

Abstract
Mutations in Pink1, a gene encoding a Ser/Thr kinase with a mitochondrial-targeting signal, are associated with Parkinson’s disease (PD), the most common movement disorder characterized by selective loss of dopaminergic neurons. The mechanism by which loss of Pink1 leads to neurodegeneration is not understood. Here we show that inhibition of Drosophila Pink1 (dPink1) function results in energy depletion, shortened lifespan, and degeneration of select indirect flight muscles and dopaminergic neurons. The muscle pathology was preceded by mitochondrial enlargement and disintegration. These phenotypes could be rescued by the wild type but not the pathogenic C-terminal deleted form of human Pink1 (hPink1). The muscle and dopaminergic phenotypes associated with dPink1 inactivation show similarity to that seen in parkin mutant flies and could be suppressed by the overexpression of Parkin but not DJ-1. Consistent with the genetic rescue results, we find that, in dPink1 RNA interference (RNAi) animals, the level of Parkin protein is significantly reduced. Together, these results implicate Pink1 and Parkin in a common pathway that regulates mitochondrial physiology and cell survival in Drosophila.
Keywords: mitochondria, Parkinson’s disease, Pten, induced kinase 1, indirect flight muscle
Parkinson’s disease (PD) is the most common movement disorder characterized pathologically by the deficiency of brain dopamine content and the selective degeneration of dopaminergic neurons in the substantia nigra. The most common forms of PD are sporadic with no known cause. Nevertheless, postmortem studies have identified common features associated with sporadic PD, such as mitochondrial complex I dysfunction, oxidative stress, and aggregation of abnormal proteins (1, 2).
Although initial studies on the etiology of PD have focused on environmental factors, recent genetic studies have firmly established the contribution of inheritable factors in PD pathogenesis (2, 3). At least ten distinct loci have been associated with rare familial forms of PD (FPD). It is anticipated that understanding the molecular lesions associated with these FPD genes will shed light on the pathogenesis of the more common forms of the disease. Dominant mutations in α-Synuclein (α-Syn) and LRRK2/dardarin and recessive mutations in parkin, DJ-1, and Pink1 have been associated with FPD (4–10). Of these five genes, α-Syn, parkin, and DJ-1 have been most intensively studied. Studies using in vivo animal models and in vitro cell culture have linked mutations of these genes to impairments of mitochondrial structure and function and oxidative stress response, reinforcing the general involvement of mitochondrial dysfunction and oxidative stress in PD pathogenesis (11–21). Consistent with this notion, these proteins have been shown to be present in mitochondria or interact with mitochondrial proteins (8, 22–24), suggesting that they may directly regulate mitochondria function.
A further link between mitochondria and PD was supported by the fact that Pink1 encodes a predicted Ser/Thr kinase of the Ca2+/calmodulin family localized to the mitochondria (8). The mitochondrial localization of Pink1 protein has been shown by using transfected cells (8, 25, 26). In vitro biochemical studies have demonstrated that Pink1 possesses autophosphorylation activity and that pathogenic mutations in Pink1 have differential effects on this activity (25, 26). The in vivo substrate(s) and molecular function of Pink1 are unknown. Cell culture studies have shown that overexpression of wild-type Pink1 can lead to a reduction of cytochrome c release from mitochondria and prevent the subsequent activation of caspases under both basal and apoptotic stress conditions (27). This result suggests that Pink1 may play a critical role in regulating mitochondrial physiology and/or the mitochondrial pathway of cell death, although the in vivo relevance of these findings remains to be determined.
To understand the physiological function of Pink1 and how its dysfunction may cause PD, we have used Drosophila as a model system. Here, we describe the phenotypes caused by inactivation of Drosophila Pink1 (dPink1). Our results indicate that dPink1 is required for maintaining proper mitochondria morphology and the integrity of subsets of muscle cells and dopaminergic neurons. Significantly, we find that Pink1 and Parkin show clear genetic and biochemical interactions. Our results suggest that Parkin and Pink1 function in a common molecular pathway that regulates mitochondria function and the survival of selective cell types.
Results
Knocking Down of dPink1 Expression by Transgenic RNAi.
In the sequenced fly genome, the one homologue of human Pink1 (hPink1) is dPink1. Although dPink1 and hPink1 share ≈26% identity at the amino acid level, their homology is quite low at the DNA level, because several sequence homology prediction algorithms failed to detect significant homology between the two. Based on the fact that loss-of-function of hPink1 is associated with familial PD, we used the transgenic RNAi approach to knockdown the expression of dPink1 in an effort to model Pink1-associated PD. To confirm that the expression of dPink1 dsRNA resulted in a down-regulation of endogenous dPink1 transcripts, we used RT-PCR to measure dPink1 mRNA levels after ubiquitous induction of RNAi. An ≈80% reduction of dPink1 mRNA was observed (Fig. 1A). We next assayed the effect of RNAi on endogenous dPink1 protein expression by using a polyclonal antibody raised against dPink1. As shown in Fig. 1B, ubiquitous dPink1 RNAi resulted in a similar degree reduction of endogenous dPink1 protein level. Taken together, these results demonstrated that our RNAi approach was effective in causing significant reduction of dPink1 mRNA and protein expression.
Fig. 1.

Inhibition of dPink1 by RNAi causes defects of abnormal wing posture, shortened lifespan, and ATP deficit. (A and B) RT-PCR and Western blot analyses of dPink1 mRNA and protein levels after RNAi. Genotypes: 1, Da-Gal4/+; 2, Da-Gal4>dPink1 RNAi. RP49 and tubulin serve as controls. (C–E) Wing posture phenotypes in control and dPink1 RNAi flies. Left, females; right, males. Flies were 7 days old and kept at 29°C. (C) Straight wing posture of control flies. (D) dPink1 RNAi flies displaying held-up wings. (E) dPink1 RNAi flies displaying drooped wings. (F) Abolished flight activities in dPink1 RNAi flies. Two independent lines were used. (G) dPink1 RNAi flies exhibit a sharper age-dependent decline of ATP content. (H) Female dPink1 flies exhibit a shortened lifespan. (A–H) RNAi was achieved by using a UAS-dPink1 RNAi construct driven by Da-Gal4. ∗, P < 0.01 in Student’s t test.
Ubiquitous Inhibition of dPink1 Results in Abnormal Wing Posture, Energy Depletion, and Shortened Lifespan.
We next analyzed the physiological consequence of inhibiting dPink1 function. First, we used daughterless (Da)-Gal4 to drive the expression of dPink1 dsRNA ubiquitously. The emergence of adult flies after ubiquitous inhibition of dPink1 suggested that either loss of dPink1 do not lead to lethality in Drosophila or that the level of reduction of dPink1 expression by this transgenic RNAi approach was not complete enough to cause lethality.
The first discernible phenotype of these dPink1 RNAi flies was abnormal wing posture: Both females and males exhibited either a held-up (Fig. 1D) or a drooped (Fig. 1E) wing posture, whereas control flies always held their wings parallel to the body axis (Fig. 1C). The penetrance of this phenotype increased with age: When raised at 29°C, ≈20% of newly eclosed flies exhibited abnormal wing posture, whereas by 7 days of age nearly 100% of them displayed this phenotype. These flies had no problem with walking, but their climbing ability was greatly reduced and their ability to fly was completely abolished by 10 days of age (Fig. 1F). This phenotype was observed in two independent dPink1 RNAi lines analyzed.
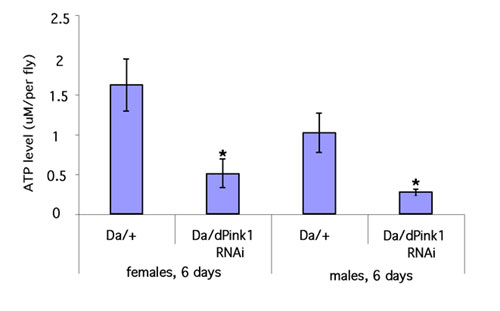
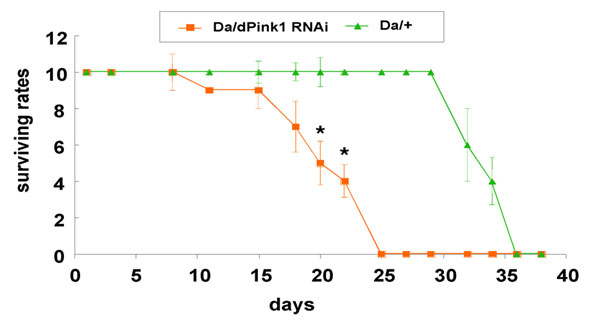
Considering that flight is a rather energy-consuming physiological process relying on mitochondrial ATP synthesis and that hPink1 has been reported to be associated with mitochondria, we then measured the energy supply of the dPink1 RNAi flies, by using ATP level as an index. Our HPLC data showed that dPink1 RNAi flies displayed an ≈70% reduction of overall ATP level compared with control flies (Fig. 6, which is published as supporting information on the PNAS web site). To test whether this ATP deficit was age-dependent, we took advantage of the fact that the level of transgene expression is temperature-dependent in the UAS-Gal4 system. We raised the flies at 18°C until eclosion to minimize RNAi effect and then shifted them to 29°C immediately after eclosion to induce stronger RNAi effect. Our HPLC analysis clearly demonstrated that, although the ATP level of the newly eclosed dPink1 RNAi flies was comparable with that of the control flies, it dropped sharply to ≈40% of the control level within a week, and the level remained low after 2 weeks under this experimental condition (Fig. 1G). We next asked whether the energy deficiency affected lifespan of dPink1 RNAi flies. We found that global inhibition of dPink1 reduced lifespan significantly (Fig. 1H and Fig. 7, which is published as supporting information on the PNAS web site). Taken together, these data suggest that Pink1 plays an important role in regulating energy metabolism and that this function has impact on lifespan.
Knocking Down of dPink1 Induced Selective Muscle Degeneration.
One possible anatomical basis for the abnormal wing posture could be defective flight muscles. Histological analysis of indirect flight muscles (IFMs), the major flight muscles, of the Da-Gal4>dPink1 RNAi flies revealed severe disruption of muscle integrity, consistent with the finding of abolished flight capacity in these flies (Fig. 2B). Disrupted muscle integrity was observed in both the wing elevator muscles [dorsal ventral muscles (DVMs)] and depressor muscles [dorsal longitudinal muscles (DLMs)]. To determine whether dPink1 plays a direct role in the flight muscles, we directed dPink1 RNAi specifically in the muscle with the myosin heavy chain (Mhc)-Gal4 driver. Despite a lower penetrance (≈40% of 7-day-old flies raised at 29°C), we observed similar abnormal wing posture and muscle disruption in the flies with muscle-specific dPink1 knockdown (Fig. 2 D and J). The lower penetrance could be because of less efficient RNAi by Mhc-Gal4 driver or contribution by nonmuscle cells to the phenotypes observed in ubiquitous RNAi animals. Several lines of evidence suggested that this muscle phenotype resulted from specific inhibition of dPink1 by RNAi. First, expression of white, DJ-1A, or DJ-1B dsRNAs driven by the same Gal4 driver had no effect on the flight muscles (data not shown). Second, we could rescue this tissue-specific RNAi phenotype with increased expression of dPink1. We reasoned that by raising the level of dPink1 transcripts, the RNAi effect could be dampened. Indeed, coexpression of a UAS-dPink1 transgene could suppress the abnormal wing and disrupted muscle phenotypes induced by dPink1 RNAi (Fig. 2 E and K). This rescuing effect is unlikely due to titration of Gal4 protein by the addition of UAS transgenes, because a UAS-GFP transgene had no effect on the phenotypes (Fig. 2 F and L). Finally, we could rescue the dPink1 RNAi phenotypes by coexpression of full-length hPink1 in the muscle (Fig. 2 G and M). However, a C-terminal truncated form of hPink1 (hPink1ΔC) was not able to rescue (Fig. 2 H and N), consistent with the findings that C-terminal truncations of hPink1 are linked to familial PD (8, 28). The fact that we used transgenic flies expressing comparable levels of full-length hPink1 and hPink1ΔC ruled out the possibility that the differential rescuing effect was because of differential expression of hPink1 (Fig. 8, which is published as supporting information on the PNAS web site). Instead, this result indicates that hPink1can functionally substitute for dPink1. The divergence at the DNA sequence level between dPink1 and hPink1 makes it unlikely that hPink1 RNA will interfere with the RNAi efficiency of dPink1. Further, the sequence corresponding to dPink1 dsRNA target sequence is wholly present in both full-length hPink1 and hPink1ΔC constructs, making hPink1ΔC as competent as full-length hPink1 in competitive saturation of the RNAi machinery, if this did happen. Taken together, we concluded that the abnormal wing/muscle phenotype is specifically caused by inactivation of dPink1.
Fig. 2.

Inhibition of dPink1 results in disrupted IFMs. (A-H) Light microscopy was used to examine IFM architecture (stars, dorsal longitudinal muscles; arrows, dorsal ventral muscles). Sections from resin-embedded thoraces of 1-week-old adult flies were stained with toluidine blue to visualize tissue morphology; anterior is to the left. (I–N) Wing posture phenotypes of control and dPink1 RNAi flies directed by Mhc-Gal4. Fly genotypes are Da-Gal4/+ (A), Da-Gal4>UAS-dPink1 RNAi (B), Mhc-Gal4/+ (C and I), Mhc-Gal4>UAS-dPink1 RNAi (D and J), Mhc-Gal4>UAS-dPink1 RNAi; UAS-dPink1 (E and K), Mhc-Gal4>UAS-dPink1 RNAi; UAS-GFP (F and L), Mhc-Gal4>UAS-dPink1 RNAi; UAS-hPink1 (G and M), Mhc-Gal4>UAS-dPink1 RNAi; UAS-hPink1ΔC (H and N).
Muscle-Specific dPink1 RNAi Flies Exhibited Mitochondria Dysfunction, DNA Fragmentation, and Nemaline-Like Myopathology.
We then used transmission electron microscopy to further investigate the nature of the muscle defects. Wild-type adult IFMs had a highly regular and compact myofibril arrangement, with many tightly packed electron-dense mitochondria interspersed between rows of sarcomeres (Fig. 3A). In both ubiquitous and muscle-specific dPink1 RNAi flies, some IFMs showed irregular and dispersed myofibril arrangement (Fig. 3B). The number of mitochondria among myofibril was reduced, whereas many of the remaining mitochondria were grossly swollen, lacking electron-dense material, and showing disintegration of cristae (Fig. 3B). Interestingly, there were electron-dense deposits within the area of IFMs that succumbed to severe disruption (Fig. 3B), reminiscent of nemaline (rod body) myopathy in humans (29). Notably, abnormally swollen mitochondria were present within morphologically normal myofibrils (Fig. 3B). Consistent with the histological observation, myofibril and mitochondrial integrity could be restored by the overexpression of full-length hPink1 (Fig. 3C) but not hPink1ΔC (Fig. 3D).
Fig. 3.

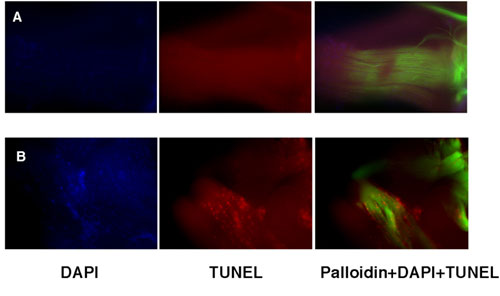
Muscle-specific dPink1 RNAi results in myopathology and age-dependent apoptosis in the IFMs. (A–D) EM analysis of IFM ultrastructure of flies with the following genotypes: Mhc-Gal4/+ (A), Mhc-Gal4>UAS-dPink1 RNAi (B), Mhc-Gal4>UAS-dPink1 RNAi; UAS-hPink1 (C), and Mhc-Gal4>UAS-dPink1 RNAi; UAS- hPink1ΔC (D). Arrows, swollen mitochondria; arrowheads, rod body-like deposits. (E and F) EM analysis of TTM ultrastructure in Mhc-Gal4/+ (E) and Mhc-Gal4>UAS-dPink1 RNAi (F) flies. Scale bars (5 μm) are shown at the lower left corner of each image. (G and H) TUNEL staining of thoracic musculatures from newly eclosed (G) and 1-week-old (H) Mhc-gal4>dPink1 RNAi flies. Left, DAPI staining; Center, TUNEL staining; Right, merged images. Arrows point to TUNEL-positive nuclei.
In contrast to IFMs, the tergotrochanteral muscles (TTMs), or the “jumping” muscles, remained fairly normal in the muscle-specific dPink1 RNAi flies (Fig. 3F, compared with Fig. 3E). These differential effects were unlikely due to the expression pattern of Mhc-GAL4, because flies with ubiquitous dPink1 RNAi also had normal TTMs. The TTMs apparently have fewer mitochondria than the IFMs. We speculate that the phenotypic difference between IFMs and TTMs may be related to their different energy demands.
To investigate whether muscle degeneration in dPink1 RNAi flies developed through a cell death mechanism, the IFMs were subjected to a terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling (TUNEL) assay. Flies with ubiquitous and continuous induction of dPink1 RNAi exhibited extensive TUNEL-positive signals in the IFMs when analyzed at 4 days of age, whereas age-matched control flies lacked such staining (Fig. 9, which is published as supporting information on the PNAS web site). To exclude any developmental effect and to test whether TUNEL-positive signals could be induced progressively, muscle-specific dPink1 RNAi flies were subjected to the temperature-shift protocol mentioned earlier. No positive TUNEL signal was detected in IFMs from flies newly emerged at 18°C (Fig. 3G). However, after shifting to 29°C and continuously kept at that temperature for 7 days, dPink1 RNAi flies readily showed many TUNEL-positive nuclei in IFMs (Fig. 3H), although the same treatment had no effect in control flies. These data suggest that muscle-specific dPink1 RNAi could lead to age-dependent muscle degeneration characterized by extensive DNA fragmentation probably indicative of cell death.
Inactivation of dPink1 in Dopaminergic Neurons Leads to Loss of Tyrosine Hydroxylase (TH)+ Neuron and Reduction of Brain Dopamine Content.
We next analyzed the effects of inhibiting dPink1 function in dopaminergic neurons by inducing dPink1 RNAi with the dopaminergic neuron-specific TH-Gal4 driver. The presence of dopaminergic neurons in the CNS was assayed by TH immunostaining of whole-mount preparations of adult fly brain. The number of TH+ neurons in the different dopaminergic clusters in control and dPink1 RNAi flies were counted and subjected to statistical analysis. As shown in Fig. 4B, 25-day-old dPink1 RNAi flies raised at 29°C showed a significant reduction of TH+ neurons in the lateral protocerebral posterior (PPL1) cluster. The dorsomedial protocerebral posterior (PPM) cluster [also known as dorsomedial cluster (DMC)] also showed a modest reduction of neuronal number, whereas the other clusters were relatively unaffected (Fig. 4F). Importantly, as observed in the muscle, coexpression of full-length hPink1, but not hPink1ΔC, was able to suppress the dopaminergic phenotype (Fig. 4 C, D, and F). Overexpression of hPink1 or hPink1ΔC alone has no effect on TH+ neuron number (data not shown).
Fig. 4.

Dopaminergic defects in dPink1 RNAi flies. (A–E) Whole-mount brain TH immunostaining of dorsolateral protocerebral posterior (PPL1) cluster neurons in 25-day-old flies of the following genotypes: TH-Gal4/+ (A), TH-Gal4/UAS-dPink1 RNAi; UAS-GFP (B), TH-Gal4/UAS-dPink1 RNAi; UAS- hPink1ΔC (C), TH-Gal4/UAS-dPink1 RNAi; UAS-hPink1 (D), and TH-Gal4/UAS-dPink1 RNAi; UAS-hParkin (E). (F) Quantification of TH+ neurons in individual dopaminergic clusters in the adult brains of the flies with the indicated genotypes. ∗, P < 0.01 in Student’s t test. (G) Quantification of head dopamine levels in TH-Gal4/+, TH-Gal4/UAS-White RNAi, or TH-Gal4/UAS-dPink1RNAi flies, with white RNAi flies serving as control. ∗, P < 0.01 in Student’s t test.
To further confirm that loss of dPink1 leads to dopaminergic dysfunction, we measured brain dopamine levels by using head extracts prepared from control and dPink1 RNAi flies. In newly eclosed flies, dopamine content was comparable between control and dPink1 RNAi flies (Fig. 4G). As the flies age, both control and dPink1 RNAi flies showed age-dependent decline of dopamine levels. However, dPink1 RNAi flies consistently exhibited a more dramatic reduction than the control (Fig. 4G). Overexpression of full-length hPink1, but not hPink1ΔC, fully restored dopamine level in dPink1 RNAi animals (Fig. 10, which is published as supporting information on the PNAS web site). Taken together, these data suggested that dPink1 plays a critical role in promoting dopaminergic neuronal function and survival.
Overexpression of Parkin Rescued the Muscle and Dopaminergic Phenotypes Caused by dPink1 Inactivation.
We noticed the similarity between our dPink1 RNAi flies and parkin mutant flies in the muscle and dopaminergic pathology (12, 30, 31). This observation prompted us to investigate whether Parkin and Pink1 might be involved in common physiological processes. Strikingly, the abnormal wing postures caused by dPink1 RNAi could be rescued by the overexpression of human Parkin (hParkin) (Fig. 5A), whereas hDJ-1 (Fig. 5B) or dDJ-1A overexpression had no rescuing effect. Histological and EM analyses indicated that the suppression of the wing phenotype was accompanied by restoration of myofibril integrity and mitochondrial morphology (Fig. 5 C and E, compared with Fig. 5 D and F). Further, overexpression of hParkin was able to restore ATP levels in dPink1 RNAi flies (Fig. 5G). Importantly, overexpression of hParkin in dPink1 RNAi background restored the number of TH+ neurons in the PPL1 and dorsomedial protocerebral posterior (PPM1) clusters (Fig. 4 E and F). However, although hParkin overexpression showed a tendency to elevate brain dopamine levels in dPink1 RNAi animals, the effect was not statistically significant (Fig. 10). Thus, overexpression of hParkin is capable of rescuing most, but not all, of the defects caused by dPink1 inactivation.
Fig. 5.

Genetic and biochemical interaction between Pink1 and Parkin. Wing posture (A and B), thorax musculature histology (C and D), and IFM EM images (E and F) of Mhc-Gal4/UAS-dPink1 RNAi; UAS-hParkin (A, C, and E), and Mhc-Gal4/UAS-dPink1 RNAi; UAS-hDJ-1 (B, D, and F) flies are shown. Scale bars (2 μm) in E and F are shown at the bottom left corner. (G) Whole-body ATP measurements of the indicated genotypes. ∗, P < 0.01 in Student’s t test. (H) Western blot analysis showing reduction of dParkin levels in dPink1 RNAi animals. Protein extracts prepared from Da-Gal4/+ and Da-Gal4/UAS-dPink1 RNAi animals were probed with anti-dParkin antibody. Actin serves as protein loading control.
To gain further insight into the molecular mechanism by which Parkin overexpression rescues dPink1 RNAi phenotypes, we examined Parkin protein level in dPink1 RNAi animals. As shown in Fig. 5H, Parkin protein level was significantly reduced in dPink1RNAi animals compared with that in the controls. Thus, the rescue of dPink1 RNAi phenotypes by Parkin overexpression is likely because of restoration of Parkin protein expression and activity.
Discussion
In this study, we show that inhibition of dPink1 in Drosophila leads to mitochondrial abnormality and degeneration of subsets of muscle fibers and dopaminergic neurons. The similar phenotypes caused by dPink1 and Drosophila Parkin (dParkin) inactivation prompted our investigation into their genetic and biochemical relationships. We found that overexpression of hParkin, but not DJ-1, could rescue both the muscle and dopaminergic pathology induced by dPink1 inactivation. Consistent with the genetic interaction results, we found that the level of dParkin was significantly reduced in dPink1 RNAi animals. Together, these results strongly suggest that Parkin and Pink1 act in a common cellular pathway that normally promotes the function and survival of select cell types including dopaminergic neurons and that Parkin may act downstream of Pink1 in this pathway.
What could be the molecular mechanism underlying the degeneration phenotypes induced by Pink1 dysfunction? At lease three possibilities could be envisioned. First, Pink1 may regulate energy metabolism. When Pink1 function is compromised, tissues that have the greatest demand for energy, presumably the IFM and dopaminergic neurons, may become particularly vulnerable. Our observation that inhibition of Pink1 leads to ATP depletion is consistent with this possibility. The second possibility is that Pink1 may be normally required to guard against the mitochondrial pathway of apoptotic cell death, as suggested earlier (27). In this regard, it is interesting to note that Parkin has also been shown to prevent mitochondrial swelling and cytochrome c release in mitochondria-dependent cell death (23). Thus, Parkin and Pink1 may participate in a common pathway that protects cells against mitochondria-dependent cell death induced by toxic insults. The abnormal mitochondrial morphology associated with Parkin and Pink1 inactivation also suggests the third possibility that they may play fundamental roles in regulating mitochondrial biogenesis or mitochondrial dynamics, such as mitochondrial fusion or fission events. A connection between aberrant mitochondrial fission/fusion and neurodegeneration has been appreciated before (32). Further studies are needed to distinguish among these possibilities.
Our in vivo rescue studies clearly showed that the C terminus of hPink1 is required for hPink1 to rescue dPink1 RNAi phenotypes. Thus, although the C-terminal deleted form of Pink1 may have higher in vitro kinase activity in terms of autophosphorylation (26), it is incapable of providing the full spectrum of Pink1’s biological activity. It is possible that C-terminal deletion may affect the binding of Pink1 to its substrates or other cofactors. Alternatively, deletion of Pink1 C terminus may contribute to disease pathogenesis by causing deregulation of Pink1 kinase activity.
Our in vivo biochemical study showed that in dPink1 RNAi animals the level of dParkin is significantly reduced. This result provides one explanation of why Pink1 and Parkin mutants give very similar mutant phenotypes and why Parkin overexpression can rescue dPink1 RNAi phenotypes. It further supports the notion that Parkin acts downstream of Pink1 in a common pathway. The biochemical mechanism by which Pink1 regulates Parkin protein level requires further investigation. In summary, the mitochondrial pathology and IFM and dopaminergic neuron degeneration phenotypes observed in dPink1 RNAi animals and the clear genetic interaction between Pink1 and Parkin suggest that further genetic analysis of the cellular pathway involving Pink1 and Parkin will reveal fundamental mechanisms governing mitochondrial and cellular maintenance. Such mechanisms will likely be applicable to mammalian systems.
Materials and Methods
Drosophila Genetics.
Fly culture and crosses were performed according to standard procedures and raised at indicated temperatures. All general fly stocks and GAL4 lines were obtained from the Bloomington Drosophila stock center. The TH-GAL4 driver was a gift from Serge Birman (Developmental Biology, Institute of Marseille, Marseille, France) (33). The other fly stocks were described earlier as follows: UAS-hParkin (34), UAS-hDJ-1 (21), and UAS-White RNAi (35). To generate UAS-dPink1 RNAi transgenics, genomic DNA/cDNA hybrid constructs were generated as described (35), with the cDNA sequence covered by the following PCR primers (5′-TTCTGCCACCACCGCCCCCACACTTC and 3′-CCGCAGCACATTGGCAGCGGTGG).
Molecular Biology.
To make UAS-hPink1, UAS-hPink1ΔC, and UAS-dPink1 transgenics, corresponding cDNAs were cloned into the pUAST vector. The plasmid containing hPink1 cDNA was a gift from M. Unoki (36). The hPink1ΔC construct contains the first 509 aa, mimicking the reported disease-linked, C-terminal truncated form of hPink1 (28). Details of the cloning steps are available on request. For RT-PCR analysis, 4-day-old adult flies from the cross between UAS-dPink1 RNAi and Da-GAL4 raised at 29°C were used to prepare total RNA by using an RNeasy Kit (Qiagen). Details of the quantitative RT-PCR procedure were essentially as described (34). Antibody against dPINK1 was elicited in rabbits with recombinant proteins purified from bacteria culture expressing pGEX-6P-1-Pink1C, which contains the C-terminal 97 aa of dPink1 (amino acids 624 to 721). Western blot analysis by using this antibody was performed as described (34), with the primary antibody used at 1:1,000 dilution. hParkin cDNA, a gift from N. Hattori (Juntendo University, Tokyo, Japan), is inserted into pcDNA3 vector with a Myc tag in C terminus. Rabbit anti-Drosophila TH antibody was raised against recombinant GST-Drosophila TH (longer isoform, 1–328 aa) produced in bacteria. The crude serum was immunoabsorbed with NHS-activated Sepharose (General Electric Biosciences) coupled with soluble bacteria proteins. Supernatant (1:500) was used for the immunostaining.
Histology and Immunohistochemistry, Muscle Histology, and Transmission Electron Microscopy Analysis.
Whole-mount immunohistochemistry for TH staining was performed as described (31). An average of eight flies for each genotype per time point were examined, and each experiment was repeated at least once. Muscle histology and transmission electron microscopy analysis were performed as described (31), except that Epon resin was used for embedding. For TUNEL analysis, adult flies were fixed in ice-cold 4% formaldehyde/PBS for 3 h. For permeation, 1% Triton and prechilled acetone were used. Head and abdomen were then dissected away, and thoraces were subject to TUNEL analysis according to the manufacturer’s instructions (Roche and Promega).
Flight and Longevity Analyses.
For flight analysis, ≈15 flies of control and experimental flies were placed in individual vials. Flight events were counted for two consecutive minutes in the late morning and then subjected to averaging. For lifespan analysis, flies were maintained on standard media at 29°C, 15 flies per vial, and transferred to new food medium daily. Mortality was scored daily.
ATP and Dopamine Measurements.
For whole-fly ATP measurement, live adult flies were dropped into liquid nitrogen for a snap freezing. Sixty microliters of dry ice-embedded acetonitrile was added to three flies, which were homogenized while adding ice-cold deionized H2O. The homogenate was centrifuged at 13,200 × g at 4°C for 15 min, and 50 μl of supernatant was mixed with 50 μl deionized H2O, centrifuged again, and readied for HPLC assay. HPLC analysis of ATP content was performed essentially as described (37, 38). HPLC analysis of catecholamine levels was performed as described (39, 40). For sample preparation, adult male fly heads were dissected out and homogenized in 0.1 M perchloric acid (generally 50 μl per four or five heads) by using a motorized hand-held tissue grinder. The homogenate was frozen immediately on dry ice and stored at −80°C before HPLC analysis.
Supplementary Material
Acknowledgments
We thank Dr. Serge Birman and the Bloomington Drosophila Stock Center for fly stocks, Dr. M. Unoki for the hPink1 cDNA, Dr. N. Hattori for hParkin cDNA, Dr. Leo Palanck for dParkin antibody, Dr. Su Guo for reading the manuscripts, and Dr. Zhuohua Zhang for communicating unpublished results. We also thank Jennifer Quach and Yali Zhang for excellent technical support and members of the Lu laboratory for discussions. This work was supported by the McKnight, Beckman, and Sloan Foundations (B.L.), Naito Foundation and Japan Society for the Promotion of Science Postdoctoral Fellowships for Research Abroad (to Y.I.), and a Stanford Bio X Graduate Fellowship (to Y.Y.).
Abbreviations
- DA
dopamine
- dPink1
Drosophila Pink1
- hPink1
human Pink1
- dParkin
Drosophila Parkin
- hParkin
human Parkin
- IFM
indirect flight muscle
- PD
Parkinson’s disease
- RNAi
RNA interference
- TH
tyrosine hydroxylase
- TTM
tergotrochanteral muscles.
Footnotes
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Dunnett S. B., Bjorklund A. Nature. 1999;399:A32–A39. doi: 10.1038/399a032. [DOI] [PubMed] [Google Scholar]
- 2.Dawson T. M., Dawson V. L. Science. 2003;302:819–822. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- 3.Bertoli-Avella A. M., Oostra B. A., Heutink P. Hum. Genet. 2004;114:413–438. doi: 10.1007/s00439-004-1097-7. [DOI] [PubMed] [Google Scholar]
- 4.Polymeropoulos M. H., Lavedan C., Leroy E., Ide S. E., Dehejia A., Dutra A., Pike B., Root H., Rubenstein J., Boyer R., et al. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 5.Kitada T., Asakawa S., Hattori N., Matsumine H., Yamamura Y., Minoshima S., Yokochi M., Mizuno Y., Shimizu N. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 6.Leroy E., Boyer R., Auburger G., Leube B., Ulm G., Mezey E., Harta G., Brownstein M. J., Jonnalagada S., Chernova T., et al. Nature. 1998;395:451–452. doi: 10.1038/26652. [DOI] [PubMed] [Google Scholar]
- 7.Bonifati V., Rizzu P., Squitieri F., Krieger E., Vanacore N., van Swieten J. C., Brice A., van Duijn C. M., Oostra B., Meco G., Heutink P. Neurol. Sci. 2003;24:159–160. doi: 10.1007/s10072-003-0108-0. [DOI] [PubMed] [Google Scholar]
- 8.Valente E. M., Abou-Sleiman P. M., Caputo V., Muqit M. M., Harvey K., Gispert S., Ali Z., Del Turco D., Bentivoglio A. R., Healy D. G., et al. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 9.Zimprich A., Biskup S., Leitner P., Lichtner P., Farrer M., Lincoln S., Kachergus J., Hulihan M., Uitti R. J., Calne D. B., et al. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 10.Paisan-Ruiz C., Jain S., Evans E. W., Gilks W. P., Simon J., van der Brug M., de Munain A. L., Aparicio S., Gil A. M., Khan N., et al. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 11.Palacino J. J., Sagi D., Goldberg M. S., Krauss S., Motz C., Klose J., Shen J. J. Biol. Chem. 2004;279:18614–18622. doi: 10.1074/jbc.M401135200. [DOI] [PubMed] [Google Scholar]
- 12.Greene J. C., Whitworth A. J., Kuo I., Andrews L. A., Feany M. B., Pallanck L. J. Proc. Natl. Acad. Sci. USA. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsu L. J., Sagara Y., Arroyo A., Rockenstein E., Sisk A., Mallory M., Wong J., Takenouchi T., Hashimoto M., Masliah E. Am. J. Pathol. 2000;157:401–410. doi: 10.1016/s0002-9440(10)64553-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hyun D. H., Lee M., Hattori N., Kubo S., Mizuno Y., Halliwell B., Jenner P. J. Biol. Chem. 2002;277:28572–28577. doi: 10.1074/jbc.M200666200. [DOI] [PubMed] [Google Scholar]
- 15.Orth M., Tabrizi S. J., Schapira A. H., Cooper J. M. Neurosci. Lett. 2003;351:29–32. doi: 10.1016/s0304-3940(03)00941-8. [DOI] [PubMed] [Google Scholar]
- 16.Sherer T. B., Betarbet R., Stout A. K., Lund S., Baptista M., Panov A. V., Cookson M. R., Greenamyre J. T. J. Neurosci. 2002;22:7006–7015. doi: 10.1523/JNEUROSCI.22-16-07006.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shen J., Cookson M. R. Neuron. 2004;43:301–304. doi: 10.1016/j.neuron.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 18.Kim R. H., Smith P. D., Aleyasin H., Hayley S., Mount M. P., Pownall S., Wakeham A., You-Ten A. J., Kalia S. K., Horne P., et al. Proc. Natl. Acad. Sci. USA. 2005;102:5215–5220. doi: 10.1073/pnas.0501282102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Menzies F. M., Yenisetti S. C., Min K. T. Curr. Biol. 2005;15:1578–1582. doi: 10.1016/j.cub.2005.07.036. [DOI] [PubMed] [Google Scholar]
- 20.Meulener M., Whitworth A. J., Armstrong-Gold C. E., Rizzu P., Heutink P., Wes P. D., Pallanck L. J., Bonini N. M. Curr. Biol. 2005;15:1572–1577. doi: 10.1016/j.cub.2005.07.064. [DOI] [PubMed] [Google Scholar]
- 21.Yang Y., Gehrke S., Haque M. E., Imai Y., Kosek J., Yang L., Beal M. F., Nishimura I., Wakamatsu K., Ito S., et al. Proc. Natl. Acad. Sci. USA. 2005;102:13670–13675. doi: 10.1073/pnas.0504610102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang L., Shimoji M., Thomas B., Moore D. J., Yu S. W., Marupudi N. I., Torp R., Torgner I. A., Ottersen O. P., Dawson T. M., Dawson V. L. Hum. Mol. Genet. 2005;14:2063–2073. doi: 10.1093/hmg/ddi211. [DOI] [PubMed] [Google Scholar]
- 23.Darios F., Corti O., Lucking C. B., Hampe C., Muriel M. P., Abbas N., Gu W. J., Hirsch E. C., Rooney T., Ruberg M., Brice A. Hum. Mol. Genet. 2003;12:517–526. doi: 10.1093/hmg/ddg044. [DOI] [PubMed] [Google Scholar]
- 24.Elkon H., Don J., Melamed E., Ziv I., Shirvan A., Offen D. J. Mol. Neurosci. 2002;18:229–238. doi: 10.1385/JMN:18:3:229. [DOI] [PubMed] [Google Scholar]
- 25.Beilina A., Van Der Brug M., Ahmad R., Kesavapany S., Miller D. W., Petsko G. A., Cookson M. R. Proc. Natl. Acad. Sci. USA. 2005;102:5703–5708. doi: 10.1073/pnas.0500617102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Silvestri L., Caputo V., Bellacchio E., Atorino L., Dallapiccola B., Valente E. M., Casari G. Hum. Mol. Genet. 2005;14:3477–3492. doi: 10.1093/hmg/ddi377. [DOI] [PubMed] [Google Scholar]
- 27.Petit A., Kawarai T., Paitel E., Sanjo N., Maj M., Scheid M., Chen F., Gu Y., Hasegawa H., Salehi-Rad S., et al. J. Biol. Chem. 2005;280:34025–34032. doi: 10.1074/jbc.M505143200. [DOI] [PubMed] [Google Scholar]
- 28.Rohe C. F., Montagna P., Breedveld G., Cortelli P., Oostra B. A., Bonifati V. Ann. Neurol. 2004;56:427–431. doi: 10.1002/ana.20247. [DOI] [PubMed] [Google Scholar]
- 29.Wallgren-Pettersson C. Semin. Pediatr. Neurol. 2002;9:132–144. doi: 10.1053/spen.2002.33804. [DOI] [PubMed] [Google Scholar]
- 30.Whitworth A. J., Theodore D. A., Greene J. C., Benes H., Wes P. D., Pallanck L. J. Proc. Natl. Acad. Sci. USA. 2005;102:8024–8029. doi: 10.1073/pnas.0501078102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pesah Y., Pham T., Burgess H., Middlebrooks B., Verstreken P., Zhou Y., Harding M., Bellen H., Mardon G. Development (Cambridge, U.K.) 2004;131:2183–2194. doi: 10.1242/dev.01095. [DOI] [PubMed] [Google Scholar]
- 32.Bossy-Wetzel E., Barsoum M. J., Godzik A., Schwarzenbacher R., Lipton S. A. Curr. Opin. Cell Biol. 2003;15:706–716. doi: 10.1016/j.ceb.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 33.Friggi-Grelin F., Coulom H., Meller M., Gomez D., Hirsh J., Birman S. J. Neurobiol. 2003;54:618–627. doi: 10.1002/neu.10185. [DOI] [PubMed] [Google Scholar]
- 34.Yang Y., Nishimura I., Imai Y., Takahashi R., Lu B. Neuron. 2003;37:911–924. doi: 10.1016/s0896-6273(03)00143-0. [DOI] [PubMed] [Google Scholar]
- 35.Kalidas S., Smith D. P. Neuron. 2002;33:177–184. doi: 10.1016/s0896-6273(02)00560-3. [DOI] [PubMed] [Google Scholar]
- 36.Unoki M., Nakamura Y. Oncogene. 2001;20:4457–4465. doi: 10.1038/sj.onc.1204608. [DOI] [PubMed] [Google Scholar]
- 37.Matthews R. T., Yang L., Jenkins B. G., Ferrante R. J., Rosen B. R., Kaddurah-Daouk R., Beal M. F. J. Neurosci. 1998;18:156–163. doi: 10.1523/JNEUROSCI.18-01-00156.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Manfredi G., Yang L., Gajewski C. D., Mattiazzi M. Methods. 2002;26:317–326. doi: 10.1016/S1046-2023(02)00037-3. [DOI] [PubMed] [Google Scholar]
- 39.Ito S., Kato T., Fujita K. Biochem. Pharmacol. 1988;37:1707–1710. doi: 10.1016/0006-2952(88)90432-7. [DOI] [PubMed] [Google Scholar]
- 40.Beal M. F., Kowall N. W., Swartz K. J., Ferrante R. J. Neurosci. Lett. 1990;108:36–42. doi: 10.1016/0304-3940(90)90702-b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}