

Short abstract
Tryptophan synthase consists of two subunits, α and β. Two subgroups of β chain exist; the major group TrpEb_1 and the minor group, TrpEb_2. Conserved amino-acid residues of TrpEb_1 that make allosteric contact with the TrpEa subunit (the α chain) are absent in TrpEb_2. Representatives of Archaea, Bacteria and higher plants all exist that possess both TrpEb_1 and TrpEb_2. at least six lineages of the Archaea are likely to use TrpEb_2 as the functional β chain.
Abstract
Background
Tryptophan synthase consists of two subunits, α and β. Two distinct subgroups of β chain exist. The major group (TrpEb_1) includes the well-studied β chain of Salmonella typhimurium. The minor group of β chain (TrpEb_2) is most frequently found in the Archaea. Most of the amino-acid residues important for catalysis are highly conserved between both TrpE subfamilies.
Results
Conserved amino-acid residues of TrpEb_1 that make allosteric contact with the TrpEa subunit (the α chain) are absent in TrpEb_2. Representatives of Archaea, Bacteria and higher plants all exist that possess both TrpEb_1 and TrpEb_2. In those prokaryotes where two trpEb genes coexist, one is usually trpEb_1 and is adjacent to trpEa, whereas the second is trpEb_2 and is usually unlinked with other tryptophan-pathway genes.
Conclusions
TrpEb_1 is nearly always partnered with TrpEa in the tryptophan synthase reaction. However, by default at least six lineages of the Archaea are likely to use TrpEb_2 as the functional β chain, as TrpEb_1 is absent. The six lineages show a distinctive divergence within the overall TrpEa phylogenetic tree, consistent with the lack of selection for amino-acid residues in TrpEa that are otherwise conserved for interfacing with TrpEb_1. We suggest that the standalone function of TrpEb_2 might be to catalyze the serine deaminase reaction, an established catalytic capability of tryptophan synthase β chains. A coincident finding of interest is that the Archaea seem to use the citramalate pathway, rather than threonine deaminase (IlvA), to initiate the pathway of isoleucine biosynthesis.
Background
Tryptophan biosynthesis is absent in mammals but is a general metabolic capability of prokaryotes, eukaryotic microorganisms and higher plants. It has been a classic system for elucidation of gene-enzyme relationships and regulation, thanks largely to the lifelong efforts of Charles Yanofsky [1]. Tryptophan is biochemically the most expensive of the amino acids to synthesize [2]. The clustered organization of the trp genes into an operon has been a model system for many years and continues to receive attention. In the current genomics era, emerging surprises reveal there is still much to learn. These surprises include the revelations that operons are being organizationally reshuffled, invaded by insertion of apparently unrelated genes, disrupted by either partial or complete dispersal of genes to extra-operon locations, or complicated by the seemingly unnecessary presence of additional operon-gene copies located outside of the operon.
Tryptophan synthase, catalyzing the final step of tryptophan biosynthesis, is one of the most rigorously documented examples of an enzyme complex [3,4,5,6]. It consists of an α subunit, which cleaves indoleglycerol phosphate to indole and glyceraldehyde 3-phosphate, and a β subunit, which condenses indole and L-serine to yield L-tryptophan. The αββα complex forms a tunnel into which enzymatically generated indole is released. The α monomers and β dimers contact one another via highly sophisticated mechanisms of allostery [7], and it is little wonder that the genes encoding these two subunits are almost always closely linked, frequently being translationally coupled.
Against this background, it seems curious that a significant number of organisms possess more than one gene encoding the β chain of tryptophan synthase. Usually, but not always, the 'extra' gene is unlinked to the gene encoding the α chain, and it also defines a distinct subgroup of the β chain. This has been recognized in the COGs database [8] as "alternative tryptophan synthase" (COG 1350). In this paper we present a detailed analysis of the distribution of the two subgroups of the β chain in prokaryotes within the context of the surprisingly dynamic past and ongoing alterations of organization for genes responsible for tryptophan biosynthesis.
Nomenclature
In recent publications involving bioinformatic analysis of aromatic amino-acid biosynthesis [9,10,11,12,13,14], we have implemented a nomenclature that attempts logical and consistent naming of genes and their gene products for different organisms. The problem is exemplified by the contemporary naming in two model organisms of 3-deoxy-D-arabino-heptulosonate 7-phosphate (DAHP) synthase, the initial enzyme step of aromatic amino-acid biosynthesis. In Bacillus subtilis the gene, appropriately enough, has been named aroA, but in Escherichia coli the equivalent function is represented by genes encoding three differentially regulated paralogs. These E. coli genes have been named aroF, aroG and aroH. In B. subtilis these latter three gene designations refer to 5-enolpyruvylshikimate-3-phosphate synthase (step 6 of chorismate synthesis), chorismate synthase (step 7) and chorismate mutase (initial step of phenylalanine and tyrosine biosynthesis). Even in B. subtilis, where the naming was intended to follow an orderly progression in terms of order of reaction steps, there is the complication that DAHP synthase is expressed as a fusion of two catalytic domains, one being a class of chorismate mutase called AroQ. This requires naming at the level of domain and the designation aroQ• aroA was implemented to denote such a fusion [9,10,11,12]. Thus, a single enzymatic function in one organism is accommodated through the cumulative expression of three paralog genes, but in another organism is only encoded by a portion of a single gene. A universal nomenclature is needed that labels at the level of domain, that labels in synchrony with order of reaction steps as much as possible, and that labels isofunctional paralogs at the same hierarchical level but with discriminating identifiers.
The status of nomenclature for the tryptophan pathway is not so chaotic as the genes in almost all prokaryotes have been named in line with E. coli, but even here distinct problems have arisen because gene fusions that exist in E. coli are often absent elsewhere. Much of the literature up to at least 1996 used five gene designations for the seven protein domains [15]. In B. subtilis these seven protein domains are encoded by separate genes [16]. Thus, E. coli trpD encodes the equivalent of B. subtilis TrpG and TrpD. The E. coli gene fusion has been re-designated with the convention of a bullet to represent a fusion: trpG• trpD [17]. Likewise, the E. coli trpC encodes the equivalent of B. subtilis TrpC and TrpF, and the E. coli gene fusion has been re-designated trpC• trpF. The nomenclature we advocate is more easily remembered because genes are named in the order of pathway reaction. Subunits for a given reaction are named at the same hierarchical level: anthranilate synthase (first reaction) consists of TrpAa and TrpAb, and tryptophan synthase (fifth reaction) consists of TrpEa and TrpEb. The implementation of a logical and consistent nomenclature, as illustrated in Figure 1, should be most helpful in the long term.
Figure 1.
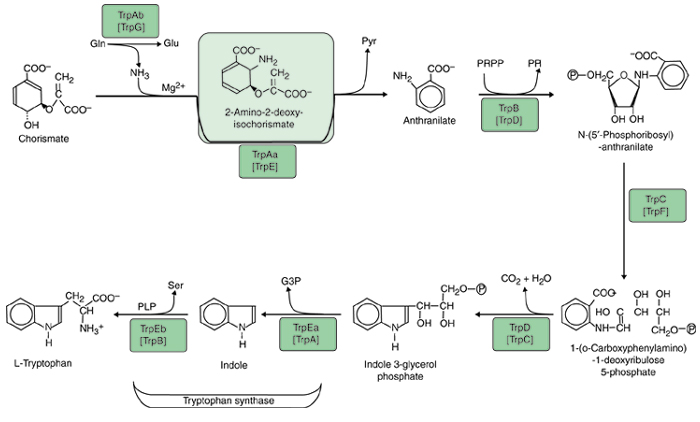
Biochemical pathway of tryptophan biosynthesis. Acronyms that are currently used for the seven monofunctional proteins of Bacillus subtilis are shown in brackets below the acronyms used in this paper. TrpAa, large aminase subunit of anthranilate synthase; TrpAb, small glutamine-binding subunit of anthranilate synthase; TrpB, anthranilate phosphoribosyl transferase; TrpC, phosphoribosyl-anthranilate isomerase; TrpD, indoleglycerol phosphate synthase; TrpEa, α subunit of tryptophan synthase; TrpEb, β subunit of tryptophan synthase.
Results and discussion
Phylogenetic tree construction
Initial amino-acid alignments were generated using ClustalW software, version 1.4 [18]. Manual adjustments were made through visual inspection to bring conserved motifs and residues into register. This was implemented by use of the BioEdit multiple alignment tool [19]. Inferences about the evolutionary relationships within the TrpEa and TrpEb protein families were made using the PHYLIP package of programs [20]. The Protpars program was used to generate a maximum parsimony tree, and the neighbor-joining and Fitch programs were used to generate a distance-based tree. The distance matrix used in the latter programs was produced using the program Protdist with a Dayoff PAM matrix. The Seqboot and Consense programs were then used to assess the statistical strength of the tree using bootstrap resampling. Neighbor-joining (PHYLIP), Fitch and Margolash (Fitch in PHYLIP), and maximum parsimony methods [21] all produced trees consistent with one another. Despite low bootstrap values at many individual internal nodes, the clusters formed and arrangement of taxa within them were largely identical. Ninety TrpEb and 63 TrpEa sequences were analyzed.
Distinctly different types of TrpEb
TrpEb proteins divide into two distinctly different groups, as illustrated by the unrooted tree shown in Figure 2. The major group, denoted TrpEb_1, includes the well-studied enzymes from such organisms as E. coli, Salmonella typhimurium, and B. subtilis. The minor group, denoted TrpEb_2, is represented heavily, but not exclusively, by archaeal proteins. Among the current inventory of completed archaeal genomes, only Methanococcus jannaschii lacks TrpEb_2. Seven archaeal genomes possess both TrpEb_1 and TrpEb_2 (Methanosarcina barkeri possesses two paralogs of TrpEb_1 in addition to one species of TrpEb_2). Six archaeal genomes possess TrpEb_2, but not TrpEb_1.
Figure 2.
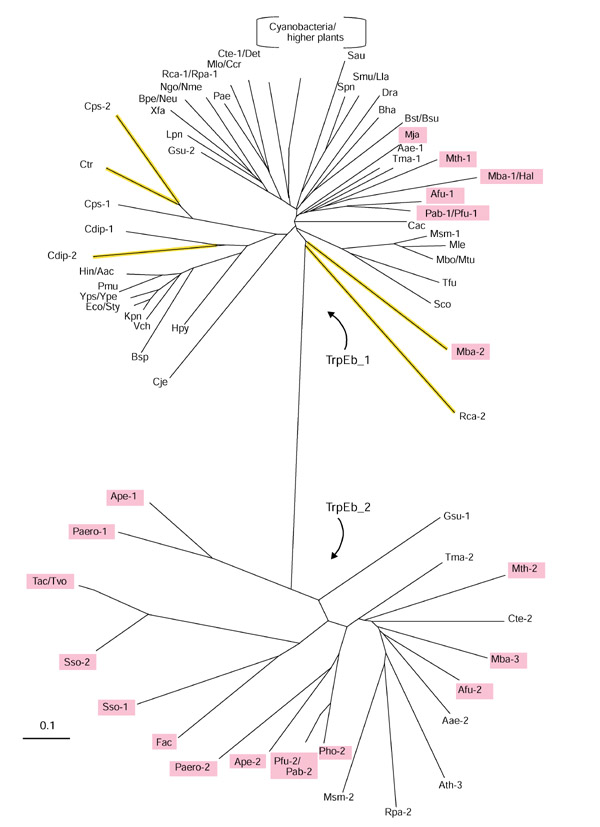
Unrooted phylogenetic tree (radial view) of the TrpEb protein family consisting of subfamily TrpEb_1 (top) and TrpEb_2 (bottom). Phylogenetic reconstruction of the inferred amino-acid sequence was accomplished by the neighbor-joining method using the PHYLIP program. Organismal acronyms are defined in Table 1. For economy of space, a single branch is used to represent proteins that have diverged very recently, for example, TrpEb_1 proteins from E. coli and S. typhimurium (Eco/Sty). Archaeal proteins are highlighted in magenta. The detailed order of branching for TrpEb_1 proteins in cyanobacteria and higher plants is shown in Figure 3. Probable pseudogenes are shown in yellow.
Bacterial TrpEb_2 proteins are thus far limited to Aquifex, Thermotoga, Mycobacterium, Geobacter, Chlorobium and Rhodopseudomonas genera. In addition, one of the multiple TrpEb proteins present in the higher plant, Arabidopsis thaliana, belongs to the TrpEb_2 subfamily. In view of the distinct divergence of TrpEb_2 from TrpEb_1, one might expect that either TrpEb_2 has lost the ability to interact allosterically with TrpEa, or perhaps that a divergent subgroup of TrpEa has coevolved with TrpEb_2. Multiple copies of TrpEb are often present in genomes. Examples include cases where two TrpEb_1 species coexist, where two TrpEb_2 species coexist, or where TrpEb_1 and TrpEb_2 coexist in the same organism. A number of organisms (M. barkeri, Rhodobacter capsulatus, Chlamydia psittaci, and Corynebacterium diphtheriae) possess two copies of TrpEb_1. In each case the trpEb_1 copy that is linked to trpEa is highly conserved, whereas the remaining copy has diverged to the extent that it may be a pseudogene (Table 2). The TrpEb_1 products of these probable pseudogenes have elongated branches (highlighted yellow) on the protein tree shown in Figure 2.
Table 2.
Exceptions to residue invariance
| Protein | Invariant residue* | Exceptions: organism (homologous residue) | ||
| TrpEa | 57P | Sso (A) | ||
| 60D | Hal (E) | |||
| 61G | Ctr (N) | |||
| 64I | Mth (V) | |||
| 183T | Ctr (R) | |||
| 184G | Hin (S) | |||
| 211G | Ctr (R) | |||
| 212F/L | Ctr (R) | |||
| 213G | Ctr (D) | Paero (A) | Ape (S) | |
| 234G | Ctr (K) | |||
| TrpEb_1 | 10G | Ctr (H) | Cps-2 (E) | Cps-1 (Y) |
| 21L | Cje (A) | |||
| 41F | Asp-1 (Y) | |||
| 55R | Mth-1 (K) | |||
| 78E | Ccr (D) | |||
| 79D | Rca-1 (E) | Paeru (E) | ||
| 80L | Mth-1 (M) | Dra (Q) | ||
| 82H | Mba-2 (Q) | Dra (F) | ||
| 94Q | Rca-2 (E) | |||
| 96L | Rpa-1 (M) | Spn (W) | Bsp (M) | |
| 97L | Cps-2 (I) | |||
| 102G | Bsp (K) | |||
| 114Q | Rpa-1 (M) | Paeru (M) | ||
| 124A | Zma-2 (R) | Cps-1 (G) | ||
| 133F/Y | Rca-2 (H) | |||
| 141R | Rca-2 (K) | |||
| 146V | Mba-1 (A) | |||
| 149M | Cps-2 (I) | |||
| 153G | Hal (D) | |||
| 156V | Cje (I) | |||
| 159V | Bha (A) | |||
| 162G | Cdip-2 (E) | |||
| 167K | Cdip-2 (S) | Sau (S) | ||
| 173A | Hal (T) | Rca-2 (C) | ||
| 177W | Cps-2 (F) | Rca-2 (Y) | ||
| 186Y | Cps-2 (F) | Rca-2 (F) | ||
| 208I | Xfa (V) | |||
| 213R/K | Bpe (L) | |||
| 224P | Cje (V) | |||
| 225D | Bha (T) | Rca-2 (A) | ||
| 267H | Rca-2 (N) | |||
| 269A | Sau (L) | Mba-1 (S) | ||
| 274G | Aac (A) | |||
| 299S | Rca-2 (T) | |||
| 310G | Mba-2 (S) | |||
| 345I | Rca-2 (V) | |||
| 383D | Yps (E) | |||
| Protein | Invariant residue† | Exceptions: organism (homologous residue) | ||
| TrpEb_2 | P10 | Paero-1 (gap) | ||
| Y14 | Aae-2 (L) | |||
| E54 | Sso-1 (Q) | |||
| L86 | Msm-2 (F) | |||
| E87 | Paero-2 (D) | |||
| E101 | Paero-2 (Q) | |||
| S108 | Paero-2 (N) | |||
| A114 | Mba-3 (S) | |||
| W138 | Sso-1 (R) | |||
| G176 | Fac (N) | |||
| S184 | Sso-1 (T) | |||
| S203 | Fac (T) | |||
| I206 | Msm-2 (M) | |||
| A207 | Fac (G) | |||
| S209 | Ape-1 (A) | |||
| E210 | Sso-2 (D) | |||
| G226 | Fac (A) | |||
| N265 | Paero-2 (S) | |||
| P272 | Mba-3 (E) | |||
| G300 | Cte-2 (A) | |||
| F/Y325 | Mba-3 (H) | |||
| F371 | Ape-1 (M) | |||
| P379 | Fac (A) | |||
| S410 | Ath-3 (C) | |||
*Residues are numbered according to the S. typhimurium sequence. †Residues are numbered according to the P. furiosus sequence.
It is noteworthy that cyanobacterial and higher plant amino-acid sequences form a cohesive cluster for TrpEb_1, as shown in phylogram form in Figure 3 (left panel). An analysis of TrpEa proteins from the same organisms yields a very similar phylogram output (Figure 3, right panel). This higher plant/cyanobacteria relationship is pleasingly consistent with the endosymbiotic hypothesis of organelle evolution. In each case Prochlorococcus marinus and Synechococcus species are the outlying sequence group, with the other cyanobacterial sequences (Nostoc punctiforme and Anabaena species) being closer to the higher plant sequences from A. thaliana and corn (Zea mays). The order of branching shown is supported by very high bootstrap values. Zma-3 is the TrpEa protein that has been proposed [22] to function independently of a TrpEb partner, producing indole for entry into a pathway other than tryptophan. In this case indole serves as a precursor for a defense metabolite that is active against insects, bacteria and fungi.
Figure 3.
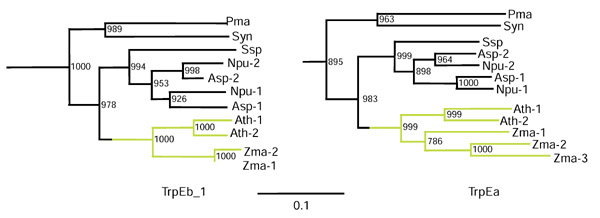
Unrooted phylogenetic tree (phylogram view) of cyanobacterial and higher plant TrpEb_1 (zoom-in expansion from Figure 2) and TrpEa (zoom-in expansion from Figure 4) protein sequences. The higher-plant lineage is shown in green. Bootstrap values (from 1,000 replications) supporting the order of branching shown are given at the nodes.
TrpEa in organisms lacking TrpEb_1
Six organisms (all Archaea) possess intact tryptophan pathways, but they lack TrpEb_1. TrpEb in Thermoplasma volcanii, T. acidophilum, and Ferroplasma acidarmanus is represented only by a single species of TrpEb_2. Although Sulfolobus solfataricus, Aeropyrum pernix and Pyrobaculum aerophilum all possess two species of TrpEb, both are the TrpEb_2 variety. Thus, in all six of these lineages TrpEa either might be unable to form a tight complex with TrpEb_2, or might have evolved different protein-protein contacts. In the latter case, distinct TrpEa subgroupings might be expected in parallel with the two TrpEb subgroupings. On the contrary, all TrpEa sequences fall into a single group (Figure 4). However, in contrast to sequences present in those Archaea that do possess TrpEb_1 (for example, Archaeoglobus, species of Pyrococcus, Methanococcus, Methanobacterium, Methanosarcina and Halobacterium), the six archaeal lineages that possess only TrpEb_2 have very distinctive elongated branches on the TrpEa tree (Figure 4). This suggests an elevated rate of evolutionary divergence, due either to selection for new productive contacts of TrpEa with TrpEb_2 or to lack of constraint to maintain TrpEa residues previously important for contacts with TrpEb_1.
Figure 4.
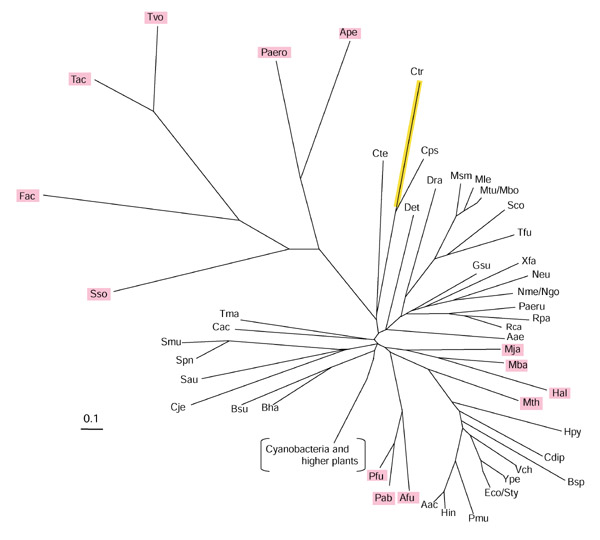
Unrooted phylogenetic tree (radial view) of the TrpEa protein family. Organismal acronyms are defined in Table 1. Phylogenetic reconstruction of the inferred amino-acid sequence was accomplished by the neighbor-joining method using the PHYLIP program. The detailed order of branching for cyanobacterial and higher-plant sequences is shown in Figure 3. Archaeal proteins are visualized in magenta, and the yellow line indicates the TrpEa protein from C. trachomatis, which is probably encoded by a pseudogene.
The long branch of the TrpEa sequence of Chlamydia trachomatis reflects its likely status as a pseudogene. This is consistent with the observation that C. trachomatis TrpEb_1 (Figure 2) also seems to be a pseudogene. One does not expect positive selection for maintenance of function in C. trachomatis as it lacks an intact tryptophan pathway. Indeed, the alteration in C. trachomatis of many otherwise invariant amino-acid residues is evident from the information given in Table 2.
Overview comparison of TrpEb_1 and TrpEb_2
Figure 5 shows an alignment of the amino-acid sequence of TrpEb_1 from S. typhimurium with TrpEb_2 from P. furiosus. Each sequence is shown as a template for its own subfamily, as extracted from a refined multiple alignment. Conserved residues deduced from a full multiple alignment (available from the author on request) are indicated, as are the gap positions present in the full alignment. Functional roles in catalysis and allosteric regulation are indicated for the S. typhimurium TrpEb_1 sequence in order to compare similarities and differences between TrpEb_1 and TrpEb_2 proteins. Residues that are ligands of pyridoxal phosphate or that interact with pyridoxal phosphate are scattered throughout the sequences, including the catalytic K87, and are highly conserved. Residue E109 has been shown to render indole more nucleophilic via proton abstraction from N1 [23]. The serine substrate-binding region is highly conserved, as is a monovalent cation (MVC) binding region [7] coordinating with G232, F/Y306, and G/A/S308. A number of indels (insertions/deletions) distinguish TrpEb_1 and TrpEb_2, and TrpEb_2 is about 50 residues longer overall than TrpEb_1. In addition to other residues conserved between both TrpEb_1 and TrpEb_2, each subgroup has its own repertoire of uniquely conserved residues. The COMM domain [24], a rigid but mobile domain as originally defined with S. typhimurium TrpEb_1 [25], differs from the corresponding region of the TrpEb_2 subfamily by the presence of an indel. Key TrpEb_1 regulatory residues (R141, K167) within this region as well as one residue near the MVC site (D305) are not conserved in the TrpEb_2 subgroup.
Figure 5.
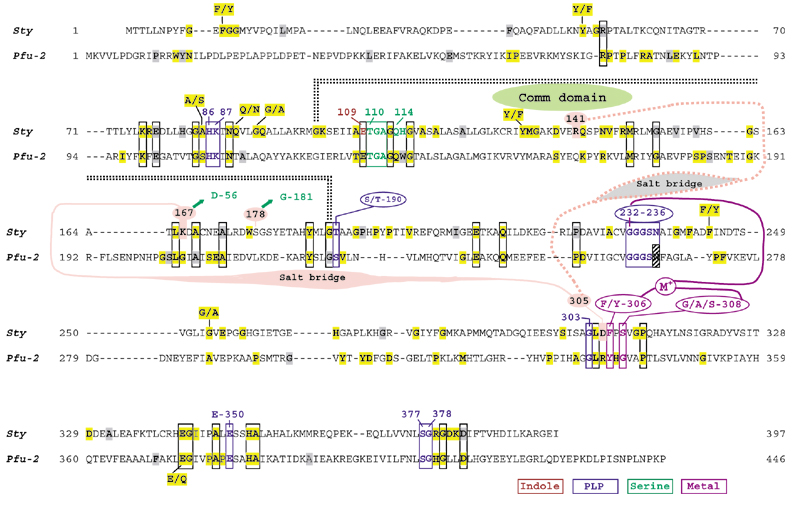
Alignment of TrpEb_1 from S. typhimurium (Sty) and of TrpEb_2 from P. furiosus (Pfu-2). The sequences are shown as they appear in a comprehensive alignment, including gaps. Invariant residues in each subfamily are highlighted and near-invariant residues (delineated in Table 2) are shaded. Invariant or near-invariant residues common to both subfamilies are boxed. Residues shown in Sty to be relevant to metal coordination or to the binding of indole, serine or PLP are color-coded as indicated at the bottom.
Loss of intersubunit contacts in TrpEa-TrpEb_2 systems
The tryptophan synthase of S. typhimurium is a rigorously documented example of substrate channeling in which indole generated as an intermediate is passed through an internalized tunnel ([7] and references therein). Ligand binding at the α-site and covalent transformations at the β-site accomplish mutually reinforcing overall allostery. The movable COMM domain is comprised of residues G102 to G189 in S. typhimurium TrpEb_2. COMM interacts with both the β-active site and with α-subunit loops 2 and 6 in response to allosteric signals. Within the COMM domain of TrpEb_1, S178 participates in intersubunit signaling with G181 of TrpEa. Competing allosteric conformations are mediated by alternative salt bridges between K167 of TrpEb_1 and D305 of TrpEb_1 on the one hand, or between K167 of TrpEb_1 and D56 of TrpEa, on the other. When D305 of TrpEb_1 is not occupied with K167, it forms an alternative salt bridge with R141, as shown in Figure 5.
As intersubunit signaling between TrpEb_2 and TrpEa is either lacking or involves different contacts, one might expect the important catalytic residues, but not the allosteric residues, to be conserved in comparison of TrpEb_2 with TrpEb_1. This comparison is shown in Figure 5. Likewise, in those TrpEa proteins that lack a TrpEb_1 partner, instead being forced to function in concert with TrpEb_2, one might expect retention of catalytic residues but loss of allosteric residues. This comparison is shown in Figure 6. Of the intersubunit signaling pair TrpEb_1 S178/TrpEa G181, S178 is not conserved within its own subfamily and equivalent residues seem to exist [26]. It is, however, striking that G181 is invariant in all TrpEa proteins, except for those belonging to the six archaeal lineages lacking TrpEb_1. It is even more striking that K167, which participates in the alternative β-β or β-α salt bridges is invariant in TrpEb_1, but absent in TrpEb_2. Likewise, the salt-bridge partner D305 is invariant in TrpEb_1, but absent in TrpEb_2. The salt-bridge partner D56 in TrpEa is invariant except for the six archaeal lineages that lack TrpEb_1. Figure 5 shows that TrpEb_2 sequences carry an insertion of 16 residues in the general region corresponding to the COMM domain of TrpEb_1 proteins.
Figure 6.
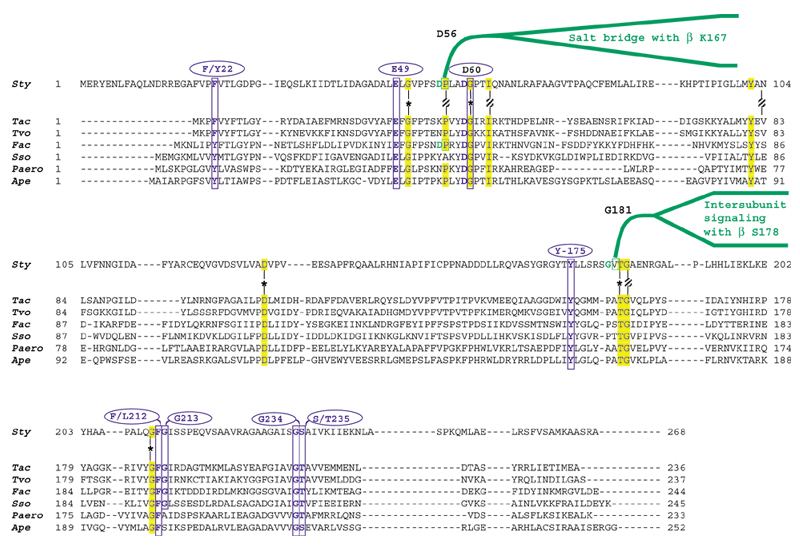
Alignment of TrpEa from S. typhimurium with TrpEa proteins restricted to co-function with a TrpEb_2 type of β subunit. Catalytic residues Sty E49 and D60, as well as other conserved residues shown to be important for indoleglycerol phosphate binding are shown in blue. Other invariant (highlighted) or near-invariant (shaded) residues are marked in yellow. Conserved residues that important for intersubunit signaling with TrpEb_1 are shown in green.
Tryptophan gene organization in TrpEb_2-containing genomes
The organization of tryptophan-pathway genes in the ten archaeal genomes and six bacterial genomes that are thus far known to possess TrpEb_2 are displayed on a 16S rRNA tree in Figure 7. Pyrococcus horikoshii has lost the entire pathway, and only trpEb_2 is present. In Aquifex and Chlorobium all trp genes are dispersed, and trpEa is not linked to either trpEb_1 or trpEb_2. In Bacteria and Archaea the gene order trpEb_1 → trpEa is one of the most highly conserved of genomic gene arrangements. Often translational coupling exists, as seen in Figure 7 for P. furiosus, P. abyssi, M. thermoautotrophicum, T. volcanium, Archaeoglobus fulgidus and T. maritima. In each of the latter genomes trpEb_2 is outside the trp operon. Both species of Thermoplasma possess an otherwise intact trp operon that lacks trpEb_1 (also not present elsewhere in the genome). Hence, by default it appears that the unlinked trpEb_2 must function for tryptophan biosynthesis in these organisms. In A. pernix and S. solfataricus all of the trp genes are adjacent, but trpEb_2 flanks trpEa, and trpEb_1 is absent from these genomes. In each case, a second unlinked copy of trpEb_2 is present. In Geobacter sulfurreducens, both trpEa and trpEb_1 (presumably partnered based on the results shown in Figure 4) are unlinked to one another, and both are outside an otherwise intact trp operon. In this case it seems curious indeed that the operon contains trpEb_2.
Figure 7.
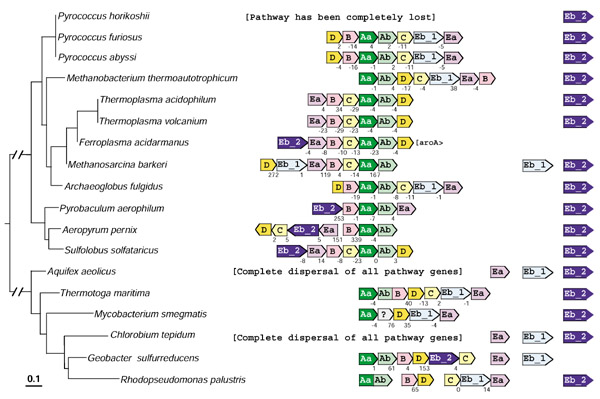
Organization of tryptophan-pathway genes in those archaea and bacteriathat possess trpEb_2. The 16S rRNA tree of the organisms that possess trpEb_2 is shown at the left. The tree was obtained from the SUBTREE program of the Ribosomal Database [41,42]. Each of the eight genes is color-coded differently. For economy of space trpD is D, trpC is C, etc. aroA in F. acidarmanus belongs to the AroAIβ grouping [12]. Flanking genes are shown as boxes touching one another with intergenic spacing indicated. Minus values indicate translational coupling. The open gray box is a conserved hypothetical protein. Pointed box ends indicate the direction of transcription. Space between boxes indicates a lack of genetic linkage.
A snapshot of the incredibly dynamic alteration of tryptophan gene organization in prokaryotes is apparent from Figure 7. Of the organisms shown in Figure 7, the most consistent linkage is that of trpAa and trpAb. In A. fulgidus trpD and trpB are fused, whereas in Rhodopseudomonas palustris trpAa and trpAb are fused. Operons are sometimes incomplete as with P. aerophilum and G. sulfurreducens, or fragmented, as with R. palustris. In A. pernix an inverted arrangement yields a divergent transcription of trpEa → trpEb_2 → trpC → trpD and trpB → trpAa → trpAb. This is also one of the very few cases where the order is trpEa → trpEb instead of the usual trpEb → trpEa.
The Ferroplasma genome illustrates a case where a non-tryptophan pathway gene, aroA encoding DAHP synthase, is a member of the operon (translationally coupled). The implied transcriptional control of DAHP synthase by L-tryptophan potentially could produce growth inhibition by L-tryptophan because of limitation of precursors needed for L-phenylalanine and L-tyrosine biosynthesis. This is because no other genes encoding DAHP synthase appear to be present in the genome. It would be interesting to know whether the Ferroplasma DAHP synthase is sensitive to allosteric control or not. The phenomenon of growth inhibition triggered by exogenous amino acids is exemplified by the effect of exogenous L-phenylalanine upon DAHP synthase in Neisseria gonorrhoeae [27] and in other organisms ([27] and references therein).
What is the function of TrpEb_2? As previously discussed, it seems clear that TrpEb_2 has sometimes been pressed into service with TrpEa to function as tryptophan synthase. What might be its function in those situations where trpEb_2 is isolated away from a typical tryptophan operon, which possesses closely linked or translationally coupled genes specifying trpEa and trpEb_1?
TrpEb_1 from S. typhimurium is the prototype member of a superfamily of pyridoxal phosphate (PLP)-dependent enzymes that are of remote relationship and that catalyze β-replacement and β-elimination reactions [28,29,30]. These include O-acetylserine sulfhydrylase, threonine deaminase, threonine synthase, cystathionine β-synthase, 1-aminocyclopropane-1-carboxylate deaminase, L-serine dehydratase, and D-serine dehydratase. Isolated TrpEb_1 does catalyze the reaction of L-serine dehydratase (deaminase) in vitro, but does not support significant levels of the other activities. It does not seem likely that TrpEb_2 catalyzes the latter reactions either, as Psi-Blast of TrpEb_2 sequences did not return hits for them any more avidly than was the case for TrpEb_1 queries.
Perhaps the most likely 'alternative' function of TrpEb_2 proteins is a catalytic activity already established as an in vitro activity of isolated TrpEb_1, of which there are two. The L-serine + indole → H2O + L-tryptophan reaction might function alone for tryptophan biosynthesis in cells that acquire indole from the environment. Little information about the availability and utilization of indole in nature seems to exist. Model organisms such as E. coli and B. subtilis transport indole poorly and it tends to be toxic, but these organisms lack TrpEb_2.
As stated above, the second established in vitro activity of isolated TrpEb_1 dimers is L-serine dehydratase (deaminase): L-serine → pyruvate + ammonia. It is indeed suggestive that the presence of TrpEb_2 correlates almost perfectly with the absence of the primary L-serine deaminase (COG1760) used in nature (Table 3). This SdaA class of serine deaminase is an iron-sulfur protein, not a pyridoxal 5'-phosphate protein [31]. It is absent from the Archaea. Although widely distributed in the Bacteria, it is not present in Thermotoga, Aquifex, Chlorobium, Geobacter and Rhodopseudomonas. Mycobacterium smegmatis is the only TrpEb_2-containing organism to possess an SdaA homolog.
Table 3.
Possible sources of serine deaminase in organisms containing TrpEb_2
| Serine deaminases | Threonine deaminases | |||
| Organisms | Fe-S* | PLP* | TdcA* | IlvA* |
| Pyrococcus horikoshii | - | - | - | - |
| Pyrococcus furiosus | - | - | - | - |
| Pyrococcus abyssi | - | - | - | - |
| Methanobacterium thermoautotrophicum | - | - | - | - |
| Thermoplasma acidophilum | - | - | + | - |
| Thermoplasma volcanium | - | - | + | - |
| Ferroplasma acidarmanus | - | - | + | - |
| Methanosarcina barkeri | - | - | - | - |
| Archaeoglobus fulgidus | - | - | - | - |
| Pyrobaculum aerophilum | - | - | + | - |
| Aeropyrum pernix | - | - | + | - |
| Sulfolobus solfataricus | - | - | + | - |
| Aquifex aeolicus | - | - | - | - |
| Thermotoga maritima | - | - | + | - |
| Mycobacterium smegmatis | + | - | + | - |
| Chlorobium tepidum | - | - | - | - |
| Geobacter sulfurreducens | - | - | + | - |
| Rhodopseudomonas palustris | - | - | + | - |
*Sequences that were used to query the databases at NCBI [38], at JGI [39] for Fac and Mba, and at ERGO [40] for Paer and Rpa were gi 2501150 (iron-sulfur (Fe-S) serine deaminases), gi 134387 (PLP-dependent serine deaminases), gi 135723 (catabolic threonine deaminase), and gi 135720 (biosynthetic threonine deaminase).
There are other candidates for carrying out the serine deaminase reaction. A serine deaminase that is PLP-dependent exists in eukaryotes, but seems to be absent or rare in prokaryotes. Threonine dehydratase has a lesser capability to carry out the serine dehydratase reaction in vitro. 'Biosynthetic' (IlvA) and 'catabolic' (TdcA) threonine deaminase are homologs, except that IlvA has a unique carboxy-terminal extension that provides an allosteric module. Most archaea lack ilvA, and only a limited number possess catabolic threonine dehydratase.
If archaea lack ilvA, what is the source of α-ketobutyrate for isoleucine biosynthesis? It appears likely that α-ketobutyrate is generated instead by the 'pyruvate' pathway in which citramalate is generated as the initial intermediate. A few tracer studies in the older literature have indicated derivation of α-ketobutyrate from pyruvate rather than from threonine [32,33], and (R)-citramalate synthase activity has been shown recently in M. jannaschii (MJ1392) [34]. This pathway was initially shown to exist in Leptospira [35], and it seems likely that more examples will surface. Indeed, it is interesting that an enteric bacterium has a latent potential to replace the threonine deaminase step with a pyruvate-derived pathway under appropriate selective conditions [36]. The citramalate pathway for conversion of pyruvate to 2-ketobutyrate involves a carbon-chain elongation mechanism that uses an initial step of condensation with acetyl-CoA, followed by rearrangement, oxidation and elimination of a carbon to produce a keto acid differing from the original substrate by the presence of an additional carbon. This mechanism is familiar in nature, analogous steps being used in the TCA cycle, the ketoadipate pathway of lysine biosynthesis, and in leucine biosynthesis. These analogous steps in different pathways were originally used in formulating the recruitment hypothesis for evolutionary acquisition of new function [37], and it is interesting that the citramalate synthase gene shown in M. jannaschii had previously been annotated as α-isopropylmalate synthase (which catalyzes the initial step of leucine biosynthesis).
Overall, the cumulative evidence indicates that established sources of L-serine dehydratase are low or absent in organisms that possess TrpEb_2, and therefore points to a plausible role for TrpEb_2 as L-serine dehydratase.
Table 1.
Key to sequence identifiers
| NCBI GI number | |||
| Species name | Acronym | trpEb-1 | trpEb-2 |
| Actinobacillus actinomycetemcomitans | Aac | N/A | |
| Aeropyrum pernix | Ape-1 | 7674393 | |
| Aeropyrum pernix | Ape-2 | 7674395 | |
| Anabaena sp. | Asp-1 | N/A | |
| Anabaena sp. | Asp-2 | N/A | |
| Aquifex aeolicus | Aae-1 | 6226273 | |
| Aquifex aeolicus | Aae-2 | 7674374 | |
| Arabidopsis thaliana | Ath-1 | 136251 | |
| Arabidopsis thaliana | Ath-2 | 1174779 | |
| Arabidopsis thaliana | Ath-3 | 10176821 | |
| Archaeoglobus fulgidus | Afu-1 | 3334387 | |
| Archaeoglobus fulgidus | Afu-2 | 7674372 | |
| Bacillus halodurans | Bha | 10174280 | |
| Bacillus stearothermophilus | Bst | 226585 | |
| Bacillus subtilis | Bsu | 136270 | |
| Bordetella pertussis | Bpe | N/A | |
| Buchnera sp. APS | Bsp | 11182450 | |
| Campylobacter jejuni | Cje | 11269304 | |
| Caulobacter crescentus | Ccr | 136272 | |
| Chlamydia trachomatis | Ctr | 6226274 | |
| Chlamydia psittaci | Cps-1 | N/A | |
| Chlamydia psittaci | Cps-2 | N/A | |
| Chlorobium tepidum | Cte-1 | N/A | |
| Chlorobium tepidum | Cte-2 | N/A | |
| Clostridium acetobutylicum | Cac | N/A | |
| Corynebacterium diphtheriae | Cdip-1 | N/A | |
| Corynebacterium diphtheriae | Cdip-2 | N/A | |
| Dehalococcoides ethenogenes | Det | N/A | |
| Deinococcus radiodurans | Dra | 7474051 | |
| Escherichia coli | Eco | 136273 | |
| Ferroplasma acidarmanus | Fac | N/A | |
| Geobacter sulfurreducens | Gsu-1 | N/A | |
| Geobacter sulfurreducens | Gsu-2 | N/A | |
| Haemophilus influenzae | Hin | 1174785 | |
| Halobacterium sp. | Hal | 14423973 | |
| Helicobacter pylori | Hpy | 7674399 | |
| Klebsiella pneumoniae | Kpn | N/A | |
| Lactococcus lactis | Lla | 267168 | |
| Legionella pneumophila | Lpn | N/A | |
| Mesorhizobium loti | Mlo | 13474230 | |
| Methanobacterium thermoautotrophicum | Mth-1 | 3334383 | |
| Methanobacterium thermoautotrophicum | Mth-2 | 7674371 | |
| Methanococcus jannaschii | Mja | 2501412 | |
| Methanosarcina barkeri | Mba-1 | N/A | |
| Methanosarcina barkeri | Mba-2 | N/A | |
| Methanosarcina barkeri | Mba-3 | N/A | |
| Mycobacterium bovis | Mbo | N/A | |
| Mycobacterium leprae | Mle | 13093205 | |
| Mycobacterium smegmatis | Msm-1 | N/A | |
| Mycobacterium smegmatis | Msm-2 | N/A | |
| Mycobacterium tuberculosis | Mtu | 3024761 | |
| Neisseria gonorrhoeae | Ngo | N/A | |
| Neisseria meningitides | Nme | 11269306 | |
| Nitrosomonas europaea | Neu | N/A | |
| Nostoc punctiforme | Npu-1 | N/A | |
| Nostoc punctiforme | Npu-2 | N/A | |
| Pasteurella multocida | Pmu | 13432266 | |
| Prochlorococcus marinus | Pma | N/A | |
| Pseudomonas aeruginosa | Paeru | 12230946 | |
| Pyrobaculum aerophilum | Paero-1 | N/A | |
| Pyrobaculum aerophilum | Paero-2 | N/A | |
| Pyrococcus abyssi | Pab-1 | 14520675 | |
| Pyrococcus abyssi | Pab-2 | 14423981 | |
| Pyrococcus furiosus | Pfu-1 | N/A | |
| Pyrococcus furiosus | Pfu-2 | N/A | |
| Pyrococcus horikoshii | Pho | 14591361 | |
| Rhodobacter capsulatus | Rca-1 | N/A | |
| Rhodobacter capsulatus | Rca-2 | N/A | |
| Rhodopseudomonas palustris | Rpa-1 | N/A | |
| Rhodopseudomonas palustris | Rpa-2 | N/A | |
| Salmonella typhimurium | Sty | 136281 | |
| Staphylococcus aureus | Sau | 13701169 | |
| Streptococcus mutans | Smu | N/A | |
| Streptococcus pneumoniae | Spn | N/A | |
| Streptomyces coelicolor | Sco | 6226276 | |
| Sulfolobus solfataricus | Sso-1 | 14424473 | |
| Sulfolobus solfataricus | Sso-2 | 13814334 | |
| Synechococcus sp. | Syn | N/A | |
| Synechocystis sp. | Ssp | 2501413 | |
| Thermomonospora fusca | Tfu | N/A | |
| Thermoplasma acidophilum | Tac | 13878841 | |
| Thermoplasma volcanium | Tvo | 13541762 | |
| Thermotoga maritima | Tma-1 | 1717761 | |
| Thermotoga maritima | Tma-2 | 7674388 | |
| Vibrio cholerae | Vch | 11269279 | |
| Xylella fastidiosa | Xfa | 11269281 | |
| Yersinia pestis | Ype | N/A | |
| Yersinia pseudotuberculosis | Yps | N/A | |
| Zea mays | Zma-1 | 1174780 | |
| Zea mays | Zma-2 | 1174778 | |
Acknowledgments
Acknowledgements
The graduate research studies of G.X. were partially supported by funding from the National Institutes of Health at Los Alamos National Laboratories.
References
- Yanofsky C. Advancing our knowledge in biochemistry, genetics, and microbiology through studies on tryptophan metabolism. Annu Rev Biochem. 2001;70:1–37. doi: 10.1146/annurev.biochem.70.1.1. [DOI] [PubMed] [Google Scholar]
- Bentley R. The shikimate pathway: a metabolic tree with many branches. Crit Rev Biochem Mol Biol. 1990;25:307–384. doi: 10.3109/10409239009090615. [DOI] [PubMed] [Google Scholar]
- Miles EW, Bauerle R, Ahmed SA. Tryptophan synthase from Escherichia coli and Salmonella typhimurium. Methods Enzymol. 1987;142:398–414. doi: 10.1016/s0076-6879(87)42051-x. [DOI] [PubMed] [Google Scholar]
- Ogasahara K, Hiraga K, Ito W, Miles EW, Yutani K. Origin of the mutual activation of the α and β2 subunits in the α2β2 complex of tryptophan synthase: Effect of alanine or glycine substitutions at proline residues in the α subunit. J Biol Chem. 1992;267:5222–5228. [PubMed] [Google Scholar]
- Ro H-S, Miles EW. Structure and function of the tryptophan synthase α2β2 complex: Roles of β subunit histidine 86. J Biol Chem. 1999;274:36439–36445. doi: 10.1074/jbc.274.51.36439. [DOI] [PubMed] [Google Scholar]
- Weyand M, Schlichting I. Crystal structure of wild-type tryptophan synthase complexed with the natural substrate indole-3-glycerol phosphate. Biochemistry. 1999;38:16469–16480. doi: 10.1021/bi9920533. [DOI] [PubMed] [Google Scholar]
- Weber-Ban E, Hur O, Bagwell C, Banik U, Yang L-H, Miles EW, Dunn MF. Investigation of allosteric linkages in the regulation of tryptophan synthase: The roles of salt bridges and monovalent cations probed by site-directed mutation, optical spectroscopy, and kinetics. Biochemistry. 2001;40:3497–3511. doi: 10.1021/bi002690p. [DOI] [PubMed] [Google Scholar]
- Clusters of Orthologous Groups http://www.ncbi.nlm.nih.gov/COG/
- Calhoun DH, Bonner CA, Gu W, Xie G, Jensen RA. The emerging periplasm-localized subclass of AroQ chorismate mutases, exemplified by those from Salmonella typhimurium and Pseudomonas aeruginosa. Genome Biol. 2001;2:0030.1–0030.16. doi: 10.1186/gb-2001-2-8-research0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosset G, Bonner CA, Jensen RA. Microbial origin of plant-type 2-keto-3-deoxy-D-arabino-heptulosonate 7-phosphate synthases, exemplified by the chorismate- and tryptophan-regulated enzyme from Xanthomonas campestris. J Bacteriol. 2001;183:4061–4070. doi: 10.1128/JB.183.13.4061-4070.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Williams DS, Aldrich HC, Xie G, Gabriel DW, Jensen RA. The AroQ and PheA domains of the bifunctional P-protein from Xanthomonas campestris in a context of genomic comparison. Microb Comp Genomics. 1997;2:141–158. doi: 10.1089/omi.1.1997.2.141. [DOI] [PubMed] [Google Scholar]
- Subramaniam PS, Xie G, Xia T, Jensen RA. Substrate ambiguity of 3-deoxy-D-manno-octulosonate 8-phosphate synthase from Neisseria gonorrhoeae in the context of its membership in a protein family containing a subset of 3-deoxy-D-arabino-heptulosonate 7-phosphate synthases. J Bacteriol. 1998;180:119–127. doi: 10.1128/jb.180.1.119-127.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie G, Bonner CA, Jensen RA. A probable mixed-function supraoperon in Pseudomonas exhibits gene organization features of both intergenomic conservation and gene shuffling. J Mol Evol. 1999;49:108–121. doi: 10.1007/pl00006523. [DOI] [PubMed] [Google Scholar]
- Xie G, Brettin TS, Bonner CA, Jensen RA. Mixed-function supraoperons that exhibit overall conservation, albeit shuffled gene organization, across wide intergenomic distances within eubacteria. Microb Comp Genomics. 1999;4:5–28. doi: 10.1089/omi.1.1999.4.5. [DOI] [PubMed] [Google Scholar]
- Pittard AJ. Biosynthesis of the aromatic amino acids. In Escherichia coli and Salmonella Cellular and Molecular Biology Edited by Neidhardt FC Washington, DC:ASM Press, 1996. pp. 458–484.
- Henner D, Yanofsky C. Biosynthesis of aromatic amino acids. In Bacillus subtilis and other Gram-positive Bacteria Biochemistry, Physiology, and Molecular Genetics Edited by Sonenshein A, Hoch J, Losick R Washington, DC:ASM Press, 1993. pp. 269–280.
- Crawford IP. Evolution of a biosynthetic pathway: the tryptophan paradigm. Annu Rev Microbiol. 1989;43:567–600. doi: 10.1146/annurev.mi.43.100189.003031. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall T. BioEdit: Biological Sequence Alignment Editor for Windows 95/98/NT, 486 edn Raleigh: North Carolina State University; 2000.
- Felsenstein J. PHYLIP - Phylogeny Inference Package (Version 3.2). Cladistics. 1989;5:164–166. [Google Scholar]
- Fitch WM. Toward defining the course of evolution: minimum change for a specific tree topology. Syst Zool. 1971;20:406–416. [Google Scholar]
- Melanson D, Chilton M-D, Masters-Moore D, Chilton WS. A deletion in an indole synthase gene is responsible for the DIMBOA-deficient phenotype of bxbx maize. Proc Natl Acad Sci USA. 1997;94:13345–13350. doi: 10.1073/pnas.94.24.13345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzovic PS, Kayastha AM, Miles EW, Dunn MF. Substitution of glutamic acid 109 by aspartic acid alters the substrate specificity and catalytic activity of the beta-subunit in the tryptophan synthase bienzyme complex from Salmonella typhimurium. Biochemistry. 1992;31:1180–1190. doi: 10.1021/bi00119a030. [DOI] [PubMed] [Google Scholar]
- Schneider TR, Gerhardt E, Lee M, Liang P-H, Anderson KS, Schlichting I. Loop closure and intersubunit communication in tryptophan synthase. Biochemistry. 1998;37:5394–5406. doi: 10.1021/bi9728957. [DOI] [PubMed] [Google Scholar]
- Rhee S, Parris DD, Hyde CC, Ahmed SA, Miles EW, Davies DR. Crystal structures of a mutant betaK87T tryptophan synthase alpha2beta2 complex with ligands bound to the active sites of the alpha- and beta-subunits reveal ligand-induced conformational changes. Biochemistry. 1997;36:7664–7680. doi: 10.1021/bi9700429. [DOI] [PubMed] [Google Scholar]
- Marabotti A, De Biase D, Tramonti A, Bettati S, Mozzarelli A. Allosteric communication of tryptophan synthase: Functional and regulatory properties of the β S178P mutant. J Biol Chem. 2001;276:17747–17753. doi: 10.1074/jbc.M011781200. [DOI] [PubMed] [Google Scholar]
- Bhatnagar RK, Berry A, Hendry AT, Jensen RA. The biochemical basis for growth inhibition by L-phenylalanine in Neisseria gonorrhoeae. Mol Microbiol. 1989;3:429–435. doi: 10.1111/j.1365-2958.1989.tb00188.x. [DOI] [PubMed] [Google Scholar]
- Alexander FW, Sandmeier E, Mehta PK, Christen P. Evolutionary relationships among pyridoxal-5'-phosphate-dependent enzymes. Regio-specific alpha, beta and gamma families. Eur J Biochem. 1994;219:953–960. doi: 10.1111/j.1432-1033.1994.tb18577.x. [DOI] [PubMed] [Google Scholar]
- Grishin NV, Phillips Ma, Goldsmith EJ. Modeling of the spatial structure of eukaryotic ornithine decarboxylases. Protein Sci. 1995;4:1291–1304. doi: 10.1002/pro.5560040705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta PK, Christen P. The molecular evolution of pyridoxal-5'-phosphate-dependent enzymes. Adv Enzymol Relat Areas Mol Biol. 2000;74:129–184. doi: 10.1002/9780470123201.ch4. [DOI] [PubMed] [Google Scholar]
- Hofmeister AEM, Textor S, Buckel W. Cloning and expression of the two genes coding for L-serine dehydratase from Peptostreptococcus asaccharolyticus : Relationship of the iron-sulfur protein to both L-serine dehydratases from Escherichia coli. J Bacteriol. 1997;179:4937–4941. doi: 10.1128/jb.179.15.4937-4941.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eikmanns B, Linder D, Thauer RK. Unusual pathway of isoleucine biosynthesis in Methanobacterium thermoautotrophicum. Arch Microbiol. 1983;136:111–113. [Google Scholar]
- Ekiel I, Smith ICP, Sprott GD. Biosynthesis of isoleucine in methanogenic bacteria: a 13C NMR study. Biochemistry. 1984;23:1683–1687. [Google Scholar]
- Howell DM, Xu H, White RH. (R)-Citramalate synthase in methanogenic archaea. J Bacteriol. 1999;181:331–333. doi: 10.1128/jb.181.1.331-333.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westfall HN, Caron NW, Peterson DE. Multiple pathways for isoleucine biosynthesis in the spirochete Leptospira. J Bacteriol. 1983;154:846–853. doi: 10.1128/jb.154.2.846-853.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisumi M, Komatsubara S, Chibata I. Pathway for isoleucine formation from pyruvate by leucine biosynthetic enzymes in leucine-accumulating isoleucine revertants of Seratia marcescens. J Biochem. 1977;82:95–103. doi: 10.1093/oxfordjournals.jbchem.a131698. [DOI] [PubMed] [Google Scholar]
- Jensen RA. Enzyme recruitment in evolution of new function. Annu Rev Microbiol. 1976;30:409–425. doi: 10.1146/annurev.mi.30.100176.002205. [DOI] [PubMed] [Google Scholar]
- Microbial genomes BLAST databases http://www.ncbi.nlm.nih.gov/Microb_blast/unfinishedgenome.html
- DOE Joint Genome Institute http://www.jgi.doe.gov/
- ERGO http://wit.integratedgenomics.com/ERGO/
- Maidak BL, Cole JR, Parker CT, Garrity GM, Larsen N, Li B, Lilburn TG, McCaughey MJ, Olsen GJ, Overbeek R, et al. A new version of the RDP (Ribosomal Database Project). Nucleic Acids Res. 1999;27:171–173. doi: 10.1093/nar/27.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribosomal Database Project II http://rdp.cme.msu.edu/html/