

Abstract
Type I interferon (IFN) gene therapy modulates the immune response leading to inflammatory heart disease following cytomegalovirus (CMV) infection in a murine model of post-viral myocarditis. Efficacy of different immunisation protocols for the IFN constructs was influenced by the dose of DNA, subtype choice, combination use, pre-medication, and timing of DNA administration. Optimal efficacy was found with bupivacaine treatment prior to DNA inoculation of 200mg IFN DNA 14 days prior to virus challenge. Maximal antiviral and antimyocarditic effects were achieved with this vaccination schedule. Furthermore, inoculation of synergistic IFN subtypes demonstrated enhanced efficacy when delivered either alone or with CMV gB DNA vaccination in the CMV model. Thus naked DNA delivery of IFN provides an avenue of immunotherapy for regulating herpesvirus-induced diseases.
Keywords: interferons, gene therapy, cytomegalovirus, DNA
Introduction
Cytomegalovirus (CMV) is a herpesvirus that causes latent and persistent infection. This virus may induce inflammation of the heart known as myocarditis, which may lead to dilated cardiomyopathy, arrhythmias and sudden death. Currently, there is no effective safe vaccine for CMV, as the vaccines studied so far, only provide partial protective immunity upon challenge with infectious virus. Vaccines for viral infections and diseases have encompassed live attenuated viruses, subunit virions, recombinant viral proteins/peptides and recently viral DNA constructs. Interest in cytokines has increased with the aim of seeking improvement of the efficacy of such viral DNA vaccines. Nonetheless, optimization of DNA delivery is an important aspect of vaccine development. Specifically in the case of DNA vaccines, factors including DNA dosage, choice of cytokine genes, pre-medication, and timing of DNA administration can influence the immune response following intramuscular (i.m.) vaccination.
Many studies of murine models have found that effective DNA vaccination occurs in a dose-dependent manner, generally with a dose range of 25-200μg plasmid DNA (1-4). Previously, we have found in vivo expression of interferon alpha (IFNA) genes following i.m. inoculation of 200μg naked DNA to be effective against murine cytomegalovirus (MCMV) infection. This approach also allows us to examine the superiority of individual and combinational usage of different IFN subtypes within the multigene family (5-9). Other groups using type I IFN-expressing plasmids have reported effective inoculation with 100μg i.m. (10) or 100μg/eye (11) in cancer and herpes simplex virus (HSV) infection, respectively. In addition, investigators have found a dose-response effect of pCMV-IFNα1 plasmid on HSV-1 replication in the range of 25-100μg/eye (11).
The choice of cytokines is very important in providing protective immunity via immunotherapy. Since type I IFNs belong to a multigene family, we examined the effect of seven individual IFN subtypes (IFNA1, A2, A4, A5, A6, A9 and B), administered by DNA inoculation, on systemic MCMV infection and myocarditis (8). We found that IFNA6 treatment reduced MCMV replication whilst IFNA5 and A2 treatment enhanced virus replication. Moreover, IFNA6, A9, and B treatment inhibited acute myocarditis whilst IFNA6 was able to also reduce chronic cardiac inflammation. In addition, viral DNA vaccines encoding the CMV glycoprotein B (gB) are currently being investigated and require improved efficacy. We have previously studied the efficacy of the gB DNA vaccine with the application of type I IFNs, IFNA6, IFNA9 and IFNB (9). CMV-induced myocarditis was preferentially reduced with either IFNA9 or IFNB but not with IFNA6, coimmunisation. However, IFNA6, IFNA9 and IFNB markedly reduced chronic myocarditis in CMV gB-vaccinated mice.
Pre-medication with bupivacaine prior to DNA inoculation is also a factor in the optimization of the DNA inoculation protocol. Bupivacaine is an anaesthetic which destroys myofibres, induces myogenesis and is reported to enhance protein expression following DNA inoculation since regenerating muscle is thought to encourage DNA uptake (12). Reports of the effect of bupivacaine pre-medication on transgene expression demonstrate conflicting evidence with some reports suggesting no effect and some observing up to 40-fold increase in gene expression (6, 12).
Previous reports have demonstrated that the timing of DNA inoculation contributes to therapeutic outcome. In particular, timing of DNA inoculation can influence the type of Th cell response induced. This was first demonstrated with plasmid encoded GM-CSF (pGM-CSF) co-administered with DNA encoding the envelope protein of HIV. Inoculation of pGM-CSF 3 days prior to DNA vaccination induced a predominantly Th2-type immune response, simultaneous administration with antigen induced both a Th1 and Th2 type response and inoculation 3 days after DNA vaccination stimulated a predominantly Th1 response (13). A similar effect has since been observed with plasmid administered IL-12 in models of HIV and Leishmania vaccination (1, 2). Similarly, previous studies in our laboratory have demonstrated variability in the immunomodulatory efficacy of type I IFN subtype transgenes, which is dependent on the timing of DNA inoculation in relation to MCMV challenge (5).
In this study, we determined the optimal inoculation schedule for type I IFN subtype DNA constructs in the MCMV model of infection and disease. We found that DNA dosage, bupivacaine pre-medication, and timing of DNA inoculation (5), were contributing factors in the therapeutic efficacy of IFN DNA therapy in regulating herpesvirus infection and disease sequelae. Furthermore, we examined the synergistic effects of gB, IFNA6 and IFNB compared to that of the gB DNA vaccine alone. We found that the combined gB/IFNA6/IFNB DNA vaccine was superior to the gB DNA vaccine in providing protection from MCMV infection. Further applications of the synergistic properties provided by type I IFNs may be used in conjunction with other viral DNA vaccines for improved vaccine efficacy.
Materials and Methods
Mice
Specific pathogen-free inbred male BALB/c mice of 4 weeks of age were supplied by the Animal Resources Centre (Murdoch, Western Australia).
Virus
The K181 strain of MCMV was prepared as a salivary gland homogenate by passage through 3-week-old female BALB/c mice and stored under liquid nitrogen. MCMV titres in target organs were quantitated by plaque assay using mouse M2-10B4 stromal cells. Virus titres are expressed as mean pfu/g tissue ± SEM (5 mice per group). The limit of detection was 100 pfu/g for liver and spleen and 6400 pfu/g for salivary gland tissue.
Expression plasmid DNA constructs
IFN subtype genes and the full-length gB MCMV gene were cloned into the mammalian expression vector, pkCMVint (VICAL, San Diego, CA) as previously described (8, 9). Large-scale plasmid preparations were obtained from terrific broth cultures of transformed E.coli (DH-5α) using standard DNA extraction procedures with LiCl precipitation. DNA integrity was checked by agarose gel electrophoresis and concentrations determined by spectrophotometric analysis.
DNA immunisation and virus challenge protocol in vivo
To induce muscle regeneration, mice were injected bilaterally with 20μL 0.5% bupivacaine 5 days prior to inoculation of DNA constructs (either 50, 100, 200 or 400μg/mouse as indicated). DNA was diluted in a 50μL volume of pyrogen-free saline and injected bilaterally in tibialis anterior (TA) muscles. Mice were either treated with the Blank expression vector or vectors expressing IFN subtypes (IFNA6, A9 and B) or the gB transgene alone or in combination. At 14 days post DNA-treatment, mice were inoculated with 100μL of 1x104 pfu MCMV, diluted in pyrogen-free saline, by the intraperitoneal (i.p.) route (5 mice/group). At days 3, 7, 10 and 56 post-infection (p.i.), tissues were collected. Deviations from this standard protocol are stated in the results and figure legends.
Myocarditis analysis
Hearts from infected mice were removed at day 7 and 56 p.i., transected along the midline and fixed overnight in Bouin’s fluid, transferred to 70% ethanol and then processed into paraffin blocks. Sections were stained with haematoxylin and eosin (H&E) and evaluated for evidence of cellular inflammation and necrosis as previously described (8). Two heart sections were scored for each animal. Myocarditis was evaluated histologically as the mean number of inflammatory foci per heart section ± SEM (5 mice/group).
Statistical analysis
Levels of significance (p<0.05) were determined by the unpaired student t-test assuming unequal variance between the means.
Results and Discussion
Results
Previously, we have demonstrated efficacy using 200μg IFN transgenes inoculated i.m. Here, we evaluated IFN transgene inoculation using lower doses of DNA for therapeutic efficacy in reducing virus titres and cardiac inflammatory foci. Mice were treated with either 50μg or 100μg Blank, IFNA6 or IFNB DNA and challenged with a sublethal dose of MCMV (1x104 pfu i.p.). The effect of low doses of IFNA6 and IFNB on early MCMV replication and acute-phase myocarditis was evaluated in comparison to the Blank vector. Acute-phase viral titres were determined by plaque assay of spleen and liver homogenates at day 3 p.i. and salivary gland homogenates at day 7 p.i. and acute phase myocarditis was evaluated at day 7 p.i.
The antiviral efficacy previously demonstrated by these IFN subtypes at 200μg per mouse (8) was suppressed at these lower doses. More specifically, IFNA6 and IFNB DNA had no significant effect on viral titres in any tissue examined (Fig. 1A, B and C). IFNA6 at 50µg reduced liver titres by only 1.6-fold and at 100µg marginally reduced both spleen (2.0-fold) and liver (1.3-fold) titres compared to Blank-vector treated mice. Whereas, treatment with IFNA6 at 200µg was most effective with significant reductions of 5.7- and 19-fold of virus titres in the spleen and liver, respectively. However, treatment with IFNB at either 50µg, 100µg or 200µg did not affect virus titres in any tissue tested. Administration of 200µg of either IFNA6 or IFNB has been previously associated with suppression of acute stage myocarditis with 4.7- and 2.4-fold reduction, repectively compared to Blank vector-treated mice (8). Here, we show that myocarditis was also significantly reduced with inoculation of 50μg/mouse of either IFNA6 (4.3-fold) or IFNB (1.7-fold) DNA in comparison to Blank vector-treated mice. However, treatment with 100μg IFN was not effective with only a 1.4-fold reduction in comparison to control groups (Fig. 1D). Similar to our previous findings, viral titre was not an indicator of disease. In brief, reducing the dose of IFN DNA to 50μg/mouse or 100μg/mouse reduced the antiviral and antimyocarditic efficacy of IFNA6 and IFNB that we have previously observed with the higher dose of 200μg/mouse in the treatment of MCMV infection and myocarditis.
Fig. 1.
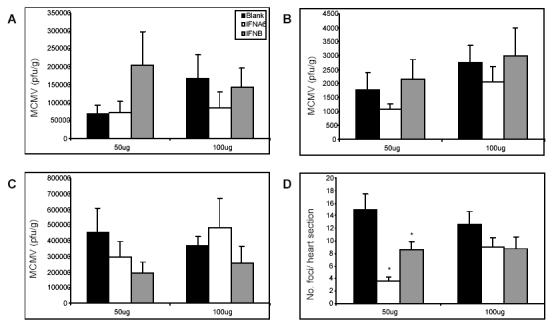
The effect of DNA dosage in IFN transgene treatment of MCMV infection and myocarditis. Bupivacaine-treated BALB/c mice were inoculated with doses of either 50μg or 100μg of Blank, IFNA6 or IFNB DNA at 14 days prior to viral challenge with 1x104 pfu of MCMV i.p. Viral titres were determined by plaque assay in (A) spleen and (B) liver at day 3 p.i. and (C) salivary glands at day 7 p.i. Results are expressed as mean pfu/g ± SEM (n=5). (D) Myocarditis was evaluated at day 7 p.i. and is expressed as the number of foci per heart section ± SEM (n=5). *, Statistical significance (p≤0.05) compared with Blank treatment groups.
Assessment of DNA dose in co-administration of synergistic IFN subtypes
The effect of combining two type I IFN subtypes was examined for determination of their possible synergistic efficacy in abrogating MCMV replication and disease. Previously we found synergistic properties of effective type I IFN subtypes when administered in combination (5). Combinations of Blank, IFNA6, IFNA9 and IFNB DNA were examined for their antiviral and antimyocarditic efficacy. In separate studies, BALB/c mice were inoculated i.m. with 200mg of each DNA construct (total 400mg DNA/mouse) or with 100mg of each DNA construct (total 200mg DNA/mouse) 14 days prior to challenge with 1x104 pfu MCMV by the i.p. route. Efficacious IFN inoculation was demonstrated only at the lower dose of 200mg total DNA, as previously described (5), while the dosage of 400mg total DNA suppressed the efficacy associated with individual administration of these IFN subtypes.
In terms of antiviral efficacy, the 400µg IFNA6/Blank combination was not as effective as 200µg IFNA6 alone. In target tissues, IFNA6/Blank treatment showed no significant change with only a 1.8-fold maximum reduction in virus titres (Fig, 2A, B, C) compared to IFNA6 alone treatment, resulting in 5.7-, 19- and 1.5-fold reductions in virus titres in the spleen, liver and salivary glands, respectively. The IFNA9 and IFNB treatments had no significant effects on viral titres with the exception of IFNB/Blank treatment at 400µg which significantly increased liver titres by 2.2-fold at day 3 p.i. (Fig. 2B) as opposed to no significant change with 200µg IFNB alone at this timepoint. Overall, the 400mg combination of IFN/Blank did not demonstrate the level of antiviral protection observed with 200mg individual and combination IFN treatment, as previously described (5, 8).
Fig. 2.
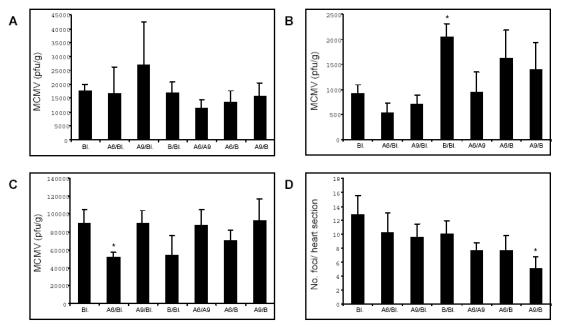
Co-treatment with 400μg IFN subtype DNA does not affect acute-phase MCMV infection or myocarditis. Bupivacaine treated BALB/c mice were inoculated with Blank vector or combinations of IFNA6/Blank, IFNA9/Blank, IFNB/Blank, IFNA6/A9, IFNA6/B, and IFNA9/B DNA at a dosage of 200μg per transgene to a total of 400μg DNA, bilaterally via the TA muscle. At 14 days post-inoculation, mice were challenged with 1x104 pfu MCMV i.p. Viral titres were determined in (A) spleen and (B) liver at day 3 p.i. and (C) salivary glands at day 7 p.i. and are expressed as pfu/g tissue ± SEM (n=5). (D) Myocarditis was evaluated at day 7 p.i. and is expressed as the number of foci per heart section ± SEM (n=5). *, Statistical significance (p≤0.05) compared with Blank treatment groups. Bl. denotes Blank expression vector.
In addition, we have previously described the synergistic properties of effective IFNs (IFNA6, A9 and B) in combination to a total of 200µg DNA. Here, we also demonstrate that doses of 400µg DNA in combination is inferior for antiviral and antimyocarditic efficacy. Specifically, in the spleen, 200µg IFNA6/A9 reduced virus titres 3.0-fold and IFNA6/B reduced virus titres 50-fold while 400µg of each of these IFN combination reduced virus titres by only 1.6-fold and 1.3-fold, respectively. Similarly, in the liver, 200µg IFNA6/B reduced virus titres 3.2-fold and IFNA9/B reduced virus titres 2.7-fold while 400µg of these DNA vaccines marginally enhanced virus titres by 1.8-fold and 1.5-fold, respectively. IFN combinations had no detectable antiviral effects in the salivary glands.
IFN treatment at 400μg DNA/mouse had no effect on acute phase MCMV-induced myocarditis with the exception of the IFNA9/B treatment combination, which reduced disease severity by 2.5-fold (Fig. 2D). However, this compared to a reduction in disease of 5.3-fold following administration of 200µg IFNA9/B and therefore is suboptimal. Notably, no significant reduction in acute myocarditis was observed with either IFNA6/Blank (1.2-fold), IFNA9/Blank (1.3-fold) or IFNB/Blank (1.3-fold, Fig. 2D) despite the protective effect previously observed following DNA treatment with these subtypes at doses of 200μg/mouse with reductions of 4.7-, 2.0- and 2.4-fold, respectively (8). Thus co-inoculation of effective type I IFNs with Blank vector at a total of 400μg/mouse resulted in abrogation of efficacy previously observed in this animal model.
Pre-medication with bupivacaine is critical for IFNA6 immunotherapy
We have previously demonstrated the superior efficacy of IFNA6 inoculation, which has potent antiviral effects and reduces disease pathogenesis in the MCMV model (8). Our standard inoculation protocol includes the treatment of mice 5 days prior to DNA inoculation with 20mL 0.5% bupivacaine given bilaterally to the TA muscles. Here, we investigated the necessity of bupivacaine treatment prior to IFNA6 administration. Mice were either treated or not treated with bupivacaine 5 days prior to IFNA6 DNA inoculation and subsequently challenged with 1x104 pfu MCMV. The effect of bupivacaine treatment on the antiviral response and myocarditis was evaluated.
Treatment with the IFNA6 transgene with bupivacaine demonstrated reduced MCMV titres in the spleen and liver at day 3 p.i. (Fig. 3A and B) and the salivary gland at day 7 p.i. (Fig. 3C). However, without the administration of bupivacaine, a significant reduction in MCMV was observed only in the liver at day 3 p.i. (Fig. 3B) with no effect observed in other tissues. Similarly, IFNA6 inoculation significantly reduced acute-phase myocarditis whilst IFNA6 administration without bupivacaine had no effect on disease severity in the myocardium (Fig. 3D). Therefore, maximal efficacy for MCMV infection was achieved with bupivacaine pre-medication i.m.
Fig. 3.
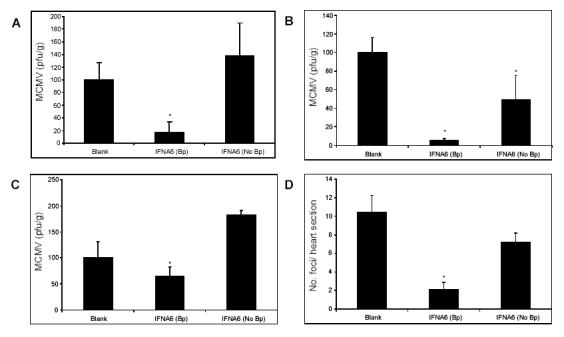
IFN DNA inoculation without bupivacaine treatment is ineffective against MCMV infection and myocarditis. BALB/c mice were either not treated (No Bp) or treated (Bp) with bupivacaine 5 days prior to DNA inoculation with 200μg of either Blank or IFNA6 vector DNA and subsequently challenged with 1x104 pfu of MCMV i.p 14 days later. Viral titres were determined by plaque assay in (A) spleen and (B) liver at day 3 p.i., and (C) salivary glands at day 7 p.i. Results expressed as % Blank treatment group ± SEM (n=5). (D) Myocarditis was evaluated at day 7 p.i. and is expressed as the number of foci per heart section ± SEM (n=5). *, Statistical significance (p≤0.05) compared with Blank treatment groups.
Combinational IFN immunotherapy for CMV gB DNA vaccination
Previous studies have revealed the capacity of specific type I IFN DNAs, when co-immunised with gB DNA, to enhance the protection afforded by the gB DNA vaccine against MCMV replication (9). Here we examine the capacity of gB/IFNA6/IFNB to enhance the protection previously observed against MCMV infection afforded by gB DNA vaccines (9), DNA encoding specific IFN subtypes (8), and combinations of IFN subtypes (5). Each mouse was infected with 1x104 PFU of MCMV 14 days post-vaccination with either Blank (200µg), gB (200µg) or combination gB/IFNA6/IFNB (50µg, 50µg, 100µg respectively) DNA constructs. The addition of the IFN combination as adjuvant to the gB vaccine did not alter the vaccines antiviral efficacy in the spleen (Fig. 4A). However, inoculation of IFNA6/IFNB DNA with gB led to a significant reduction in MCMV replication in the liver at day 7 p.i. (Fig. 4B). In addition, the combined gB/IFNA6/IFNB DNA vaccine significantly decreased both acute (Fig. 4C) and chronic (Fig. 4D) myocarditis in comparison to the Blank and gB DNA vaccinated groups.
Fig. 4.
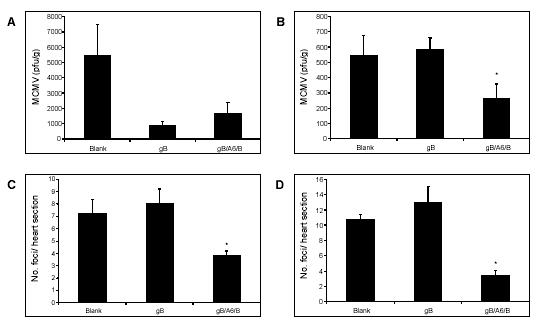
Combinational IFN DNA co-administration with gB improves vaccine efficacy in vivo. BALB/c mice were vaccinated with either Blank, gB, or gB/IFNA6/IFNB DNA (200μg/mouse) 14 days prior to i.p. inoculation with 1x104 pfu of MCMV. Viral titres were determined by plaque assay in (A) spleen at day 3 p.i. and (B) liver at day 7 p.i. Myocarditis was evaluated at (C) day 10 p.i. and (D) day 56 p.i. Myocarditis is expressed as the number of foci per heart section ± SEM (n=5). *, Statistical significance (p≤0.05) compared with gB DNA vaccinated group.
Discussion
Here we describe a detailed study focusing on optimization of the inoculation protocol for type I IFN subtype DNA constructs in the MCMV model. Delivery of either individual or combination IFN subtypes allows for investigation of the biological function and synergy of IFN in vivo. In addition, the delivery of IFN DNA subtypes can be examined for enhancement of protective immunity afforded by viral vaccination of mice.
Contributing factors for effective DNA treatment examined in this study included optimal dose determination for IFN DNA inoculation. Previous studies in our laboratory using mammalian expression plasmid administered IFNα have effectively used inoculations of 200μg DNA (5-8). Data presented in this study demonstrates that lower doses of 50μg and 100μg type I IFN DNA, inoculated 14 days prior to virus, were ineffective in reducing viral replication. Interestingly, 50μg DNA effectively reduced acute myocarditis while 100μg IFN had no effect. Co-inoculation of effective type I IFNs (total 400μg/ mouse) resulted in abrogation of the antiviral efficacy and suppression of disease previously observed in this model (5, 8). It may be that excess doses of IFN DNA (400μg) render the IFN DNA treatment ineffective at reducing antiviral titres and myocarditis and altering cytokine profiles. The exception to this was treatment with IFNA9/B, which reduced disease (2.5-fold) and circulating IFNg titres (data not shown), however, this was not as effective as the 200μg combination IFNA9/B which reduced myocarditis by 5.3-fold (5). Therefore, dosage of DNA was found to be crucial for therapeutic outcome. In this study, we have demonstrated that both insufficient and excess DNA suppressed the efficacy of type I IFN DNA treatment. Dose-response curves from previous studies demonstrate optimal DNA doses at 100-200μg with decline in efficacy at higher concentrations (14). Effective gene immunotherapy has previously been demonstrated with doses of 200μg/mouse for IFNγR-Fc (15) and IL-4/IgG1 (16) and type I IFNs (7). Similarly, we have shown that type I IFN DNA inoculation is most effective at 200μg/mouse, administered both as individual subtypes and as combination treatments. This indicates that effective type I IFN DNA inoculation is dose-dependent which is consistent with findings in other models of DNA vaccination (1, 13).
It is widely believed that inoculation of DNA constructs into regenerating muscle contributes to enhanced uptake and expression of transgenes (12, 17). Bupivacaine treatment is an efficient inducer of myogenesis prior to plasmid inoculation (12), however, evidence for and against this treatment is conflicting. In this study, plasmid administration without bupivacaine treatment was generally ineffective for all aspects evaluated with the exception that IFNA6 reduced viral titres in the liver. Despite this, viral titres in the liver were 9.4-fold greater in the group not treated with bupivacaine compared to the bupivacaine treated group. Similarly, viral titres following IFNA6 inoculation without bupivacaine treatment increased 7.8-fold and 2.8-fold in the spleen and salivary gland respectively compared to the bupivacaine treated group. Furthermore, myocarditis increased 3.5-fold without bupivacaine treatment compared with bupivacaine treatment following IFNA6 treatment. Therefore, we demonstrated that bupivacaine treatment is a necessary component of the optimal inoculation program in DNA inoculation in the MCMV model.
We previously investigated the timing of DNA administration and demonstrated that optimal efficacy was achieved when mice were treated with IFN DNA 14 days prior to viral challenge (5). Treatment with IFNA6 and IFNB 2 days before and 2 days after viral infection proved ineffective during the acute phase of viral infection and disease. Interestingly, IFN DNA treatment 2 days prior to viral infection reduced the chronic phase of myocarditis. The limited efficacy of IFN treatment at these time-points, however, is largely expected given that systemic IFN expression peaks at day 28 following IFN treatment and is not significantly increased in comparison to Blank groups until day 14 (5). In addition, endogenous IFN, produced as part of the immune response to viral invasion, peaks 2 days following LCMV infection and may therefore mask IFN transgene expression at this time (18). We postulate that type I IFN administrated at later time-points is insufficiently expressed prior to endogenous cytokine production in response to virus. Surprisingly, IFNB treatment 2 days prior to virus contributed to a pathogenic immune response. Indeed, another study has demonstrated a distinct role for IFNβ as an “immediate-early response gene” following viral infection. This group suggests that IFNβ must be synthesized for induction of a delayed IFN gene set (including α2, α5, α6 and α8) (19). Potentiation of IFNα cytokines by IFNβ may exacerbate the immune response and lead to detrimental responses in this model.
Finally, the optimization of the IFN DNA inoculation protocol was trialed as adjuvant immunotherapy for a CMV-specific DNA vaccine. Clearly, this study confirms the superiority of the combined viral gB/IFNA6/IFNB DNA vaccine to that of the viral gB DNA vaccine in providing protection from MCMV infection and myocarditis. Strikingly, the synergistic effects of the gB/IFNA6/IFNB DNA vaccine were found to significantly decrease inflammation within the myocardium during both the acute and chronic phases to levels previously seen in MCMV-infected C57BL/6 mice which are resistant to the development of post-viral myocarditis (20). This research into the synergistic effects of the gB/IFNA6/IFNB DNA vaccine has revealed the potential of specific IFNs and immunogenic proteins to act as an enhanced immunotherapy when combined and administered as a single DNA vaccine.
Our observations using 200µg DNA/mouse show that treatment with gB/IFNA6/IFNB DNA was ineffectual in comparison to IFNA6/IFNB (5) and gB/IFNB (9), at decreasing MCMV replication in target organs. However, results from these previous studies revealed no correlation between virus load and subsequent development of acute myocarditis. Co-immunisation of IFNA6/IFNB reduced viral titres in both spleen and liver, with no improvement in acute phase myocarditis, but reduced myocarditis in the chronic phase (5). Whereas, co-immunisation with gB/IFNB caused significantly reduced MCMV titres in target organs as well as reduced acute and chronic phase myocarditis (9). Therefore results from this study and previous experiments follow the speculation of viral load in target organs not affecting acute phase myocarditis, but possibly being a factor in the latter stage development of autoimmune myocarditis. Furthermore, the gB/IFNA6/IFNB vaccine was found to be of similar efficacy to the gB/IFNB vaccine in the general suppression of MCMV-induced myocarditis. Thus viral gB/IFNB vaccination using our optimised delivery of DNA constructs would be the preferred treatment of choice.
Therapeutic application of combined IFN subtypes to DNA vaccines may provide an extensive array of new possibilities for future research into protection from diseases that were otherwise thought incurable or exhaustive of any further applications. Clinical trials into DNA viral vaccines for HIV and influenza virus have arisen due to the urgent need for a protective vaccine for these infectious diseases. Trials have used DNA vaccines encoding viral proteins such as influenza-specific hemagglutinin (21), and HIV-specific gp120 and gp160 (22), shown only to provide partial protection. However, with the knowledge gained on type I IFNs and their synergistic properties from our studies, the application of combined IFNs could possibly be used in improving the efficacy of these DNA vaccines.
In summary, we describe in this study the optimization of intramuscular expression of DNA transgenes encoding IFNα/β subtypes for improved protection of mice from MCMV-induced myocarditis. We have successfully utilised the DNA delivery method to examine the therapeutic antiviral and antimyocarditic effects of both individual and combinational IFN subtypes in vivo. This experimental system can also be applied in DNA vaccine trials to study the therapeutic efficacy of type I IFN subtypes in host protection from other virus infections.
Acknowledgments
This work is supported by the National Health and Medical Research Council of Australia (No. 990393) with funding from the Western Australian Biomedical Research Institute, Murdoch University. The authors thank Vical Inc. (CA) for the generous donation of the plasmid vector, pkCMVint.
Appendix
Protocols
Protocol 1: Large-scale preparation of plasmid DNA
Reagents:
Terrific broth (Solution A: 1.3% Tryptone Peptone (w/v) [Difco #211705]; 2.67% Yeast extract [Becton Dickinson #211929]; 0.44% Glycerol (v/v) [Univar #242-500ML]; Solution B: 719mM K2HPO4[BDH AnalaR #104363A]; 169mM KH2PO4[Univar #391-500G]). Autoclave solution A and B separately. Combine the two solutions at a ratio of 9:1 respectively.
TEG swelling buffer (25mM Tris [Invitrogen #15504-020], pH8.0; 10mM EDTA [BDH AnalaR 10093.5V]; 0.9% Glucose (w/v) [Univar #783-500G]). Store at 4oC.
Lysis Buffer (0.2M NaOH [Univar #482-500G]; 1% SDS [BDH GPR #301754L]). Prepare just prior to use.
High Salt Neutralisation buffer (2.5M Potassium acetate [BDH #295814P]; 5% Formic acid (v/v) [Univar #2471-500]).
LiCl solution (5M LiCl [BDH GPR #290674B]; 1% MOPS (w/v) [Sigma #M-9024], pH to 8.0). Autoclave and store at 4oC.
Tris-buffered phenol. Melt phenol [Sigma # P-1037] at 65oC and extract with 1M Tris, pH 8.0 by stirring O/N. Change buffer 3 times over a 24h period or until pH of the aqueous phase is >7.6. Extract once more with 0.1M Tris, pH 8.0 and store under equilibrium buffer @ 4oC in the dark.
Phenol/ chloroform/isoamyl alcohol (25:24:1). Add an equal volume of chloroform [Sigma #C-2432]/isoamyl alcohol [Sigma #1-3643] (24:1 v/v) to Tris-buffered phenol and store under 0.1M Tris, pH8.0 at 4oC in the dark.
Isopropanol [Univar #425-2.5]
Ethanol [Sigma #E-7023]
RNase, DNase-free [Roche #1119915]
Sodium acetate [Univar #679-500G]
Protocol:
Prepare bacterial cultures containing the required plasmid by inoculating a loopful of glycerol stock plasmid into 1L Terrific Broth (containing appropriate antibiotic). Incubate O/N at 37oC with shaking.
Centrifuge at 2,500g, 4oC, 10min.
Resuspend pellet in 50mL TEG swelling buffer at 4oC. Add 100mL Lysis buffer and incubate on ice for 10min.
Add 75mL High Salt Neutralisation buffer (a white precipitate will form). Centrifuge at 2,500g, 4oC, 10min.
Pour the supernatant through sterile gauze and add an equal volume of isopropanol. Centrifuge at 2,500g, 4oC, 10min.
Pour off the supernatant and resuspend the pellet in 20mL double-deionised H2O (dd H2O). Transfer the solution to a polypropylene tube.
Add 20mL LiCl solution at 4oC, mix and stand on ice for 10min. Centrifuge at 2,500g, 4oC, 15min. Transfer the supernatant to a fresh polypropylene tube and discard pellet.
Extract the supernatant with an equal volume of phenol/ chloroform/ isoamyl alcohol. Centrifuge at 1,800g, 20oC, 5min.
Transfer the aqueous phase to a polypropylene tube and add 40mL isopropanol. Mix by inversion. Centrifuge at 2,500g, 4oC, 10min. Discard the supernatant.
Briefly dry the pellet and dissolve in 0.4mL ddH2O. Transfer to an eppendorf.
Add 4mL RNase and incubate at 37oC for 15min.
Add 40mL 3M sodium acetate (pH 4.8) and 1mL ethanol. Leave on dry ice for 15min (or at -20oC overnight). Centrifuge at 13,000g, 30min at 20oC.
Wash pellet 1mL 70% ethanol and centrifuge at 13,000g, 5min at 20oC.
Briefly dry pellet and resuspend in ddH2O.
Read OD @ 260nm and 280nm.
Protocol 2: Intramuscular immunisation of DNA constructs
Reagents:
Insulin syringes, Ultra fine needle 29 Gauge (0.33mm x 12.7mm) [Becton Dickinson #326769]
Marcain, 0.5% Bupivacaine [Astra #6641000]
Pyrogen-free saline, 0.9% sodium chloride [Astra Zeneca #217904]
Protocol:
Mice (4 week old male BALB/c) are restrained by grasping the skin behind the ears with the thumb and index finger and tucking the tail underneath the little finger (as per i.p. injections). The legs are then sprayed with 70% ethanol to enable clear identification of the tibialis anterior (TA) muscle for intramuscular inoculation.
20μL 0.5% bupivacaine inoculated bilaterally via TA muscle.
5 days later, DNA constructs (200µg/mouse) are inoculated by the intramuscular route. Dilute DNA in 50μL volume of sterile pyrogen-free saline (4μg/μL).
Inoculate mice with 25μL DNA preparation bilaterally via the TA muscle.
Allow 14 days for intramuscular protein expression from the DNA constructs prior to viral challenge.
References
- Gherardi MM, Ramirez JC, Esteban M. Interleukin-12 (IL-12) enhancement of the cellular immune response against human immunodeficiency virus type 1 Env antigen in a DNA prime/ vaccinia virus boost vaccine regimen is time and dose dependent: suppressive effects of IL-12 boost are mediated by nitric oxide. J Virol. 2000;74:6278–6286. doi: 10.1128/JVI.74.14.6278-6286.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noormohammadi AH, Hochrein H, Curtis JM, Baldwin TM, Handman E. Paradoxical effects of IL-12 in leishmaniasis in the presence and absence of vaccinating antigen. Vaccine. 2001;19:4043–4052. doi: 10.1016/S0264-410X(01)00132-3. [DOI] [PubMed] [Google Scholar]
- Prud’homme GJ, Lawson BR, Chang Y, Theofilopoulos AN. Immunotherapeutic gene transfer into muscle. Trends Immunol. 2001;22:149–155. doi: 10.1016/s1471-4906(00)01822-6. [DOI] [PubMed] [Google Scholar]
- Min W, Lillehoj HA, Burnside J, Weining KC, Staeheli P, Zhu JJ. Adjuvant effects of IL-1b, IL-2, IL-8, IL-15, IFNa, IFNg, TGFb4 and lymphotactin on DNA vaccination against Eimeria acervulina. Vaccine. 2002;20:267–274. doi: 10.1016/S0264-410X(01)00270-5. [DOI] [PubMed] [Google Scholar]
- Bartlett EJ, Cull VS, Brekalo NL, Lenzo JC, James CM. Synergy of type I interferon-A6 and interferon-B naked DNA immunotherapy for cytomegalovirus infections. Immunol Cell Biol. 2002;80:425–435. doi: 10.1046/j.1440-1711.2002.01103.x. [DOI] [PubMed] [Google Scholar]
- Lawson CM, Yeow WS, Lee CM, Beilharz MW. In vivo expression of an interferon-a gene by intramuscular injection of naked DNA. J Interferon Cytokine Res. 1997;17:255–261. doi: 10.1089/jir.1997.17.255. [DOI] [PubMed] [Google Scholar]
- Yeow WS, Lawson CM, Beilharz MW. Antiviral activities of interferon-a subtypes in vivo. J Immunol. 1998;160:2932–2939. [PubMed] [Google Scholar]
- Cull VS, Bartlett EJ, James CM. Type I interferon gene therapy protects against cytomegalovirus-induced myocarditis. Immunol. 2002;106:428–437. doi: 10.1046/j.1365-2567.2002.01423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull VS, Broomfield S, Bartlett EJ, Brekalo NL, James CM. Coimmunisation with type I IFN genes enhances protective immunity against cytomegalovirus and myocarditis in gB DNA-vaccinated mice. Gene Therapy. 2002;9:1369–1378. doi: 10.1038/sj.gt.3301809. [DOI] [PubMed] [Google Scholar]
- Horton HM, Anderson D, Hernandez P, Barnhart KM, Norman JA, Parker SE. A gene therapy for cancer using intramuscular injection of plasmid DNA encoding interferon a. Proc Nat Acad Sci USA. 1999;96:1553–1558. doi: 10.1073/pnas.96.4.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noisakran S, Campbell IL, Carr DJJ. Ectopic expression of DNA encoding IFN-a1 in the cornea protects mice from herpes simplex virus type 1-induced encephalitis. J Immunol. 1999;162:4184–4190. [PubMed] [Google Scholar]
- Danko I, Wolff JA. Direct Gene Transfer into Muscle. Vaccine. 1994;12:1499–1502. doi: 10.1016/0264-410X(94)90072-8. [DOI] [PubMed] [Google Scholar]
- Kusakabe K, Xin K, Katoh H, Sumino K, Hagiwara E, Kawamoto S, Okuda K, Miyagi Y, Aoki I, Nishioka K, Klinman D, Okuda K. The timing of GM-CSF expression plasmid administration influences the Th1/ Th2 response induced by an HIV-1-specific DNA vaccine. J Immunol. 2000;164:3102–3111. doi: 10.4049/jimmunol.164.6.3102. [DOI] [PubMed] [Google Scholar]
- Moelling K. DNA for genetic vaccination and therapy. Cytokines, Cell Molec Therapy. 1997;3:127–136. [PubMed] [Google Scholar]
- Prud'homme GJ, Chang Y. Prevention of autoimmune diabetes by intramuscular gene therapy with a nonviral vector encoding an interferon-g receptor/ IgG1 fusion protein. Gene Therapy. 1999;6:771–777. doi: 10.1038/sj.gt.3300879. [DOI] [PubMed] [Google Scholar]
- Chang Y, Prud'homme GJ. Intramuscular administration of expression plasmids encoding interferon-g-receptor/ IgG1 or IL-4/ IgG1 chimeric proteins protects from autoimmunity. J Gene Med. 1999;1:415–423. doi: 10.1002/(SICI)1521-2254(199911/12)1:6<415::AID-JGM66>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Vitadello M, Schiaffino MV, Picard A, Scarpa M, Schiaffino S. Gene transfer in regenerating muscle. Human Gene Therapy. 1994;5:11–18. doi: 10.1089/hum.1994.5.1-11. [DOI] [PubMed] [Google Scholar]
- Biron CA. Role of early cytokines, including alpha and beta interferons (IFN-a/b), in innate and adaptive immune responses to viral infections. Sem Immunol. 1998. pp. 383–390. [DOI] [PubMed]
- Marié I, Durbin JE, Levy DE. Differential viral induction of distinct interferon-a genes by positive feedback through interferon regulatory factor-7. EMBO J. 1998;17:6660–6669. doi: 10.1093/emboj/17.22.6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenzo JC, Fairweather D, Cull V, Shellam GR, James (Lawson) CM. Characterisation of murine cytomegalovirus myocarditis: cellular infiltration of the heart and virus persistence. J Mol Cell Cardiol. 2002;34:629–640. doi: 10.1006/jmcc.2002.2003. [DOI] [PubMed] [Google Scholar]
- Ulmer JB. Influenza DNA vaccines. Vaccine. 2002;20:S74–76. doi: 10.1016/S0264-410X(02)00136-6. [DOI] [PubMed] [Google Scholar]
- MacGregor RR, Ginsberg R, Ugen KE, Baine Y, Kang CU, Tu XM, Higgins T, Weiner DB, Boyer JD. T-cell responses induced in normal volunteers immunized with a DNA-based vaccine containing HIV-1 env and rev. AIDS. 2002;16:2137–2143. doi: 10.1097/00002030-200211080-00005. [DOI] [PubMed] [Google Scholar]