

Abstract
The mammalian homolog of the Schizosaccharomyces pombe Rad9 is involved in checkpoint signaling and the induction of apoptosis. While the mechanisms responsible for the regulation of human Rad9 (hRad9) are not known, hRad9 is subject to hyperphosphorylation in the response of cells to DNA damage. The present results demonstrate that protein kinase Cδ (PKCδ) associates with Rad9 and that DNA damage induces this interaction. PKCδ phosphorylates hRad9 in vitro and in cells exposed to genotoxic agents. The functional significance of the interaction between hRad9 and PKCδ is supported by the finding that activation of PKCδ is necessary for formation of the Rad9–Hus1–Rad1 complex. We also show that PKCδ is required for binding of hRad9 to Bcl-2. In concert with these results, inhibition of PKCδ attenuates Rad9-mediated apoptosis. These findings demonstrate that PKCδ is responsible for the regulation of Rad9 in the Hus1–Rad1 complex and in the apoptotic response to DNA damage.
Keywords: apoptosis/DNA damage/PKCδ/Rad9/RNAi
Introduction
The cellular response to genotoxic stress includes cell cycle arrest, activation of DNA repair and, in the event of irreparable damage, induction of apoptosis. The signaling mechanisms responsible for regulation of the DNA damage response are largely unknown. Certain insights have been derived from the finding that isoforms of the protein kinase C (PKC) family are activated in response to DNA damage. The calcium-independent PKCδ isoform is cleaved to a 40 kDa catalytically active fragment by caspase-3 in cells treated with DNA-damaging agents (Emoto et al., 1995, 1996). The finding that overexpression of the PKCδ catalytic fragment (PKCδCF) induces chromatin condensation and DNA fragmentation has supported a role for PKCδ cleavage in the induction of apoptosis (Ghayur et al., 1996). Interaction of PKCδCF with the nuclear DNA-dependent protein kinase catalytic subunit (DNA-PKcs) inhibits the function of DNA-PKcs to form complexes with DNA and to phosphorylate its downstream target, p53 (Bharti et al., 1998). In addition, cells deficient in DNA-PK are resistant to apoptosis induced by overexpressing PKCδCF (Bharti et al., 1998). Other studies have shown that PKCδ interacts with the c-Abl tyrosine kinase in cells treated with DNA-damaging agents (Yuan et al., 1998). c-Abl-mediated phosphorylation activates PKCδ and induces translocation of PKCδ to the nucleus (Yuan et al., 1998). In concert with these findings, tyrosine phosphorylation of PKCδ is necessary for its nuclear translocation and subsequent caspase-dependent cleavage in the apoptotic response to DNA damage (Blass et al., 2002).
In Schizosaccharomyces pombe, cell cycle arrest in response to incomplete DNA replication or DNA damage is dependent on expression of spRad9, spHus1 and spRad1 (al-Khodairy and Carr, 1992; al-Khodairy et al., 1994). Studies in human cells have demonstrated that this checkpoint pathway is conserved through expression of hRad9, hHus1 and hRad1 (Freire et al., 1998; Kostrub et al., 1998; Parker et al., 1998; Udell et al., 1998). As found in yeast, hRad9 forms a complex with hHus1 and hRad1 (St Onge et al., 1999; Volkmer and Karnitz, 1999). Nuclear hRad9 is expressed constitutively as multiple phosphorylated forms and is phosphorylated in response to DNA damage (St Onge et al., 1999, 2001; Volkmer and Karnitz, 1999; Chen,X. et al., 2001). The product of the gene mutated in ataxia telangiectasia (ATM) has been shown to phosphorylate Rad9 in response to DNA damage (Chen,M.J. et al., 2001). Other studies have shown that hRad9 localizes to extraction-resistant nuclear complexes in response to DNA lesions (Burtelow et al., 2000). The finding that hRad9, hHus1 and hRad1 share regions of similarity with the proliferating cell nuclear antigen (PCNA) has suggested a potential role for their involvement in the DNA replication machinery (Thelen et al., 1999). In this context, hRad17, a clamp loader protein, associates with the hRad9–hHus1–hRad1 complex, and DNA damage induces dissociation of these nuclear complexes (Rauen et al., 2000; Lindsey-Boltz et al., 2001). Another nuclear function for hRad9 may be related to the demonstration that recombinant hRad9 exhibits 3′–5′ exonuclease activity (Bessho and Sancar, 2000). Furthermore, the recent finding that hRad9 interacts with anti-apoptotic Bcl-2 family proteins through a BH3-like region has supported a role for hRad9 in the apoptotic response to DNA damage (Komatsu et al., 2000a,b).
The present studies demonstrate that PKCδ inter acts with hRad9. We show that phosphorylation of hRad9 by PKCδ is necessary for formation of the hRad9–hHus1–hRad1 complex. The results also show that PKCδ regulates the interaction of hRad9 with Bcl-2 and the hRad9-mediated apoptotic response to DNA damage.
Results
PKCδ associates with hRad9
To determine whether PKCδ associates with hRad9, lysates from human U-937 cells were subjected to immunoprecipitation with anti-PKCδ. Analysis of the immunoprecipitates with anti-Rad9 demonstrated binding of PKCδ and Rad9 (Figure 1A). Moreover, treatment with the genotoxic agent, 1-β-d-arabinofuranosylcytosine (ara-C), was associated with increased formation of PKCδ–Rad9 complexes (Figure 1A). In the reciprocal experiment, immunoblot analysis of anti-Rad9 immunoprecipitates with anti-PKCδ demonstrated a low level of binding between PKCδ and Rad9 in control cells that was increased by ara-C treatment (Figure 1B). To confirm the association of PKCδ and Rad9, nuclear extracts from control and ara-C-treated U-937 cells were analyzed for co-sedimentation in glycerol gradients. Immunoblot analysis of the gradient fractions demonstrated co-sedimentation of PKCδ and Rad9 in the nuclear lysates from ara-C-treated cells (Figure 1C). In contrast, there was little if any PKCδ in the nuclear lysates from control U-937 cells (data not shown). In concert with these results, treatment with ara-C was associated with targeting of PKCδ to the nucleus (Figure 1D). Purity of the nuclear fraction was confirmed by immunoblotting with antibodies against the cytosolic IκBα protein and the membrane-associated platelet-derived growth factor (PDGF) receptor (Figure 1D). Based on these findings, nuclear lysates were subjected to immunoprecipitation with anti-Rad9. Immunoblot analysis of the precipitates with anti-PKCδ demonstrated that PKCδ associates with Rad9 in cells treated with ionizing radiation (IR) or ara-C, but not in control cells (Figure 1E). These findings indicate that PKCδ translocates to the nucleus and associates with nuclear Rad9 in the response of cells to genotoxic stress.
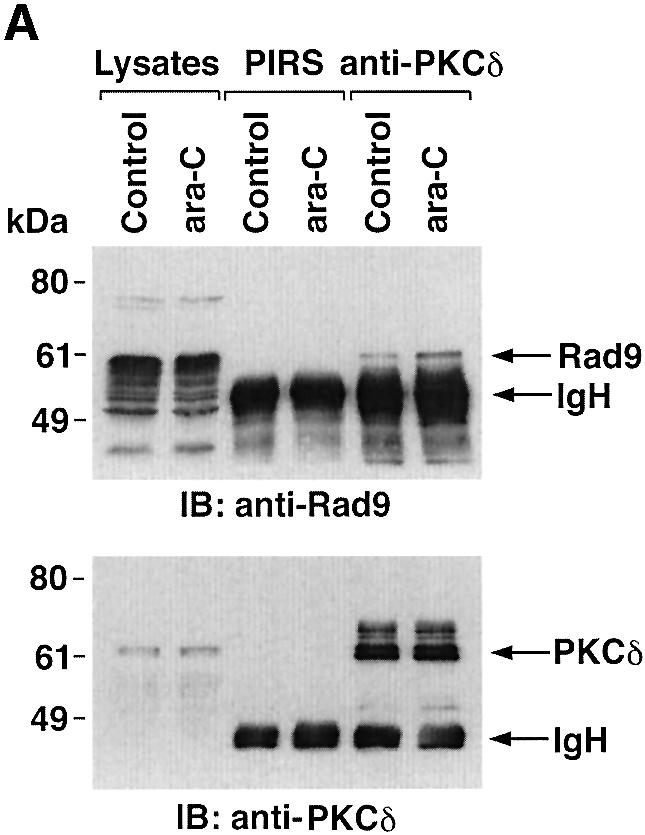
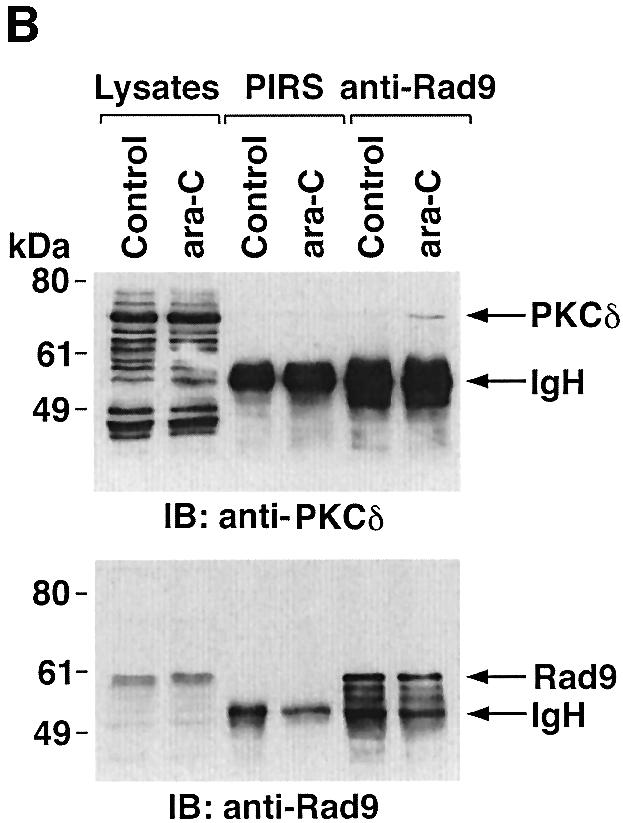


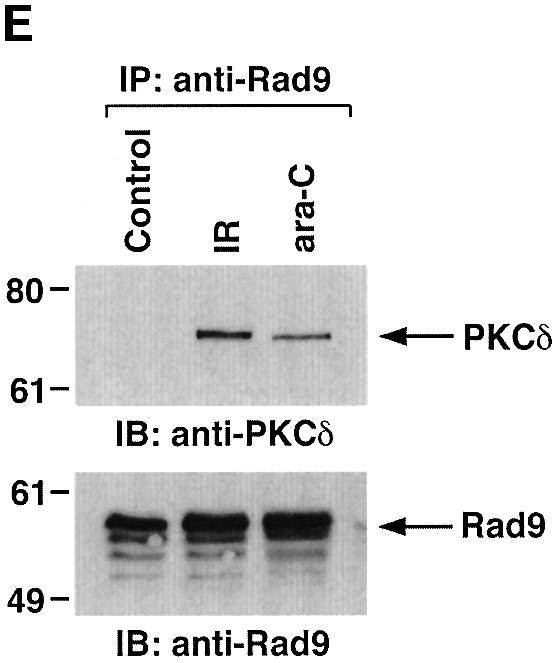
Fig. 1. Association between PKCδ and Rad9. (A and B) U-937 cells were left untreated or treated with 10 µM ara-C for 2 h. Cell lysates were subjected to immunoprecipitation with pre-immune rabbit serum (PIRS), anti-Rad9 (A) or anti-PKCδ (B). Cell lysates and immunoprecipitates were subjected to immunoblot analysis (IB) with anti-PKCδ and anti-Rad9. (C) Nuclear extracts from U-937 cells treated with 10 µM ara-C for 2 h were layered onto a 10–30% glycerol gradient. The indicated fractions were subjected to SDS–PAGE and immunoblotting with anti-PKCδ (upper panel) or anti-Rad9 (lower panel). (D) U-937 cells were treated with 10 µM ara-C for the indicated times. Lysates from the nuclear, cytosolic and membrane fractions were subjected to immunoblot analysis with anti-PKCδ (top panel), anti-laminB (second panel), anti-IκBα (third panel) or anti-PDGF receptor (bottom panel). (E) U-937 cells were treated with 20 Gy of IR and harvested at 2 h or treated with 10 µM ara-C for 2 h. Anti-Rad9 immunoprecipitates (IP) from nuclear lysates were analyzed by immunoblotting with anti-PKCδ (upper panel) and anti-Rad9 (lower panel).
PKCδ phosphorylates hRad9
To determine whether PKCδ phosphorylates hRad9 in vitro, kinase-active recombinant PKCδ was incubated with His-tagged hRad9 and [γ-32P]ATP. Analysis of the reaction products demonstrated that, as shown previously (Yoshida and Kufe, 2001), the recombinant kinase-active PKCδ undergoes autophosphorylation and that hRad9 functions as a substrate for PKCδ (Figure 2A). To assess the effects of PKCδ-mediated phosphorylation on the electrophoretic mobility of hRad9, recombinant PKCδ was incubated with His-hRad9 in the absence and presence of ATP. Immunoblot analysis of the reaction products with anti-His showed that phosphorylation of hRad9 by PKCδ is associated with a decrease in electrophoretic mobility (Figure 2B). To extend these findings, 293T cells were transfected to express green fluorescent protein (GFP) or GFP–PKCδCF. Immunoblot analysis with anti-hRad9 demonstrated that PKCδCF also induces a decrease in the electrophoretic mobility of hRad9 (Figure 2C). These findings demonstrate that PKCδ phosphorylates hRad9 in vitro and that PKCδ can induce Rad9 phosphorylation above that found constitutively in cells.
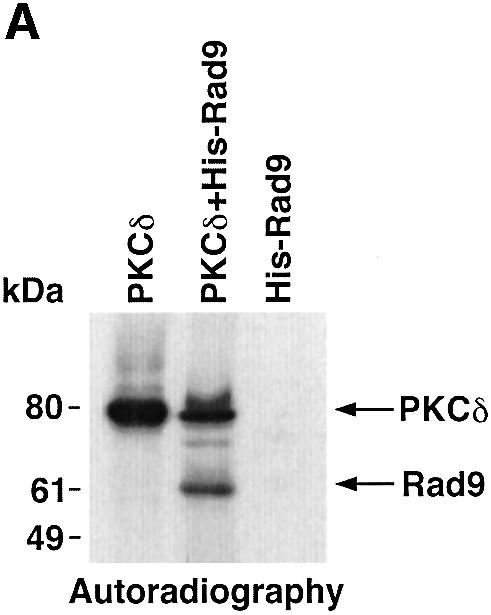
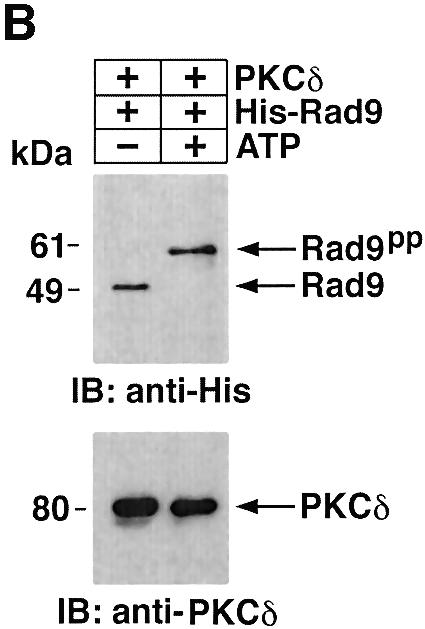
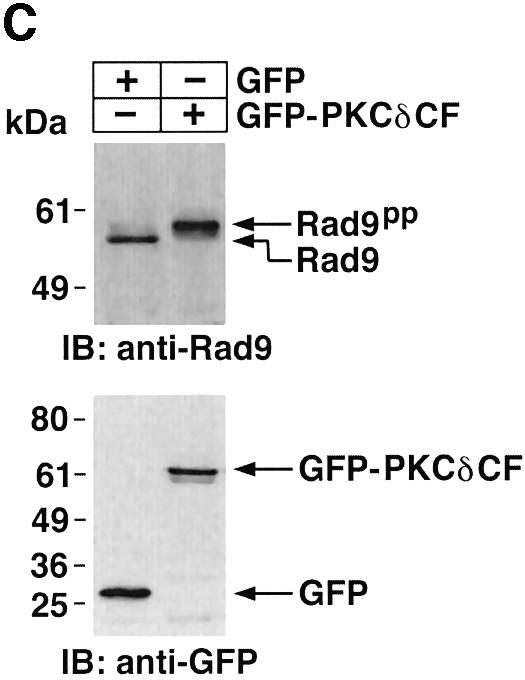
Fig. 2. Phosphorylation of Rad9 by PKCδ. (A) Recombinant PKCδ was incubated with purified His-Rad9 and [γ-32P]ATP for 15 min at 30°C. The reaction products were analyzed by SDS–PAGE and autoradiography. (B) Recombinant PKCδ was incubated with His-Rad9 in the presence or absence of ATP. The protein complexes were subjected to immunoblot analysis with anti-His (upper panel) or anti-PKCδ (lower panel). (C) 293T cells were transfected with GFP vector or GFP–PKCδCF. Cell lysates were analyzed by immunoblotting with anti-Rad9 (upper panel) or anti-GFP (lower panel).
To determine whether treatment of cells with ara-C is associated with a shift in electrophoretic mobility of hRad9, U-937 cells were exposed to different ara-C concentrations for 2 h and then analyzed by immunoblotting with anti-hRad9. The results demonstrate that treatment with 1 µM ara-C causes a decrease in electrophoretic mobility of hRad9 (Figure 3A). Moreover, maximal decreases in electrophoretic mobility were observed following treatment with 10 µM ara-C (Figure 3A). The effects of 10 µM ara-C were detectable at 1 h and persisted through 8 h of treatment (data not shown). Similar results were obtained when DNA damage was induced with IR or cisplatin (CDDP) (Figure 3B). To assess whether PKCδ contributes to the phosphorylation of hRad9 in response to genotoxic stress, cells were exposed to the PKCδ inhibitor, rottlerin (Gschwendt et al., 1994; Yoshida and Kufe, 2001). Immunoblot analysis with anti-hRad9 demonstrated that rottlerin increases the electrophoretic mobility of hRad9 in control and ara-C-treated cells (Figure 3C, left). In contrast, treatment with the PKCα, β and γ inhibitor, Gö 6976, had no apparent effect on electrophoretic mobility of hRad9 in either control or ara-C-treated cells (Figure 3C, middle). To confirm that the electrophoretic mobility shift induced by DNA damage is a consequence of phosphorylation, anti-hRad9 immunoprecipitates were prepared from lysates of ara-C-treated cells. Incubation of the immunoprecipitates with calf intestinal phosphatase (CIP) alone resulted in an increase in electrophoretic mobility (Figure 3C, right). In contrast, incubation with CIP and the phosphatase inhibitor, β-glycerophosphate, attenuated this effect (Figure 3C, right). Whereas rottlerin inhibits other kinases (Davies et al., 2000; Soltoff, 2001), 293T cells were treated with small intefering RNA (siRNA) duplexes that target PKCδ. The results demonstrate that PKCδsiRNA1 and PKCδsiRNA2 downregulate PKCδ expression (Figure 3D, left). As a control, treatment with GFPsiRNA had little effect (Figure 3D, left). Significantly, treatment with PKCδsiRNA1, and not GFPsiRNA, increased the electrophoretic mobility of Rad9 (Figure 3D, left). Moreover, PKCδsiRNA1, and not GFPsiRNA, blocked DNA damage-induced decreases in electrophoretic mobility of Rad9 (Figure 3D, right). These findings indicate that PKCδ contributes to both constitutive and DNA damage-induced phosphorylation of hRad9.
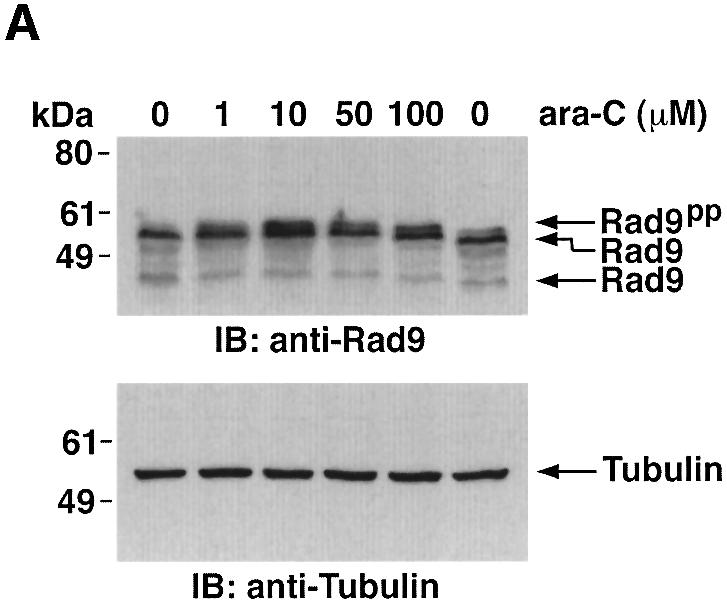
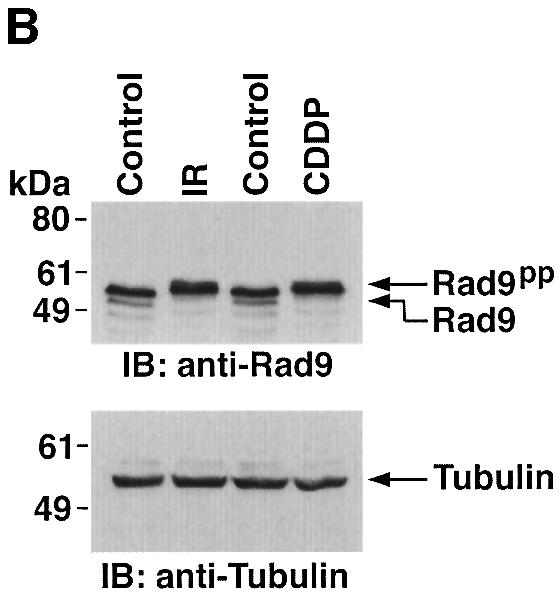


Fig. 3. Phosphorylation of Rad9 in response to DNA damage. (A) U-937 cells were treated with the indicated concentrations of ara-C for 2 h. Cell lysates were subjected to immunoblot analysis with anti-Rad9 (upper panel) or anti-tubulin (lower panel). (B) U-937 cells were treated with 15 Gy of IR or 50 µM CDDP for 2 h. Cell lysates were subjected to immunoblot analysis with anti-Rad9 (upper panel) or anti-tubulin (lower panel). (C) U-937 cells were left untreated or treated with 5 µM rottlerin (left panel) or 50 nM Gö 6976 (middle panel) for 30 min followed by treatment with 10 µM ara-C for 2 h. Cell lysates were analyzed by immunoblotting with anti-Rad9 or anti-tubulin. U-937 cells were treated with 10 µM ara-C for 2 h (right panel). Anti-Rad9 immunprecipitates from cell lysates were left untreated or treated with CIP in the presence or absence of 25 mM β-glycerophosphate (β-GP). Immune complexes were subjected to immunoblotting with anti-Rad9. (D) 293T cells were transfected with GFPsiRNA, PKCδsiRNA1 or PKCδsiRNA2. Cell lysates were analyzed by immunoblotting with anti-PKCδ, anti-Rad9 or anti-tubulin (left panel). 293T cells transfected with GFPsiRNA or PKCδsiRNA1 were treated with 20 Gy of IR and harvested at 2 h. Lysates were analyzed by immunoblotting with anti-Rad9, anti-PKCδ or anti-tubulin (right panel).
Activation of PKCδ is necessary for targeting nuclear hRad9
To determine whether activation of PKCδ is necessary for the interaction with nuclear hRad9, anti-PKCδ immunoprecipitates from nuclear lysates of ara-C-treated cells were first analyzed for PKCδ activity. Using histone H1 as substrate (Konishi et al., 1997), the results demonstrate that nuclear PKCδ activity is increased in response to ara-C (Figure 4A). In concert with these findings, nuclear levels of full-length PKCδ (PKCδFL) were increased at 1 and 2 h of ara-C treatment (Figure 4A). Longer periods of ara-C exposure were associated with proteolytic cleavage of PKCδFL to the catalytic fragment (PKCδCF) (Figure 4A) and shuttling of PKCδCF to the cytosol (data not shown). Similar findings were obtained in IR- and CDDP-treated cells (Figure 4A; data not shown). Inhibition of PKCδ with rottlerin attenuated nuclear targeting of PKCδ and increased the electrophoretic mobility of nuclear hRad9 in control and ara-C-treated cells (Figure 4B). In concert with these results, rottlerin attenuated ara-C-induced binding of PKCδ to nuclear hRad9 (Figure 4B). To confirm these findings, immunofluorescence microscopy was performed to assess the intracellular distribution of PKCδ. The reactivity of anti-PKCδ was predominantly extranuclear in control cells and nuclear in ara-C-treated cells (Figure 4C). Moreover, treatment with rottlerin blocked nuclear targeting of PKCδ in the response to ara-C (Figure 4C). In contrast, localization of hRad9 was predominantly nuclear in control cells, and treatment with ara-C and/or rottlerin had no apparent effect (data not shown). These findings demonstrate that activation of PKCδ is necessary for its nuclear targeting and interaction with hRad9.

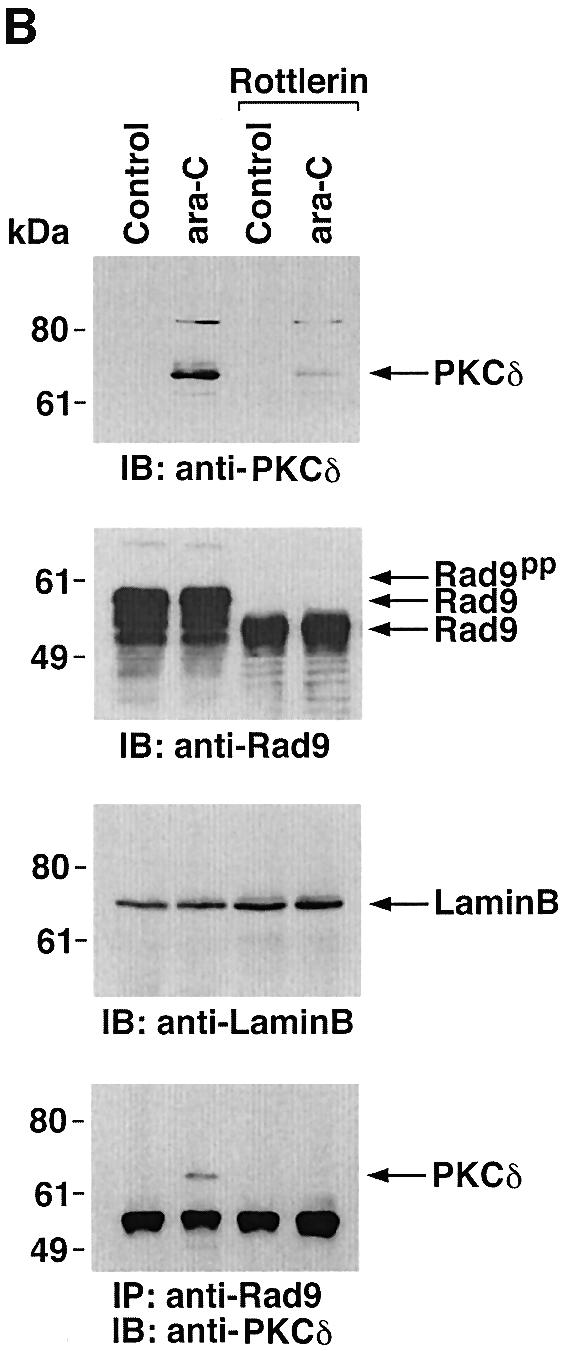

Fig. 4. Nuclear translocation of PKCδ in response to DNA damage. (A) U-937 cells were treated with 10 µM ara-C or 15 Gy of IR for the indicated times. Nuclear lysates were subjected to immunoprecipitation with anti-PKCδ. The immunoprecipitates were incubated with histone H1 and [γ-32P]ATP for 15 min at 30°C. Reaction products were resolved by SDS–PAGE and analyzed by autoradiography (upper panel). Nuclear lysates were also subjected to immunoblot analysis with anti-PKCδ (lower panel). (B) U-937 cells were left untreated or treated with 5 µM rottlerin for 30 min followed by the treatment with 10 µM ara-C for 2 h. Nuclear lysates were analyzed by immunoblotting with anti-PKCδ (top panel), anti-Rad9 (second panel) or anti-laminB (third panel). Nuclear lysates were also subjected to immunoprecipitation with anti-Rad9. The immunoprecipitates were analyzed by immunoblotting with anti-PKCδ (bottom panel). (C) U-937 cells were left untreated or treated with 5 µM rottlerin for 30 min followed by the treatment with 10 µM ara-C for 2 h. After fixation and blocking, cells were incubated with anti-PKCδ or anti-Rad9 (data not shown) and counterstained with DAPI. PIRS was used as a negative control (data not shown).
Phosphorylation of hRad9 by PKCδ is required for formation of the hRad9–hHus1–hRad1 complex
Previous work has demonstrated that hRad9 forms a complex with hHus1 and hRad1 in the response to DNA damage (St Onge et al., 1999; Volkmer and Karnitz, 1999). To define the functional significance of the interaction between PKCδ and hRad9, cells were treated with ara-C and rottlerin or Gö 6976. Anti-Rad9 immunoprecipitates from cell lysates were subjected to immunoblot analysis with anti-Rad1 or anti-Hus1. The results demonstrate that ara-C treatment is associated with binding of hRad9 to hHus1 and hRad1 (Figure 5A). In concert with involvement of PKCδ, treatment with rottlerin blocked ara-C-induced binding of hRad9 to hHus1 and hRad1 (Figure 5A). In contrast, treatment with Gö 6976 had little effect (Figure 5A). To confirm these findings, nuclear lysates from cells treated with ara-C alone, ara-C + Gö 6976 or ara-C + rottlerin were subjected to sedimentation in glycerol gradients. Immunoblot analysis of the fractions demonstrated co-sedimentation of hRad9 with hHus1 and hRad1 in cells treated with ara-C alone or ara-C + Gö 6976 (Figure 5B; data not shown). In contrast, rottlerin attenuated the formation of hRad9–-hHus1–hRad1 complexes (Figure 5C). Rottlerin also attenuated binding of hRad9 to hHus1–hRad1 in cells treated with IR or CDDP (data not shown).
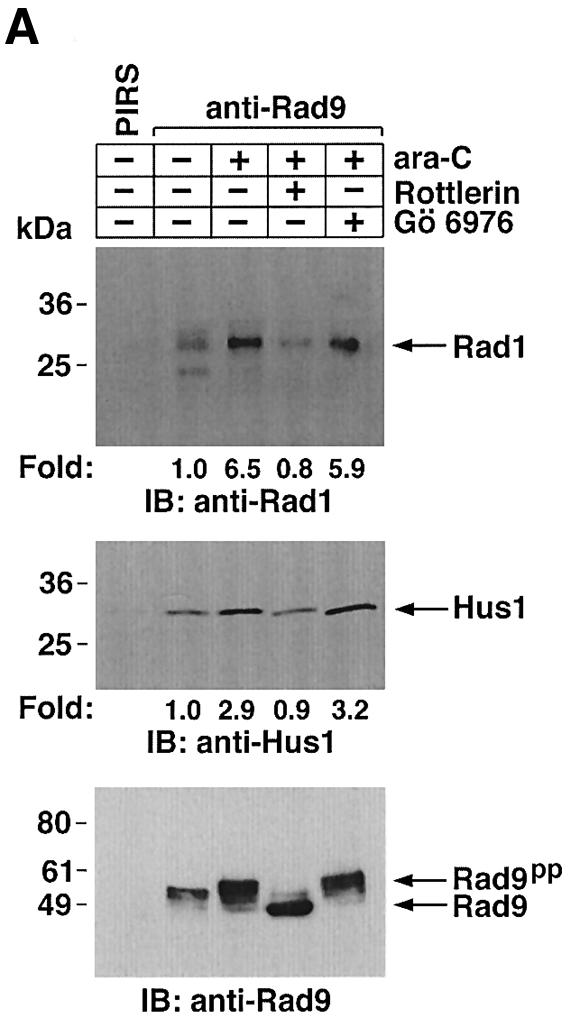

Fig. 5. Dissociation of the hRad9–hHus1–hRad9 complex by inhibition of PKCδ. (A) U-937 cells were left untreated or treated with 5 µM rottlerin or 50 nM Gö 6976 for 30 min followed by treatment with 10 µM ara-C for 2 h. Cell lysates were subjected to immunoprecipitation with PIRS or anti-Rad9. The immunoprecipitates were analyzed by immunoblotting with anti-Rad1 (upper panel), anti-Hus1 (middle panel) or anti-Rad9 (lower panel). (B and C) U-937 cells were treated with 50 nM Gö 6976 (B) or 5 µM rottlerin (C) for 30 min, followed by the treatment with 10 µM ara-C for 2 h. Nuclear extracts were layered onto a 10–40% glycerol gradient. After fractionation, the indicated fractions were subjected to SDS–PAGE and immunoblotting with anti-Rad9 (upper panel), anti-Hus1 (middle panel) or anti-Rad1 (lower panel).
ATM functions upstream of PKCδ activation
Previous studies have demonstrated that ATM phos phorylates Rad9 after exposure to IR (Chen,M.J. et al., 2001). To assess the involvement of ATM in PKCδ-mediated regulation of Rad9, we transfected 293T cells with siRNAs that target ATM. The results show that ATMsiRNA1 and ATMsiRNA2 decrease ATM expression (Figure 6A). In contrast, GFPsiRNA had no apparent effect (Figure 6A). To determine whether ATM regulates PKCδ activation in response to DNA damage, 293T cells transfected with ATMsiRNA1 were treated with IR or ara-C. Analysis of PKCδ immunoprecipitates from nuclear lysates demonstrated that ATM is required in part for PKCδ activation (Figure 6B). In this regard, PKCδ activation is required for the nuclear targeting of PKCδ (Figure 4; Yuan et al., 1998). Consistent with these findings, cells deficient in ATM exhibited less PKCδ in the nucleus (Figure 6B). Downregulation of ATM expression was also associated with attenuation of DNA damage-induced decreases in the electrophoretic mobility of Rad9 (Figure 6C). ATM functions upstream of c-Abl in the DNA damage response (Baskaran et al., 1997; Shafman et al., 1997) and c-Abl interacts with Rad9 (Yoshida et al., 2002). Moreover, c-Abl and PKCδ interact in response to DNA damage (Yuan et al., 1998). To assess the respective roles of PKCδ and c-Abl in the regulation of Rad9, studies were performed on wild-type and c-Abl–/– mouse embro fibroblasts (MEFs). As shown previously (Yoshida et al., 2002), ara-C-induced binding of Rad9 to Bcl-2 was dependent on c-Abl expression (Figure 6D). The findings also show that the formation of Rad9–Bcl-2 complexes is inhibited by rottlerin (Figure 6D). These results indicate that Rad9 is regulated by both PKCδ and c-Abl in the interaction between Rad9 and Bcl-2.
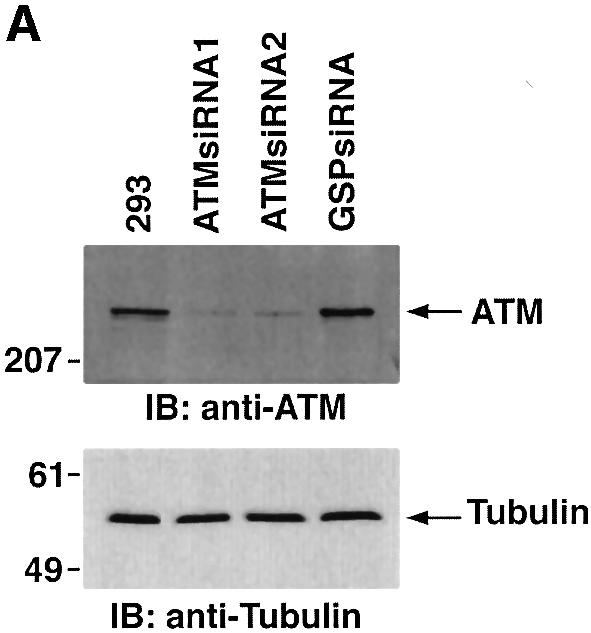
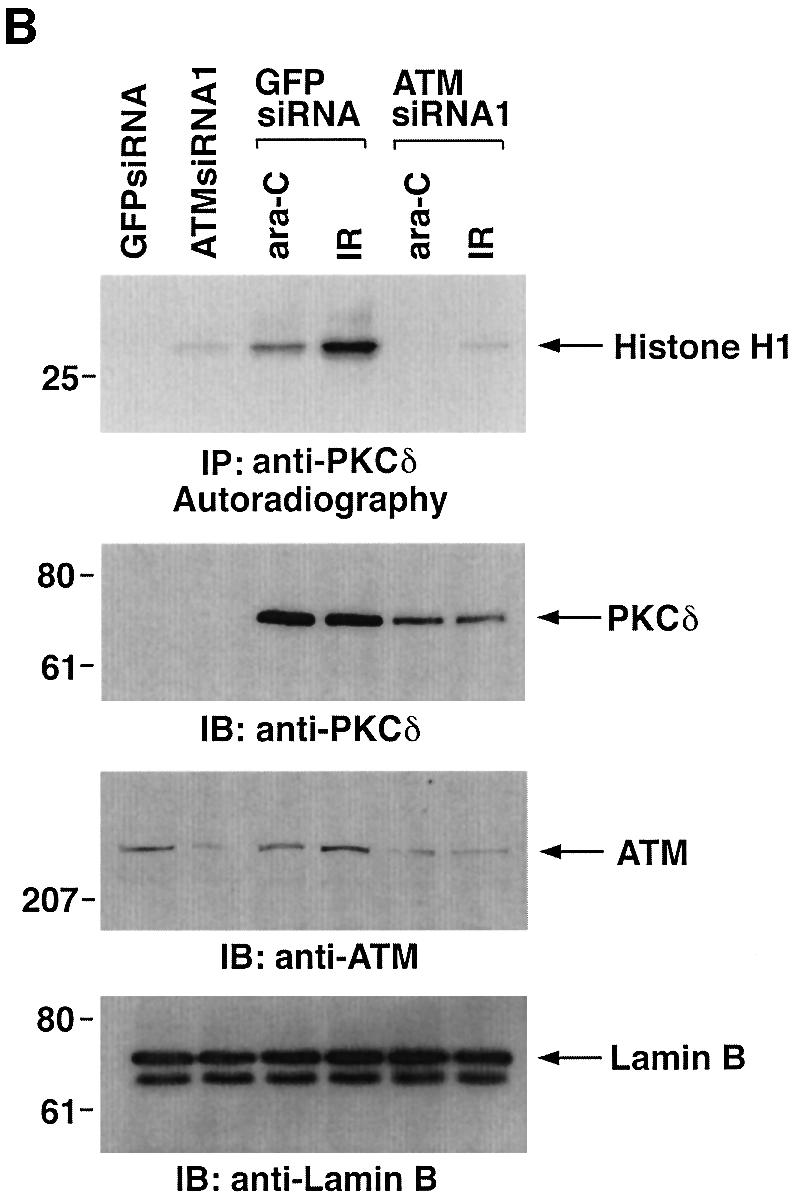
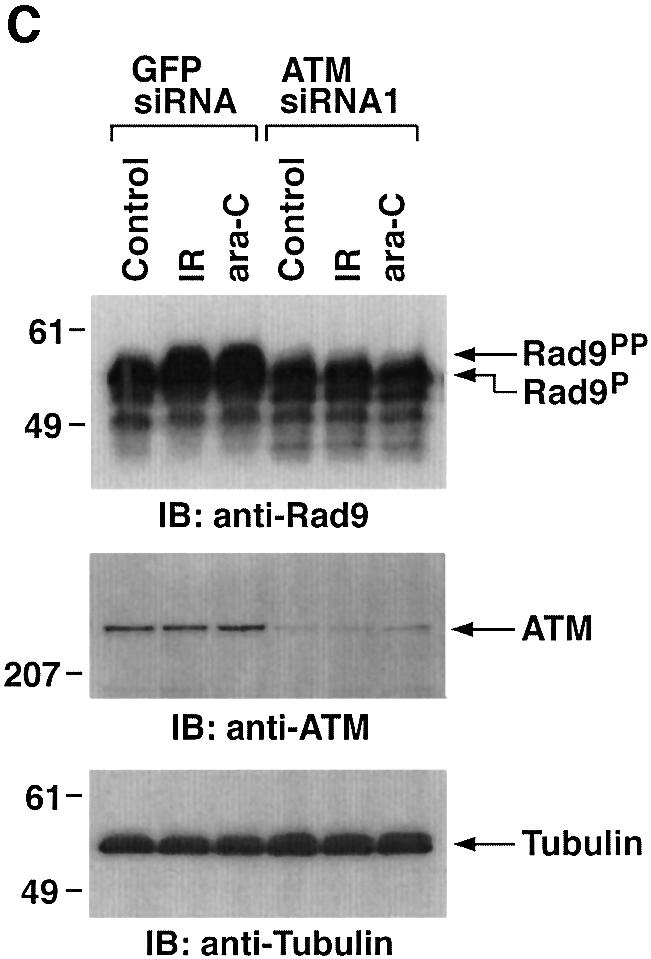
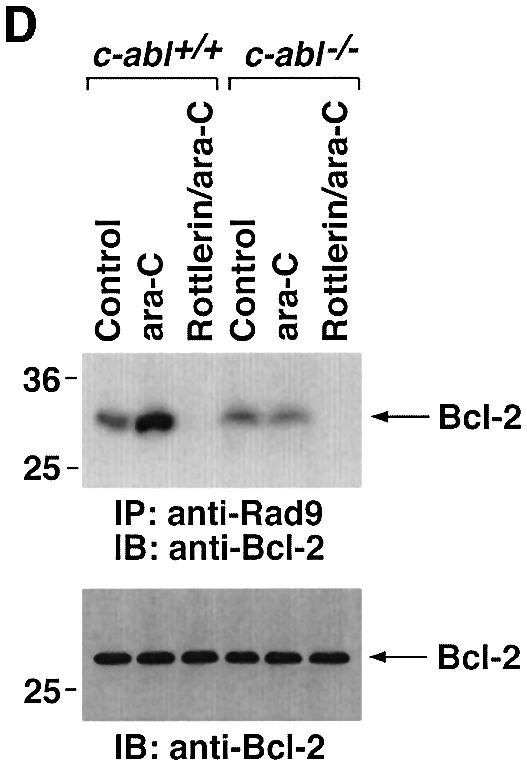
Fig. 6. Involvement of ATM in PKCδ-mediated regulation of Rad9. (A) 293T cells were transfected with GFPsiRNA, ATMsiRNA1 and ATMsiRNA2. Cell lysates were analyzed by immunoblotting with anti-ATM (upper panel) or anti-tubulin (lower panel). (B) 293T cells transfected with GFPsiRNA or ATMsiRNA1 were treated with 20 Gy of IR or 10 µM ara-C and harvested at 2 h. Anti-PKCδ immunoprecipitates from nuclear lysates were analyzed for phosphorylation of histone H1 as a substrate (top panel). Nuclear lysates were also subjected to immunoblot analysis with anti-PKCδ (second panel), anti-ATM (third panel) or anti-laminB (bottom panel). (C) 293T cells transfected with GFPsiRNA or ATMsiRNA1 were treated with 20 Gy of IR or 10 µM ara-C and harvested at 2 h. Lysates were analyzed by immunoblotting with anti-Rad9 (upper panel), anti-ATM (middle panel) or anti-tubulin (lower panel). (D) Wild-type and c-abl–/– MEFs were left untreated or treated with 5 µM rottlerin for 30 min, followed by exposure to 10 µM ara-C for 2 h. Anti-Rad9 immunoprecipitates were subjected to immunoblot analysis with anti-Bcl-2 (upper panel). Lysates were also analyzed by immunoblotting with anti-Bcl-2 (lower panel).
PKCδ regulates hRad9-mediated cell death
To assess further the functional role of PKCδ in the regulation of hRad9, U-2 OS cells were stably transfected to express kinase-inactive PKCδCF(K-R) under the control of the metallothionein-II promoter. Expression of PKCδCF(K-R) was absent in control cells and induced in the presence of cadmium sulfate (Figure 7A). Exposure to cadmium sulfate for 24 h was associated with attenuation of constitutive and ara-C-induced PKCδ activity (Figure 7B). Induction of PKCδCF(K-R) expression was also associated with an increase in electrophoretic mobility of hRad9 in control and ara-C-treated cells (Figure 7B).



Fig. 7. Attenuation of Rad9-mediated apoptosis by inhibition of PKCδ. (A) U-2 OS/PKCδCF(K-R) cells were treated with 2 µM cadmium sulfate (CdSO4) for the indicated times. Cell lysates were analyzed by immunoblotting with anti-PKCδ. Lysates from U-2 OS cells were used as a control. (B) U-2 OS/PKCδ(K-R) cells were left untreated or treated with 2 µM CdSO4 for 24 h, followed by the treatment with 10 µM ara-C for 2 h. Cell lysates were subjected to immunoprecipitation with anti-PKCδ (top panel). Anti-PKCδ immunoprecipitates were incubated with histone H1 and [γ-32P]ATP and analyzed by SDS–PAGE and autoradiography (top panel). Cell lysates were analyzed by immunoblotting with anti-Rad9 (second panel) or anti-Bcl-2 (bottom panel). Anti-Rad9 immunoprecipitates were also subjected to immunoblot analysis with anti-Bcl-2 (third panel). (C and D) U-2 OS/PKCδCF(K-R) cells were transfected with GFP vector or GFP–Rad9. At 24 h post-transfection, cells were left untreated or treated with 10 µM ara-C for 24 h in the presence or absence of 2 µM CdSO4. DNA content in cells positive for green fluorescence was analyzed by FACscan. The flow cytometry patterns represent DNA content from GFP–Rad9-transfected cells (C). The results (mean ± SD of three independent experiments) are presented as the percentage of apoptotic cells with sub-G1 DNA (D). Control cells (open bars); ara-C-treated cells (closed bars).
To determine whether PKCδ regulates the interaction between hRad9 and Bcl-2, U-2 OS cells were studied in the absence and presence of PKCδCF(K-R) expression. Immunoblot analysis of anti-hRad9 immunoprecipitates with anti-Bcl-2 demonstrated that constitutive binding of hRad9 to Bcl-2 is increased in response to ara-C treatment (Figure 7B). Expression of PKCδCF(K-R) abrogated the formation of hRad9–Bcl-2 complexes in both control and ara-C-treated cells (Figure 7B). To determine whether phosphorylation of hRad9 by PKCδ is associated with induction of apoptosis, U-2 OS cells were transfected with a GFP or GFP–hRad9 vector. At 24 h after transfection, cells were treated in the absence and presence of cadmium sulfate for 24 h. Analysis of GFP-positive cells for sub-G1 DNA demonstrated that, in the absence of cadmium sulfate, expression of hRad9 induces apoptosis (Figure 7C). In contrast, expression of PKCδCF(K-R) attenuated hRad9-induced apoptosis (Figure 7C). Treatment with ara-C potentiated the induction of apoptosis in hRad9-expressing cells (Figure 7C). Moreover, the apoptotic response to treatment of hRad9-expressing cells with ara-C was substantially attenuated by PKCδCF(K-R) (Figure 7C). Similar results obtained in multiple experiments confirm that PKCδCF(K-R) expression attenuates hRad9-induced apoptosis (Figure 7D). These findings provide support for involvement of hRad9 phosphorylation by PKCδ in the apoptotic response to DNA damage.
Discussion
Rad9 is a nuclear target of PKCδ
Previous work has shown that PKCδ is expressed predominantly in the cytoplasm and to a lesser extent in the nucleus (Bharti et al., 1998; Yuan et al., 1998). While nuclear PKCδ associates constitutively with DNA-PKcs (Bharti et al., 1998), the nuclear targets of PKCδ otherwise are largely unknown. The present findings demonstrate that nuclear PKCδ also associates constitutively at low levels with hRad9. Targeting of PKCδ to the nucleus in response to DNA damage was associated with increases in the formation of PKCδ–hRad9 complexes. Nuclear hRad9 forms a ternary complex with hHus1 and hRad1 (St Onge et al., 1999; Volkmer and Karnitz, 1999). Moreover, DNA damage increases the formation of hRad9–hHus1– hRad1 complexes (Volkmer and Karnitz, 1999; Burtelow et al., 2000). In concert with the involvement of PKCδ, treatment with rottlerin to inhibit PKCδ activation (Yoshida and Kufe, 2001) blocked DNA damage-induced formation of hRad9–hHus1–hRad1 complexes. In contrast, inhibition of the cPKCs (α, β and γ) had no effect on nuclear hRad9 complexes in the DNA damage response. Inducible expression of PKCδCF(K-R) as another approach to inhibit PKCδ activation also blocked the effects of DNA damage on hRad9–hHus1–hRad1 complex formation (data not shown). Cleavage of PKCδ to PKCδCF could also contribute to the regulation of nuclear hRad9. The generation of PKCδCF by caspase-3 is, however, a later event (4–6 h), while nuclear targeting of PKCδ and the regulation of hRad9–hHus1–hRad1 complexes occurs within 2 h of treatment with ara-C and other genotoxic agents. These findings thus support a model in which activation of PKCδ and thereby nuclear targeting of PKCδ in the DNA damage response is necessary for induction of the interaction of hRad9 with hHus1 and hRad1.
PKCδ phosphorylates hRad9
Nuclear hRad9 is constitutively detectable as multiple phosphorylated forms (St Onge et al., 1999). Moreover, hRad9 is subject to additional phosphorylation in the response to DNA damage (Volkmer and Karnitz, 1999). Whereas recent findings have indicated that ATM contributes to the phosphorylation of hRad9 (Chen,M.J. et al., 2001), other work has shown that c-Abl binds directly to the C-terminal region of hRad9 and phosphorylates the hRad9 BH3 domain on Y28 (Yoshida et al., 2002). The present results demonstrate that PKCδ phosphorylates hRad9 in vitro and that such modification is associated with a decrease in electrophoretic mobility. hRad9 contains nine potential consensus PKCδ phosphorylation sites (S/TXXR/K). In vitro studies using PKCδ fragments indicate that most if not all of these sites are subject to PKCδ phosphorylation. The results of our studies in cells provide further support for PKCδ-mediated phosphorylation of hRad9. Significantly, inhibition of PKCδ with rottlerin or siRNA resulted in a substantial increase in electrophoretic mobility. The finding that CIP has no apparent effect on mobility of hRad9 from rottlerin-treated cells indicates that constitutive phosphorylation of hRad9 is mediated predominantly by a PKCδ-dependent mechanism (data not shown). The demonstration that rottlerin or siRNA also blocks DNA damage-induced decreases in hRad9 mobility provides further support for involvement of PKCδ in phosphorylation of hRad9 in response to DNA damage. In concert with these observations, similar results were obtained with knock-down of PKCδ expression with siRNA and with inducible expression of PKCδCF(K-R) to inhibit PKCδ activity. These findings indicate that PKCδ regulates both constitutive and DNA damage-induced phosphorylation of hRad9.
PKCδ regulates the interaction between hRad9 and Bcl-2
The hRad9 BH3 domain binds to the pro-apoptotic Bcl-2 and Bcl-xL proteins (Komatsu et al., 2000b). Phosphorylation of the hRad9 BH3 domain by c-Abl induces the interaction of hRad9 and Bcl-xL (Yoshida et al., 2002). In contrast, c-Abl-mediated phosphorylation of hRad9 had no effect on binding of hRad9 to hHus1 or hRad1 (Yoshida et al., 2002). The present results demonstrate that, in addition to regulating formation of hRad9– hHus1–hRad1 complexes, PKCδ-mediated phosphorylation of hRad9 is involved in binding of hRad9 to Bcl-2. Inhibition of PKCδ activity with rottlerin or PKCδCF(K-R) expression blocked constitutive binding of hRad9 to Bcl-2. The results also demonstrate that inhibition of PKCδ activation blocks binding of hRad9 to Bcl-2 in response to DNA damage. These results and the demonstration that c-Abl is necessary for DNA damage-induced binding of hRad9 to Bcl-xL (Yoshida et al., 2002) indicate that both PKCδ and c-Abl may be required for interactions of hRad9 and Bcl-2/Bcl-xL. In addition to the c-Abl phosphorylation site at Y28, the hRad9 BH3 domain contains two potential consensus PKCδ phosphorylation sites. The findings with ATMsiRNA-mediated interference indicate that DNA damage-induced PKCδ activation is in part dependent on ATM. Whereas ATM activates c-Abl (Baskaran et al., 1997; Shafman et al., 1997), and c-Abl activates PKCδ (Yuan et al., 1998), a potential explanation is that DNA damage induces a ATM→c-Abl→PKCδ pathway. Alternatively, ATM may directly activate PKCδ in the DNA damage response. Our data also show that inhibition of PKCδ activity with rottlerin or siRNA is associated with decreases in both constitutive and DNA damage-induced phosphorylation of Rad9.
PKCδ potentiates hRad9-mediated apoptosis
Overexpression of hRad9 induces apoptosis by a mechanism that is blocked by Bcl-2 or Bcl-xL (Komatsu et al., 2000b). Moreover, the demonstration that downregulation of hRad9 expression by an antisense vector suppresses cell death by the genotoxic agent methyl methanesulfonate has supported a role for hRad9 in the apoptotic response to DNA damage (Komatsu et al., 2000b). Our results demonstrate that hRad9-mediated apoptosis is attenuated by inhibition of PKCδ activity. The results also show that hRad9 potentiates DNA damage-induced apoptosis, and these effects of hRad9 are attenuated by inducible expression of PKCδCF(K-R). Our other studies have demonstrated that the interaction between c-Abl and hRad9 also potentiates hRad9-mediated apoptosis (Yoshida et al., 2002). Therefore, in concert with the involvement of both c-Abl and PKCδ in regulating the interaction of hRad9 with Bcl-2/Bcl-xL, these kinases may exhibit independent or cooperative effects on signaling of hRad9 in the induction of apoptosis. Recent work has shown that knock-down of hRad9 with siRNA partially (∼50%) abrogates the G2 checkpoint in response to genotoxic stress (Hirai and Wang, 2002). Thus, PKCδ-mediated regulation of hRad9 could contribute, at least in part, to control of the G2 checkpoint. Defining the role of PKCδ in regulation of the hRad9 checkpoint function may, however, be complicated by interactions between PKCδ and nuclear effectors of cell cycle progression, such as c-Abl and p73 (Yuan et al., 1998; Ren et al., 2002). In this regard, further studies are needed to define the nature of the interaction, if any, between c-Abl and PKCδ in regulating both hRad9-mediated apoptosis and G2 arrest in the response to DNA damage. Based on the present findings, activation of PKCδ by genotoxic stress contributes to binding of hRad9 to Bcl-2 and thereby potentiation of hRad9-induced apoptosis.
Materials and methods
Cell culture
Human U-937 myeloid leukemia cells and human U-2 OS osteogenic sarcoma cells were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 U/ml penicillin, 100 µg/ml streptomycin and 2 mM l-glutamine. 293T embryonal kidney cells, MEFs and c-abl–/– MEFs (Tybulewicz et al., 1991) were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% FBS and antibiotics. Cells were treated with ara-C (Sigma-Aldrich), CDDP (Sigma-Aldrich), rottlerin (Calbiochem) or Gö 6976 (Calbiochem). Irradiation was performed at room temperature with a Gammacell 1000 (Atomic Energy of Canada) and a 137Cs source emitting at a fixed rate of 0.21 Gy/min.
Cell transfections
PKCδCF(K-R) cDNA (Yoshida and Kufe, 2001) was cloned into a pMEP4 vector (Invitrogen) which has an expression cassette downstream from a metallothionein-II promoter. The pMEP4-PKCδCF(K-R) vector was stably expressed in U-2 OS cells by electroporation (Gene Pulser; Bio-Rad) and selection in hygromycin B (Roche Molecular Biochemicals). For induction of PKCδCF(K-R) expression, transfected cells were treated with cadmium sulfate (Sigma-Aldrich) at a final concentration of 2 µM. 293T cells were transiently transfected by the calcium phosphate method.
Immunoprecipitation and immunoblot analysis
Cell lysates were prepared as described (Yoshida et al., 2000) and cleared by centrifugation at 12 000 g for 15 min. Soluble proteins were incubated with anti-Rad9 [sc-8324; Santa-Cruz Biotechnology (SCBT)] or anti-PKCδ (sc-937; SCBT) antibodies for 2–6 h at 4°C followed by a 1 h incubation with protein A/G–Sepharose beads (SCBT). The immune complexes were washed three times with lysis buffer. Cell lysates or immunoprecipitates were separated by SDS–PAGE and transferred to nitrocellulose filters. The filters were incubated with anti-Rad9 (Oncogene), anti-PKCδ, anti-ATM (sc-7230; SCBT), anti-tubulin (Sigma-Aldrich), anti-Bcl-2 (Zymed), anti-His (Novagen) or anti-GFP (Roche Molecular Buochemicals) for 1–4 h at room temparature. After washing, the filters were incubated with anti-rabbit or anti-mouse IgG–peroxidase conjugate (SCBT). The antigen–antibody complexes were visualized by chemiluminescence (NEN Life Science Products). In certain experiments, immune complexes were treated with 25 U of CIP (New England Biolabs) for 30 min at 30°C in the presence or absence of 25 mM β-glycerophosphate before immunoblot analysis.
Glycerol gradient sedimentation analysis
Nuclear extracts were prepared as described (Dignam et al., 1983). A 200 µl aliquot of nuclear extracts was layered on top of a glycerol gradient (5 ml) formed in 50 mM Tris–HCl pH 7.3, 0.1 M KCl, 0.2 mM EDTA, 10 mM β-mercaptoethanol and 0.1% NP-40. After centrifugation in a Beckman SW55Ti rotor at 250 000 g for 12 h at 4°C, fractions (150 µl) were collected from the bottom of the tube. Equal volumes (20 µl) of each sample were analyzed by SDS–PAGE and immunoblotting with anti-PKCδ, anti-Rad9, anti-Hus1 (sc-8323; SCBT) or anti-Rad1 (NeoMarkers, Inc).
siRNA transfections
siRNA duplexes (siRNAs) were synthesized and purified by Japan Bio Service (Saitama, Japan). The siRNA sequences for targeting PKCδ were PKCδsiRNA1 (5′-GAUGAAGGAGGCGCUCAGTT-3′) and PKCδsiRNA2 (5′-GGCUGAGUUCUGGCUGGACTT-3′). The siRNA sequences for targeting ATM were ATMsiRNA1 (5′-GCGCCUGAUU CGAGAUCCUTT-3′) and ATMsiRNA2 (5′-UGGUGCUAUUUACG GAGCUTT-3′). GFPsiRNA was used as a negative control (Hirai and Wang, 2002). Transfection of siRNAs was performed as described (Elbashir et al., 2002).
Immunofluorescence assays
Cells were harvested by centrifugation at 1000 g for 5 min, washed once with phosphate-buffered saline (PBS) and spun onto glass slides. Cells were fixed in 3.2% paraformaldehyde and permeabilized in 0.5% Triton X-100 in PBS for 10 min. Fixed cells were washed twice in PBS and blocked with 2% bovine serum albumin in PBS-T for 1 h. After washing with PBS-T three times, cells were incubated with anti-Rad9 or anti-PKCδ for 3 h. Immune complexes were then stained with fluorescein-conjugated anti-rabbit antibodies (SCBT). Stained cells were mounted with Vectashield Mounting Medium with 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories) and analyzed with a Nikon Eclipse TE2000-U microscope.
In vitro kinase assays
In vitro kinase assays were performed as described (Yoshida and Kufe, 2001) using His-Rad9 as a substrate. For analysis of PKCδ activity in the anti-PKCδ immunoprecipitates, the immune complexes kinase assays were performed using histone H1 as a substrate (Yoshida and Kufe, 2001).
Subcellular fractionation
Subcellular fractionation was performed as described (Kharbanda et al., 1996). Purity of the fractions was monitored by immunoblot analysis with anti-laminB (Oncogene), anti-IκBα (sc-37; SCBT) and anti-PDGF receptor (Oncogene).
Assessment of apoptosis
DNA content was assessed by staining ethanol-fixed cells with propidium iodide and monitoring by FACScan (Becton Dickinson). The numbers of cells positive for green fluorescence with sub-G1 DNA content were determined with a CELLQuest program (Becton Dickinson).
Acknowledgments
Acknowledgements
The authors acknowledge Kamal Chauhan and Tomoko Yamaguchi for excellent technical support. This work was supported by grants CA55241 and CA29431 (D.K.) and CA90315 (H.-G.W.) awarded by the National Cancer Institute.
References
- al-Khodairy F. and Carr,A.M. (1992) DNA repair mutants defining G2 checkpoint pathways in Schizosaccharomyces pombe. EMBO J., 11, 1343–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- al-Khodairy F., Fotou,E., Sheldrick,K.S., Griffiths,D.J., Lehmann,A.R. and Carr,A.M. (1994) Identification and characterization of new elements involved in checkpoint and feedback controls in fission yeast. Mol. Biol. Cell, 5, 147–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskaran R. et al. (1997) Ataxia telangiectasia mutant protein activates c-abl tyrosine kinase in response to ionizing radiation. Nature, 387, 516–519. [DOI] [PubMed] [Google Scholar]
- Bessho T. and Sancar,A. (2000) Human DNA damage checkpoint protein hRAD9 is a 3′ to 5′ exonuclease. J. Biol. Chem., 275, 7451–7454. [DOI] [PubMed] [Google Scholar]
- Bharti A. et al. (1998) Inactivation of DNA-dependent protein kinase by protein kinase Cδ: implications for apoptosis. Mol. Cell. Biol., 18, 6719–6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blass M., Kronfeld,I., Kazimirsky,G., Blumberg,P. and Brodie,C. (2002) Tyrosine phosphorylation of protein kinase Cδ is essential for its apoptotic effect in response to etoposide. Mol. Cell. Biol., 22, 182–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burtelow M.A., Kaufmann,S.H. and Karnitz,L.M. (2000) Retention of the human Rad9 checkpoint complex in extraction-resistant nuclear complexes after DNA damage. J. Biol. Chem., 275, 26343–26348. [DOI] [PubMed] [Google Scholar]
- Chen M.J., Lin,Y.T., Lieberman,H.B., Chen,G. and Lee,E.Y. (2001) ATM-dependent phosphorylation of human Rad9 is required for ionizing radiation-induced checkpoint activation. J. Biol. Chem., 276, 16580–16586. [DOI] [PubMed] [Google Scholar]
- Chen X., Zheng,Y., Zhu,J., Jiang,J. and Wang,J. (2001) p73 is transcriptionally regulated by DNA damage, p53 and p73. Oncogene, 20, 769–774. [DOI] [PubMed] [Google Scholar]
- Davies S.P., Reddy,H., Caivano,M. and Cohen,P. (2000) Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J., 351, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dignam J.D., Lebovitz,R.M. and Roeder,R.G. (1983) Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res., 11, 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbashir S.M., Harborth,J., Weber,K. and Tuschl,T. (2002) Analysis of gene function in somatic mammalian cells using small interfering RNAs. Methods, 26, 199–213. [DOI] [PubMed] [Google Scholar]
- Emoto Y. et al. (1995) Proteolytic activation of protein kinase Cδ by an ICE-like protease in apoptotic cells. EMBO J., 14, 6148–6156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emoto Y., Kisaki,H., Manome,Y., Kharbanda,S. and Kufe,D. (1996) Activation of protein kinase Cδ in human myeloid leukemia cells treated with 1-β-d-arabinofuranosylcytosine. Blood, 87, 1990–1996. [PubMed] [Google Scholar]
- Freire R., Murguia,J.R., Tarsounas,M., Lowndes,N.F., Moens,P.B. and Jackson,S.P. (1998) Human and mouse homologs of Schizosaccharomyces pombe rad1(+) and Saccharomyces cerevisiae RAD17: linkage to checkpoint control and mammalian meiosis. Genes Dev., 12, 2560–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghayur T. et al. (1996) Proteolytic activation of protein kinase Cδ by an ICE/CED 3-like protease induces characteristics of apoptosis. J. Exp. Med., 184, 2399–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gschwendt M., Muller,H., Keilbassa,K., Zang,R., Kittstein,W., Rinke,G. and Marks,F. (1994) Rottlerin, a novel protein kinase inhibitor. Biochem. Biophys. Res. Commun., 199, 93–98. [DOI] [PubMed] [Google Scholar]
- Hirai I. and Wang,H.G. (2002) A role of the C-terminal region of human Rad9 (hRad9) in nuclear transport of the hRad9 checkpoint complex. J. Biol. Chem., 277, 25722–25727. [DOI] [PubMed] [Google Scholar]
- Kharbanda S., Saleem,A., Yuan,Z.-M., Kraeft,S., Weichselbaum,R., Chen,L.B. and Kufe,D. (1996) Nuclear signaling induced by ionizing radiation involves colocalization of the activated p56/p53lyn tyrosine kinase with p34cdc2. Cancer Res., 56, 3617–3621. [PubMed] [Google Scholar]
- Komatsu K., Hopkins,K.M., Lieberman,H.B. and Wang,H. (2000a) Schizosaccharomyces pombe Rad9 contains a BH3-like region and interacts with the anti-apoptotic protein Bcl-2. FEBS Lett., 481, 122–126. [DOI] [PubMed] [Google Scholar]
- Komatsu K., Miyashita,T., Hang,H., Hopkins,K.M., Zheng,W., Cuddeback,S., Yamada,M., Lieberman,H.B. and Wang,H.G. (2000b) Human homologue of S.pombe Rad9 interacts with BCL-2/BCL-xL and promotes apoptosis. Nat. Cell Biol., 2, 1–6. [DOI] [PubMed] [Google Scholar]
- Konishi H., Tanaka,M., Takemura,Y., Matsuzaki,H., Ono,Y., Kikkawa,U. and Nishizuka,Y. (1997) Activation of protein kinase C by tyrosine phosphorylation in response to H2O2. Proc. Natl Acad. Sci. USA, 94, 11233–11237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostrub C.F., Knudsen,K., Subramani,S. and Enoch,T. (1998) Hus1p, a conserved fission yeast checkpoint protein, interacts with Rad1p and is phosphorylated in response to DNA damage. EMBO J., 17, 2055–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey-Boltz L.A., Bermudez,V.P., Hurwitz,J. and Sancar,A. (2001) Purification and characterization of human DNA damage checkpoint Rad complexes. Proc. Natl Acad. Sci. USA, 98, 11236–11241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker A.E., Van de Weyer,I., Laus,M.C., Oostveen,I., Yon,J., Verhasselt,P. and Luyten,W.H. (1998) A human homologue of the Schizosaccharomyces pombe rad1+ checkpoint gene encodes an exonuclease. J. Biol. Chem., 273, 18332–18339. [DOI] [PubMed] [Google Scholar]
- Rauen M., Burtelow,M.A., Dufault,V.M. and Karnitz,L.M. (2000) The human checkpoint protein hRad17 interacts with the PCNA-like proteins hRad1, hHus1 and hRad9. J. Biol. Chem., 275, 29767–29771. [DOI] [PubMed] [Google Scholar]
- Ren J., Datta,R., Shioya,H., Li,Y., Oki,E., Biedermann,V., Bharti,A. and Kufe,D. (2002) p73β is regulated by protein kinase Cδ catalytic fragment generated in the apoptotic response to DNA damage. J. Biol. Chem., 277, 33758–33765. [DOI] [PubMed] [Google Scholar]
- Shafman T. et al. (1997) Interaction between ATM protein and c-Abl in response to DNA damage. Nature, 387, 520–523. [DOI] [PubMed] [Google Scholar]
- Soltoff S.P. (2001) Rottlerin is a mitochondrial uncoupler that decreases cellular ATP levels and indirectly blocks protein kinase Cδ tyrosine phosphorylation. J. Biol. Chem., 276, 37986–37992. [DOI] [PubMed] [Google Scholar]
- St Onge R.P., Udell,C.M., Casselman,R. and Davey,S. (1999) The human G2 checkpoint control protein hRAD9 is a nuclear phosphoprotein that forms complexes with hRAD1 and hHUS1. Mol. Biol. Cell, 10, 1985–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Onge R.P., Bestley,B.D., Park,M.Y., Casselman,R. and Davey,S. (2001) DNA damage-dependent and -independent phosphorylation of the hRad9 checkpoint protein. J. Biol. Chem., 276, 41898–41905. [DOI] [PubMed] [Google Scholar]
- Thelen M.P., Venclovas,C. and Fidelis,K. (1999) A sliding clamp model for the Rad1 family of cell cycle checkpoint proteins. Cell, 96, 769–770. [DOI] [PubMed] [Google Scholar]
- Tybulewicz V.L.J., Crawford,C.E., Jackson,P.K., Bronson,R.T. and Mulligan,R.C. (1991) Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell, 65, 1153–1163. [DOI] [PubMed] [Google Scholar]
- Udell C.M., Lee,S.K. and Davey,S. (1998) RAD1 and MRAD1 encode mammalian homologues of the fission yeast rad1+ cell cycle checkpoint control gene. Nucleic Acids Res., 26, 3971–3976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkmer E. and Karnitz,L.M. (1999) Human homologs of Schizosaccharomyces pombe rad1, hus1 and rad9 form a DNA damage-responsive protein complex. J. Biol. Chem., 274, 567–570. [DOI] [PubMed] [Google Scholar]
- Yoshida K. and Kufe,D. (2001) Negative regulation of the SHPTP1 protein tyrosine phosphatase by protein kinase Cδ in response to DNA damage. Mol. Pharmacol., 60, 1431–1438. [DOI] [PubMed] [Google Scholar]
- Yoshida K., Weichselbaum,R., Kharbanda,S. and Kufe,D. (2000) Role for Lyn tyrosine kinase as a regulator of stress-activated protein kinase activity in response to DNA damage. Mol. Cell. Biol., 20, 5370–5380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K., Komatsu,K., Wang,H.-G. and Kufe,D. (2002) c-Abl tyrosine kinase regulates the human Rad9 checkpoint protein in response to DNA damage. Mol. Cell. Biol., 22, 3292–3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Z.-M., Utsugisawa,T., Ishiko,T., Nakada,S., Huang,Y., Kharbanda,S., Weichselbaum,R. and Kufe,D. (1998) Activation of protein kinase Cδ by the c-Abl tyrosine kinase in response to ionizing radiation. Oncogene, 16, 1643–1648. [DOI] [PubMed] [Google Scholar]