatoma cells encountering hypoxia and lack of nutrients and thereby may suppress activation of adjacent inflammatory cells that secrete cytokines that promote tumor growth. In addition, the absence of JNK1 may result in lower AP-1 transcriptional activity and hence lower levels of cyclin D proteins within cancer cells.
Inflammatory angiogenesis is an important contributor to the tumor growth response (49). We found that disruption of JNK1 decreased expression of VEGF, a potent proangiogenic substance responsible for tumor neovascularization. The VEGF promoter contains four potential AP-1-binding sites (50). Thus, JNK1 may regulate VEGF transcription through AP-1, but it is also possible that the effect is exerted at the level of mRNA turnover (51). Furthermore, the effect of JNK1 could be quite indirect and exerted through factors such as IL-1β and oxygen tension (26, 52). The mRNA level of IL-1β was markedly reduced at 4 h after DEN treatment in Jnk1−/− mice, but at 24 h posttreatment, no difference between Jnk1−/− mice and wild-type controls was detected. Less hypoxic areas were found in Jnk1−/− tumors than in control tumors (data not shown), but this may simply be a reflection of the smaller average size of JNK1-deficient HCCs, indicating a slower growth rate. Notably, the faster a tumor grows, the more likely it is to undergo hypoxic stress and express high levels of VEGF. It is therefore not surprising that VEGF levels show direct correlation with tumor size. Thus the exact cellular and molecular mechanism through which JNK1 promotes tumor growth, progression, and angiogenesis requires further investigation. Despite the uncertainties in its molecular mode of action, our results strongly suggest that JNK1 is an important target for the development of chemopreventive and therapeutic measures for reducing the emergence of HCC in the context of chronic liver injury and slowing the progression of preexisting tumors.
Materials and Methods
Animals.
Jnk1−/−, IkkβF/F, IkkβF/F:Alb-Cre (referred to as IkkβΔhep), and Itch−/− mice were described (3, 17) and were maintained in filter-topped cages on autoclaved food and water at the University of California at San Diego according to National Institutes of Health guidelines.
Tumor Induction and Analysis.
Fifteen-day-old mice and littermates on either a C57BL/6 (Jnk1+/+ and Jnk1−/−) or a mixed C57BL/6/129 background (IkkβΔhep, IkkβF/F, and IkkβΔhep/Jnk1−/−) were intercrossed at least six times and injected i.p. with 5 or 25 mg/kg DEN (Sigma), as indicated. After 8 or 10 months on normal chow, mice were killed, and their livers were removed, separated into individual lobes, and analyzed for the presence of HCCs, histology, and histochemical parameters, as described (3).
Biochemical and Immunohistochemical Analyses.
JNK assays, electrophoretic mobility-shift assays, immunoblotting, and immunohistochemistry have been described (3, 53, 54). Antibodies used were: anti-phospho-c-Jun, anti-phospho-ATF2 (Cell Signaling Technology, Beverly, MA); anti-c-Myc, anti-p53, anti-CDK1, anti-CDK2, anti-c-Jun, anti-VEGF, anti-JunB, anti-JunD (Santa Cruz Biotechnology); anti-PCNA, anti-JNK1, anti-JNK1/2, anti-TNF-α (PharMingen); anti-cFLIP (Stressgen Biotechnologies, San Diego); anti-F4/80 (Caltag, Burlingame, CA); and anti-cyclin D (Upstate Biotechnology, Charlottesville, VA). Analysis of mRNA expression by real-time quantitative PCR was described (3).
To measure cytokine production in vitro, bone marrow was recovered from femurs and dispensed in culture dishes as described (55). C57BL/6 hepatocytes were purified as described (13). Necrotic hepatocytes (lysed by three cycles of freezing and thawing, 5 × 105 cells per ml) were added onto 6-cm dishes with bone marrow-derived macrophages (1 ml per dish) and incubated at 37°C for 4 h.
Immunohistochemistry was performed by using the ABC staining kit (Vector Laboratories) according to the manufacturer’s recommendations. To determine intratumoral microvascular density, cryopreserved tumor sections were immunostained with anti-CD31 antibody (PharMingen) and incubated with a biotinylated anti-rat IgG antibody (Santa Cruz Biotechnology) and streptavidine peroxidase. Enzymatic activity was developed by using diaminobenzidine (Vector Laboratories) as a substrate, and sections were counterstained with Mayer’s hematoxylin. Intratumoral CD31-positive cells, indicative of endothelial cells, were counted for quantification when showing a brownish cytoplasmatic staining. The hypoxyprobe kit (Chemicon) was used for detection of tissue hypoxia.
Statistical Analysis.
Data were expressed as means ± SE. Differences were analyzed by Student’s t test. P values ≤ 0.05 were considered significant.
Supplementary Material
Acknowledgments
We thank Randall Johnson and Hyam L. Leffert for extensive discussions, advice, and critical reading of the manuscript. T.S. was supported by a fellowship from the Japan Society for the Promotion of Science and YASUDA Medical Research Foundation. S.M. was supported by the Naito Foundation. Research was supported by grants from the National Institutes of Health (ES04151, ES06376, and AI043477) and the Superfund Basic Research Program (National Institutes of Health/National Institute on Environmental Health Sciences ES010337). M.K. is an American Cancer Society Research Professor.
Abbreviations
- IKKβ
IκB kinase β
- DEN
diethylnitrosamine
- JNK
c-Jun N-terminal kinase
- HCC
hepatocellular carcinoma
- HGF
hepatocyte growth factor
- PCNA
proliferating cell nuclear antigen.
Footnotes
Conflict of interest statement: No conflicts declared.
This contribution is part of the special series of Inaugural Articles by members of the National Academy of Sciences elected on May 3, 2005.
References
- 1.Thorgeirsson S. S., Grisham J. W. Nat. Genet. 2002;31:339–346. doi: 10.1038/ng0802-339. [DOI] [PubMed] [Google Scholar]
- 2.Bosch F. X., Ribes J., Diaz M., Cleries R. Gastroenterology. 2004;127:S5–S16. doi: 10.1053/j.gastro.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 3.Maeda S., Kamata H., Luo J. L., Leffert H., Karin M. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 4.Hansen L. J., Tennant B. C., Seeger C., Ganem D. Mol. Cell. Biol. 1993;13:659–667. doi: 10.1128/mcb.13.1.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fausto N. Semin. Liver Dis. 1999;19:243–252. doi: 10.1055/s-2007-1007114. [DOI] [PubMed] [Google Scholar]
- 6.Calvisi D. F., Thorgeirsson S. S. Toxicol. Pathol. 2005;33:181–184. doi: 10.1080/01926230590522095. [DOI] [PubMed] [Google Scholar]
- 7.Lee J. S., Thorgeirsson S. S. Semin. Liver Dis. 2005;25:125–132. doi: 10.1055/s-2005-871192. [DOI] [PubMed] [Google Scholar]
- 8.Greten F. R., Eckmann L., Greten T. F., Park J. M., Li Z. W., Egan L. J., Kagnoff M. F., Karin M. Cell. 2004;118:285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 9.Pikarsky E., Porat R. M., Stein I., Abramovitch R., Amit S., Kasem S., Gutkovich-Pyest E., Urieli-Shoval S., Galun E., Ben-Neriah Y. Nature. 2004;431:461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 10.Ghosh S., Karin M. Cell. 2002;109:S81–S96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- 11.Karin M., Greten F. R. Nat. Rev. Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 12.Mauad T. H., van Nieuwkerk C. M., Dingemans K. P., Smit J. J., Schinkel A. H., Notenboom R. G., van den Bergh Weerman M. A., Verkruisen R. P., Groen A. K., Oude Elferink R. P., et al. Am. J. Pathol. 1994;145:1237–1245. [PMC free article] [PubMed] [Google Scholar]
- 13.Maeda S., Chang L., Li Z. W., Luo J. L., Leffert H., Karin M. Immunity. 2003;19:725–737. doi: 10.1016/s1074-7613(03)00301-7. [DOI] [PubMed] [Google Scholar]
- 14.Lavon I., Goldberg I., Amit S., Landsman L., Jung S., Tsuberi B. Z., Barshack I., Kopolovic J., Galun E., Bujard H., Ben-Neriah Y. Nat. Med. 2000;6:573–577. doi: 10.1038/75057. [DOI] [PubMed] [Google Scholar]
- 15.Deng Y., Ren X., Yang L., Lin Y., Wu X. Cell. 2003;115:61–70. doi: 10.1016/s0092-8674(03)00757-8. [DOI] [PubMed] [Google Scholar]
- 16.Kamata H., Honda S., Maeda S., Chang L., Hirata H., Karin M. Cell. 2005;120:649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 17.Chang L., Kamata H., Solinas G., Luo J. L., Maeda S., Venuprasad K., Liu Y. C., Karin M. Cell. 2006;124:601–613. doi: 10.1016/j.cell.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 18.Schwabe R. F., Bradham C. A., Uehara T., Hatano E., Bennett B. L., Schoonhoven R., Brenner D. A. Hepatology. 2003;37:824–832. doi: 10.1053/jhep.2003.50135. [DOI] [PubMed] [Google Scholar]
- 19.Shaulian E., Karin M. Nat. Cell. Biol. 2002;4:E131–136. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- 20.Sabapathy K., Hochedlinger K., Nam S. Y., Bauer A., Karin M., Wagner E. F. Mol. Cell. 2004;15:713–725. doi: 10.1016/j.molcel.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 21.Liu J., Minemoto Y., Lin A. Mol. Cell. Biol. 2004;24:10844–10856. doi: 10.1128/MCB.24.24.10844-10856.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sarma D. S., Rao P. M., Rajalakshmi S. Cancer Surv. 1986;5:781–798. [PubMed] [Google Scholar]
- 23.Eferl R., Ricci R., Kenner L., Zenz R., David J. P., Rath M., Wagner E. F. Cell. 2003;112:181–192. doi: 10.1016/s0092-8674(03)00042-4. [DOI] [PubMed] [Google Scholar]
- 24.Lin A. Bioassays. 2003;25:17–24. doi: 10.1002/bies.10204. [DOI] [PubMed] [Google Scholar]
- 25.Fausto N., Riehle K. J. J. Hepatobiliary Pancreat. Surg. 2005;12:181–189. doi: 10.1007/s00534-005-0979-y. [DOI] [PubMed] [Google Scholar]
- 26.Voronov E. Proc. Natl. Acad. Sci. USA. 2003;100:2645–2650. doi: 10.1073/pnas.0437939100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dinarello C. A. J. Exp. Med. 2005;201:1355–1359. doi: 10.1084/jem.20050640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jung Y. J., Isaacs J. S., Lee S., Trepel J., Neckers L. FASEB J. 2003;17:2115–2117. doi: 10.1096/fj.03-0329fje. [DOI] [PubMed] [Google Scholar]
- 29.Sica A., Wang J. M., Colotta F., Dejana E., Mantovani A., Oppenheim J. J., Larsen C. G., Zachariaem C. O., Matsushima K. J. Immunol. 1990;144:3034–3038. [PubMed] [Google Scholar]
- 30.Villungar A, Michalak E. M., Coultas F., Mullauer G., Bock M. J., Ausserlechner M. J., Adams J. M., Strasser A. Science. 2003;302:1036–1038. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- 31.De Smaele E., Zazzeroni F., Papa S., Nguyen D. U., Jin R., Jones J., Cong R., Franzoso G. Nature. 2001;414:308–313. doi: 10.1038/35104560. [DOI] [PubMed] [Google Scholar]
- 32.Tang G., Minemoto Y., Dibling B., Purcell N. H., Li Z., Karin M., Lin A. Nature. 2001;414:313–317. doi: 10.1038/35104568. [DOI] [PubMed] [Google Scholar]
- 33.Minden A., Karin M. Biochim. Biophys. Acta. 1997;1333:F85–F104. doi: 10.1016/s0304-419x(97)00018-8. [DOI] [PubMed] [Google Scholar]
- 34.Smeal T., Binetruy B., Mercola D. A., Birrer M., Karin M. Nature. 1991;354:494–496. doi: 10.1038/354494a0. [DOI] [PubMed] [Google Scholar]
- 35.Lu X. P., Koch K. S., Lew D. J., Dulic V., Pines J., Reed S. I., Hunter T., Leffert H. L. J. Biol. Chem. 1992;267:2841–2844. [PubMed] [Google Scholar]
- 36.Zhang Y. J., Jiang W., Chen C. J., Lee C. S., Kahn S. M., Santella R. M., Weinstein I. B. Biochem. Biophys. Res. Commun. 1993;196:1010–1016. doi: 10.1006/bbrc.1993.2350. [DOI] [PubMed] [Google Scholar]
- 37.Brooks A. R., Shiffman D., Chan C. S., Brooks E. E., Milner P. G. J. Biol. Chem. 1996;271:9090–9099. doi: 10.1074/jbc.271.15.9090. [DOI] [PubMed] [Google Scholar]
- 38.Verna L., Whysner J., Williams G. M. Pharmacol. Ther. 1996;71:57–81. doi: 10.1016/0163-7258(96)00062-9. [DOI] [PubMed] [Google Scholar]
- 39.Farber J. L., Gerson R. J. Pharmacol. Rev. 1984;36:71S–75S. [PubMed] [Google Scholar]
- 40.Schirmacher P., Geerts A., Jung W., Pietrangelo A., Rogler C. E., Dienes H. P. Exp. Suppl. 1993;65:285–299. [PubMed] [Google Scholar]
- 41.Fausto N. J. Hepatol. 2000;32:19–31. doi: 10.1016/s0168-8278(00)80412-2. [DOI] [PubMed] [Google Scholar]
- 42.Yamada Y., Kirillova I., Peschon J. J., Fausto N. Proc. Natl. Acad. Sci. USA. 1997;94:1441–1446. doi: 10.1073/pnas.94.4.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Horiguchi N., Takayama H., Toyoda M., Otsuka T., Fukusato T., Merlino G., Takagi H., Mori M. Oncogene. 2002;21:1791–1799. doi: 10.1038/sj.onc.1205248. [DOI] [PubMed] [Google Scholar]
- 44.Schulte-Hermann R., Bursch W, Grasl-Kraupp B., Mullauer L., Ruttkay-Nedecky B. Mutat. Res. 1995;333:81–87. doi: 10.1016/0027-5107(95)00134-4. [DOI] [PubMed] [Google Scholar]
- 45.Snibson K. J., Bhathal P. S., Hardy C. L., Brandon M. R., Adams T. E. Liver. 1999;19:242–252. doi: 10.1111/j.1478-3231.1999.tb00042.x. [DOI] [PubMed] [Google Scholar]
- 46.Gao M., Labuda T., Xia Y., Gallagher E., Fang D., Liu Y. C., Karin M. Science. 2004;306:271–275. doi: 10.1126/science.1099414. [DOI] [PubMed] [Google Scholar]
- 47.Gallagher E., Gao M., Liu Y. C., Karin M. Proc. Natl. Acad. Sci. USA. 2006;103:1717–1722. doi: 10.1073/pnas.0510664103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vakkila J., Lotze M. T. Nat. Rev. Immunol. 2004;4:641–648. doi: 10.1038/nri1415. [DOI] [PubMed] [Google Scholar]
- 49.Tang N., Wang L., Esko J., Giordano F. J., Huang Y., Gerber H. P., Ferrara N., Johnson R. S. Cancer Cell. 2004;6:485–495. doi: 10.1016/j.ccr.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 50.Tischer E., Mitchell R., Hartman T., Silva M., Gospodarowicz D., Fiddes J. C., Abraham J. A. J. Biol. Chem. 1991;266:11947–11954. [PubMed] [Google Scholar]
- 51.Dibbens J. A., Miller D. L., Damert A., Risau W., Vadas M. A., Goodall G. J. Mol. Biol. Cell. 1999;10:907–919. doi: 10.1091/mbc.10.4.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pages G., Pouyssegur J. Cardiovasc. Res. 2005;65:564–573. doi: 10.1016/j.cardiores.2004.09.032. [DOI] [PubMed] [Google Scholar]
- 53.Hibi M., Lin A., Smeal T., Minden A., Karin M. Genes Dev. 1993;11:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- 54.Sakurai T., Itoh K., Higashitsuji H., Nagao T., Nonoguchi K., Chiba T., Fujita J. J. Biol. Chem. 2004;278:10668–10674. doi: 10.1074/jbc.M206104200. [DOI] [PubMed] [Google Scholar]
- 55.Park J. M., Greten F. R., Li Z. W., Karin M. Science. 2002;297:2048–2051. doi: 10.1126/science.1073163. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}