

Abstract
Myasthenia gravis (MG) is an autoimmune neuromuscular transmission disorder characterized by loss of acetylcholine receptors (AChR’s) due primarily to the production of anti-AChR autoantibodies. In this study we investigated whether the presence of decay-accelerating factor (DAF or CD55), an intrinsic complement regulator, protects against the development of disease. Experimental autoimmune MG was induced in Daf1–/– mice (devoid of neuromuscular DAF protein) and their Daf1+/+ littermates by injection of rat anti-AChR mAb McAb-3. After twenty-four hours, grip strength assessment revealed that Daf1–/– mice exhibited hold times of less than 30 seconds, compared with more than 8 minutes for the Daf1+/+ controls. The weakness was reversed by edrophonium, consistent with a myasthenic disorder. Immunohistochemistry revealed greatly augmented C3b deposition localized at postsynaptic junctions, and radioimmunoassays showed more profound reductions in AChR levels. Electron microscopy demonstrated markedly greater junctional damage in the Daf1–/– mice compared with the Daf1+/+ littermates. Control studies showed equivalent levels of other cell surface regulators, i.e., Crry and CD59. The results demonstrate that mice that lack DAF are markedly more susceptible to anti-AChR–induced MG, which simulates the primary mechanism in the human disease, and strongly suggest that in disease flares complement inhibitors might have therapeutic value.
Introduction
Myasthenia gravis (MG) is a syndrome characterized by fatiguing skeletal muscle weakness. A large body of research on MG patients and on experimental autoimmune MG (EAMG) in animals has shown that the disease is Ab-mediated, producing loss of or compromised function of skeletal muscle nicotinic acetylcholine receptors (AChR’s). Three mechanisms have been implicated: (a) autoantibodies against AChR cross-link surface AChR and induce their endocytosis, resulting in their depletion from the postjunctional membrane; (b) the autoantibodies themselves interfere directly with AChR function by blocking acetylcholine-binding sites; and (c) the autoantibodies contribute to destruction of the endplate with consequent AChR loss (1–4).
Several lines of evidence indicate that complement activation resulting from autoantibody binding to AChR is a key effector mechanism in the pathogenesis of MG. C3 activation fragments, C9, and the membrane attack complex (MAC) can be detected at motor endplates in patients and EAMG animals (5–7). Depletion of C3 by cobra venom factor protects rats against passively or actively induced disease (8–9). As a result of diminished AChR density following initial induction, animals become resistant to a second induction of passive transfer EAMG because of insufficient Ab deposition to activate complement or induce other effector responses (10–11). Administration of anti-C6 Ab prevents the development of EAMG in rats (12). In actively induced EAMG, C5-deficient mice develop less severe disease (13). Finally, treatment with soluble CR1, a complement inhibitor, can protect against EAMG in rats (14). These data, taken together, indicate that at the motor endplate, C3b deposition and MAC assembly with consequent membrane perturbation damage the postsynaptic surface of the neuromuscular junction, compromising neuromuscular transmission.
Host tissues are protected from autologous complement-mediated injury by cell surface regulators that function intrinsically in their plasma membranes [reviewed in ref. 15]. These regulators consist of the decay-accelerating factor (DAF or CD55), the membrane cofactor protein (MCP or CD46), and the membrane inhibitor of reactive lysis (MIRL or CD59). Collectively, these control proteins accelerate the decay of autologous C3 convertases (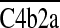 and
and 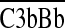 ) and C5 convertases (
) and C5 convertases (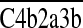 and
and 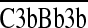 ) that inappropriately assemble on self cell surfaces (16), promote the cleavage of uncomplexed autologous cell–bound C4b and C3b fragments (17), and inhibit the formation of autologous MACs, which brings about cell lysis (18–20).
) that inappropriately assemble on self cell surfaces (16), promote the cleavage of uncomplexed autologous cell–bound C4b and C3b fragments (17), and inhibit the formation of autologous MACs, which brings about cell lysis (18–20).
In this study, we examined the role of DAF in protecting against AChR damage in passively induced EAMG in mice. To accomplish this, we used DAF knockout mice (21). Mice differ from humans in that there are two Daf genes rather than one. While the second Daf gene, termed Daf2, is constitutively expressed only in testis and in dendritic cells of the spleen (21), the first, termed Daf1, is expressed in almost all tissues (21, 22) and thus is considered to be the counterpart of the human DAF gene (22). Previous studies have shown that neuromuscular DAF protein in mice derives from the Daf1 gene (21). Consequently, we used mice targeted at this gene.
We found that following anti-AChR Ab administration, Daf1–/– mice devoid of neuromuscular DAF protein became dramatically sicker than their Daf1+/+ littermates. Greater postjunctional membrane damage was documented by electron microscopy (EM) in the Daf1–/– mice, more marked reduction of AChR levels was measured by specific immunoradiometric assay, and more C3 deposition specifically directed at motor endplates was found by immunohistological staining. The results strongly suggest that DAF plays a critical role in protecting the motor endplate and its surface AChR molecules against autoantibody-initiated, complement-mediated injury.
Methods
Daf1 knockout mice.
Daf1–/– mice were prepared by Cre/loxP-mediated deletion as previously described (21). Embryonic stem cell strain GK129 was used, and chimeric mice were bred into the C57BL/6 strain. The mice were backcrossed for four generations, and homozygous Daf1–/– mice and their Daf1+/+ littermates were used at 8–10 weeks of age.
Induction of EAMG.
EAMG was passively induced using rat anti-mouse muscle AChR mAb McAb-3 (a gift of Vanda Lennon, Mayo Clinic, Rochester, Minnesota, USA), which binds to mouse skeletal muscle AChR (23) (see Discussion). At time zero, 50 μl of purified McAb-3 (4.6 mg/ml) or ascites fluid containing an equivalent amount of mAb was injected intraperitoneally.
Assessment of muscle weakness.
Weakness was quantitated as described by Karachunski et al. (24) by hanging mice three times from a grid and measuring the time it took for them to release their hold and fall (holding time). Although weakness produced by EAMG in mice is often not obvious (25) and the hang-time test requires sensitization of animals with pancuronium bromide, in this investigation the effect was so profound that this step proved unnecessary. Twenty-four hours after Ab administration, the mice were placed upside-down on a grid placed 3 feet above ground level and the holding time was measured. Mice were then kept under close observation and sequentially analyzed at 24 and 48 hours, and videotape documentation was performed. Some animals received intraperitoneal edrophonium injections to evaluate them for a neuromuscular transmission defect.
Assay of mouse muscle AChR content.
AChR content of mouse muscles was measured by a solid-phase Ab-capture immunoradiometric assay (26). Increasing volumes (in triplicate) of muscle extract labeled with [125I]α-bungarotoxin (αBTX) (27–28) were used, and anti–rodent muscle AChR mAb 210 (26) was used as the capturing Ab. Nonspecific [125I]αBTX binding was determined using muscle extracts, preincubated for 1 hour with 1 mM unlabeled αBTX, from five randomly selected mice. The amount of AChR in the muscles was determined from the slopes of each sample and the specific activity of [125I]αBTX on the day of the assay.
EM of nerve junctions.
EM studies were performed as described by Schneiter et al. (29), with exceptions as noted. After overnight fixation in 2% glutaraldehyde (with 0.1 M sodium cacodylate), the diaphragm was isolated at the synaptic region by cutting a 4-mm strip along the course of the phrenic nerve. The strip was washed and postfixed in 1% osmium tetroxide for 30 minutes. Muscle was transferred to 1% uranyl acetate for 1 hour at room temperature, then dehydrated with ethanol and embedded in LX112 resin (Polysciences Inc., Warrington, Pennsylvania, USA). Silver sections were cut and stained with 2% uranyl acetate followed by Reynolds lead citrate. Sections were examined with a JEM-100 CX11 electron microscope (JEOL USA Inc., Peabody, Massachusetts, USA) at 80 kV. Photographs were recorded using Kodak EM film (Eastman Kodak Co., Rochester, New York, USA).
Immunohistochemistry.
For analysis of C3b deposition at neuromuscular junctions, cryosections of muscles from mice injected with McAb-3 mAb were prepared and double-stained with FITC-labeled goat anti-mouse C3 Ab (ICN Biomedicals Inc., Costa Mesa, California, USA) and Texas red–labeled αBTX (2 μg/ml; Molecular Probes Inc., Eugene, Oregon, USA) to identify endplates. Sections were examined with a Nikon Diaphot fluorescence microscope (Nikon Instruments Inc., Melville, New York, USA) and analyzed using ImagePro software (Media Cybernetics, Silver Spring, Maryland, USA). For analysis of Crry and CD59 expression at the neuromuscular junctions, staining was performed using 1:50 dilutions of rabbit anti-Crry and anti-CD59 antisera (kindly provided, respectively, by M. Holers, University of Colorado, Denver, Colorado, USA, and B.P. Morgan, University of Cardiff, Cardiff, United Kingdom) or nonimmune serum. Secondary staining of the washed cells was done with a 1:50 dilution of FITC-labeled anti-rabbit Ig (Pharmingen, La Jolla, California, USA), followed by fluorescence microscopy as above.
Results
Daf1–/– mice develop markedly increased weakness in experimental MG.
In initial experiments, two Daf1–/– mice and their littermates were injected intraperitoneally with 50 μl of anti-rat AChR mAb McAb-3. Twenty-four hours later, hang times were measured (see Methods). The two Daf1–/– mice fell from the grid in 10 seconds and 30 seconds, while both of the Daf1+/+ controls held firmly for more than 8 minutes.
In two further experiments, one using purified McAb-3 IgG and the other its corresponding ascites fluid (each with five pairs of Daf1–/– mice and their Daf1+/+ controls), the knockout groups exhibited dramatically greater weakness than the control groups at 24 hours, as observed in the initial studies. Analyses at later timepoints showed that the weakness increased even further, reaching a maximum at 48 hours. At this timepoint in both experiments (Figure 1a), the Daf1–/– mice could not even stand, but crouched at the corners of their cages, and most died within 1–4 hours. In contrast, the Daf1+/+ mice exhibited no signs of weakness (Figure 1b).
Figure 1.

Daf1–/– and Daf1+/+ mice were injected with McAb-3 24 hours prior to videotaping (photographs shown were acquired from video images). (a) The Daf1–/– mouse had a hunched posture and could not extend its digits. Its hind limbs could not support its weight. This mouse had a hang time of less than 10 seconds. (b) The Daf1+/+ littermate was normally active (shadow under belly, turning body), showing normal posture and leg position. It had a hang time of more than 8 minutes.
In a third set of studies using the purified McAb-3 mAb and three Daf1–/– mice, the disabled Daf1–/– mice were given edrophonium intraperitoneally at the 48-hour timepoint. In all three Daf1–/– mice, the drug reversed the weakness in the appropriate temporal fashion, confirming that the debility was derived from impaired activation of the postsynaptic endplate potential.
Daf1–/– mice show greater endplate AChR loss and increased C3b deposition.
EM analyses of muscles from the treated mice showed that there was dramatically greater postsynaptic junction membrane damage in the Daf1–/– animals than in the Daf1+/+ controls. In the Daf1+/+ controls, 20 junctions from two mice were identified. Seventeen of these exhibited normal architecture as depicted in Figure 2a; three junctions had widening of the synaptic cleft and a reduction in synaptic folds. This contrasted with 20 junctions examined from two Daf1–/– mice, all of which demonstrated simplified junctional architecture and widening of the synaptic cleft as shown in Figure 2, b and c. Compared with the wild-type mice shown in Figure 2a, the junction folds were totally simplified and the synaptic clefts were widened. Within the clefts, there was electron-dense material (Figure 2, c and d), most likely representing globular fragments of the junctional folds shed into the synaptic space. By contrast, the Daf1+/+ mice identically injected with McAb-3 mAb showed normal morphology with normally complex junctional folding (Figure 2a).
Figure 2.

Electron micrographs of diaphragm neuromuscular junctions following injection with McAb-3 mAb. (a) Junction typical of those observed in Daf1+/+ animals. It appears completely normal with entirely normal synaptic architecture. ×25,000. (b) Junction from a Daf1–/– mouse that was obviously weak, shows reduced synaptic folds and a widened synaptic cleft. ×25,000. (c) Three neuromuscular junctions from Daf1–/– mice, all exhibiting simplified junctional folds. ×8,000. The electron-dense material seen in the synaptic clefts of the junctions in b and c is likely postsynaptic membrane shed from the muscle by complement-mediated destruction. This material is seen in (d) at a magnification of ×20,000.
Comparative measurements of AChR levels in whole-mouse homogenates of the Daf1–/– and Daf1+/+ mice by specific immunoradiometric assay 48 hours after injection of McAb-3 mAb showed more profound reductions in the Daf1–/– animals. As shown in Figure 3, while AChR levels in Daf1+/+ mice were reduced to approximately 55% of noninjection levels, levels in Daf1–/– mice were reduced to 33% of preinjection values, although the difference in the number of mice studied (Daf1+/+, n = 4; Daf1–/–, n = 8) did not reach statistical significance (see Discussion).
Figure 3.

Whole-animal AChR levels in Daf1–/– and Daf1+/+ mice 48 hours after McAb-3 mAb injection. The bars (NI, noninjected; I, injected) show changes in levels normalized to noninjection levels. Daf1–/–, n = 8; Daf1+/+, n = 4. *P > 0.05. The noninjection levels in the Daf1–/– mice were not decreased relative to wild-type controls (0.213 vs. 0.160 pmol/g muscle).
Immunohistochemical analyses showed that compared with wild-type, strikingly more junctions from Daf1–/– animals injected with McAb-3 demonstrated C3b deposition (Figure 4, a and b). The staining was intense and remarkably was specifically localized to the motor endplate as assessed by αBTX staining.
Figure 4.

Diaphragm muscles from the Daf1–/– (a and b) and Daf1+/+ (c and d) mice 48 hours after injection with McAb-3 mAb. αBTX stain demonstrates the endplate regions (a and c). Double-labeling with FITC-labeled anti-C3 Ab (1:3,000 dilution) demonstrates C3 deposition at almost all junctions from the Daf1–/– animal (b), while no complement deposition is observed in junctions of muscles from the Daf1+/+ animal. Scale bar = 100 μm.
With the low dilution of C3b Ab used, no sections of the Daf1+/+ animals exhibited C3b deposits at the neuromuscular junction (Figure 4, c and d). These findings are consistent with the EM studies that showed only rare damage of junctions from Daf1+/+ mice.
Daf1–/– and Daf1+/+ mice have identical levels of other cell surface complement regulators.
Since self tissues are protected by other cell surface regulators in addition to DAF, studies were done to verify that changes in these molecules were not involved in the clinical and pathological differences observed in the Daf1–/– mice. Immunohistochemical staining for CD59 and Crry was not different in distribution or degree in the muscles of Daf1+/+ and Daf1–/– animals (data not shown).
Discussion
In this study we examined passively induced EAMG in Daf1–/– mice devoid of neuromuscular DAF protein. We found that following anti-AChR Ab injection, the Daf1–/– mice showed dramatically greater muscle weakness than their Daf1+/+ littermates. Reversal of the weakness with edrophonium confirmed that the weakness was due to a neuromuscular transmission disorder. EM showed markedly greater destruction of postsynaptic architecture. Specific immunoradiometric assay showed more loss of AChR protein, and immunohistochemistry revealed markedly enhanced C3b deposition, specifically localized at the motor endplate. These findings demonstrate that DAF is critical in conferring protection against the development of weakness in passively induced EAMG. McAb-3 mAb is an IgG2b isotype and a known activator of complement that allows evaluation of the role of DAF in protecting against the pathogenic component of MG that is complement-mediated. This Ab is directed against extracellular epitopes shared by Torpedo and mammalian AChR, but does not include the acetylcholine binding site (9, 23).
Further definition of the epitope of McAb-3 has not been performed, to our knowledge. However, previous studies have shown that this Ab produces myasthenic weakness and reduces miniature endplate potential amplitudes as well as muscle AChR content in rats and guinea pigs. In rats, the effect has been shown to be complement-dependent (9). Notably, in all past studies in mice, although miniature endplate potential amplitudes and total AChR are reduced, the animals usually show no discernable weakness clinically. Studies of active and passively transferred EAMG in mice have repeatedly demonstrated that the resistance of mice to development of clinical weakness is due to their high safety factor for neuromuscular transmission (24, 25, 30). Our results therefore are all the more striking. The Daf1–/– mice, which have normal AChR content and a similar distribution of other complement regulatory proteins, developed profound EAMG. In human MG the Ab response is polyclonal, and other disease effector mechanisms (as noted in the Introduction), in addition to complement, could produce neuromuscular transmission failure. Nevertheless, numerous previous studies implicating complement argue that DAF and possibly other intrinsic complement regulatory proteins would have a critical protective role in the human disease.
Our ultrastructural studies of diaphragms of Daf1–/– mice with EAMG demonstrated the classic findings of EAMG and human MG (31, 32). The damage to the postsynaptic architecture contributes to the neuromuscular transmission defect not only by reducing AChR density but also by destroying the secondary synaptic fold architecture that serves to focus current flow on voltage-gated sodium channels at the depths of the folds. Loss of synaptic membrane also leads to loss of voltage-gated sodium channels. All these factors lead to a compromise of the endplate potential necessary to generate an action potential and thereby induce muscle contraction (30). The severity of the ultrastructural damage was profoundly greater at the endplates of Daf1–/– mice, strongly indicative of much greater complement-mediated damage.
Edrophonium reversed the muscle weakness produced by EAMG in the Daf1–/– mice, verifying a defect of neuromuscular transmission. Edrophonium inhibits, for a short period of time, acetylcholinesterase activity that is concentrated at the neuromuscular junction (33). This inhibition transiently increases the amount of acetylcholine available to bind AChR, thereby elevating the endplate potential above threshold for action potential generation. The reversal of weakness by edrophonium confirmed the myasthenic nature of the weakness in the Daf1–/– mice.
Whole-animal AChR levels were reduced following McAb-3 administration in both the Daf1–/– and Daf1+/+ mice. Previous studies have demonstrated a reduction of AChR levels in mice treated with this mAb despite an absence of weakness (9, 19). Although there was greater reduction in the Daf1–/– mice than in the Daf1+/+ mice, the difference was not impressive. Relevant to this issue, the debris observed in the synaptic cleft by EM is likely to be postjunctional membrane containing AChR. Consequently, the AChR lost from the muscle would contribute to the total AChR content measured, and despite much of the AChR in Daf1–/– mice being nonfunctional, would thereby limit the difference observed between Daf1–/– and Daf1+/+ mice. By immunohistochemical measures, C3b deposition was markedly greater in Daf1–/– mice. It is possible that C3b activation fragments that bind covalently could block AChR function in addition to causing its loss. In this situation, AChR would still be detected by radioimmunoassay but would not be functional. Studies to investigate this proposed mechanism of AChR inactivation are beyond the scope of the present investigation.
A hallmark of human MG is the propensity for involvement of the extraocular muscles (34). In the mouse, gene expression profiling by DNA microarray has shown that Daf1 gene expression is five- to sevenfold lower in extraocular muscles than in other skeletal muscles (35). There are several contributing factors to the susceptibility of extraocular muscles to MG. The present results, coupled with the mRNA expression data, strongly suggest that the reduction of DAF renders these muscles more susceptible to complement-mediated damage, a fact never appreciated previously. As stated in the Introduction, MAC and C3 activation fragments can be detected at motor endplates in both MG patients and EAMG animals. This suggests that MAC formed on the postsynaptic membrane damages the membrane, leading to loss of secondary synaptic folds and reduced AChR density. Of relevance to the present study, the absence of DAF not only disables regulation of autologous C3 convertases, but also disables regulation of autologous C5 convertases, leading to increased uptake of autologous C5b and ultimately to the assembly of autologous C5b9 complexes (MAC). If expression of complement regulatory proteins is reduced in human extraocular muscle as in mice, this would make these muscles more susceptible to complement-mediated damage.
In mice, in addition to DAF, there is another cell surface C3 convertase inhibitor, termed Crry, that is uniquely present in rodents. It is ubiquitously expressed and possesses functions that overlap those of both DAF and MCP in humans. Because blocking Crry function leads to spontaneous cell surface C3b deposition (36, 37), and because Crry knockout embryos are destroyed in utero by maternal complement (36, 37), it has been proposed that in rodents, Crry function can substitute for the function of DAF. Our experiments show that despite identical expression levels of Crry on skeletal muscle of Daf1–/– mice and Daf1+/+ controls, Daf1–/– mice are much more susceptible to passively induced MG. The findings provide further evidence that although Crry may be more dominant for regulation of activation mediated by the alternative pathway, DAF may be more essential for control of classical pathway–mediated activation. This result is in accordance with our previous findings in Daf1–/– mice of much more severe renal damage in nephrotoxic serum–induced nephritis (38).
It has been shown previously (14) that daily administration of soluble human CR1, an extrinsic C3/C5 convertase inhibitor, can protect against the development of EAMG in rats, suggesting that complement inhibitors might be considered as a therapy against MG. Here, our results provide strong evidence in mice that failed C3 regulation can dramatically predispose to the manifestations of EAMG, strengthening this proposition. Complement inhibitors might be useful for the treatment of clinical MG, possibly in acute flares. Additionally, Daf1–/– mice should provide a valuable animal model for further studies of MG, since as indicated above, wild-type mice are extremely resistant to the induction of the disease.
Acknowledgments
This work was supported by NIH grants R01 AI-23598, R01 EY-11288 (both to M.E. Medof), R01 EY-13238 (to H.J. Kaminski), and P30 EY-11373 (to M.E. Medof and H.J. Kaminski). The authors thank Vanda Lennon for providing McAb-3 mAb, Michael Holers and B. Paul Morgan for providing anti-Crry and anti-CD59 antisera, and Sara Cechner for manuscript preparation.
Footnotes
Conflict of interest: No conflict of interest has been declared.
Nonstandard abbreviations used: myasthenia gravis (MG); experimental autoimmune MG (EAMG); acetylcholine receptor (AChR); membrane attack complex (MAC); decay-accelerating factor (DAF); electron microscopy (EM).
References
- 1.Lindstrom JM. Acetylcholine receptors and myasthenia. Muscle Nerve. 2002;23:453–477. doi: 10.1002/(sici)1097-4598(200004)23:4<453::aid-mus3>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 2.Drachman DB, Angus CW, Adams RN, Michelson JD, Hoffman GJ. Myasthenic antibodies cross-link acetylcholine receptors to accelerate degradation. N Engl J Med. 1978;298:1116–1122. doi: 10.1056/NEJM197805182982004. [DOI] [PubMed] [Google Scholar]
- 3.Drachman DB, Adams RN, Josifek LF, Self SG. Functional activities of autoantibodies to acetylcholine receptors and the clinical severity of myasthenia gravis. N Engl J Med. 1982;307:769–775. doi: 10.1056/NEJM198209233071301. [DOI] [PubMed] [Google Scholar]
- 4.Richman DP, et al. Antibody effector mechanisms in myasthenia gravis. The complement hypothesis. Ann NY Acad Sci. 1998;841:450–465. doi: 10.1111/j.1749-6632.1998.tb10962.x. [DOI] [PubMed] [Google Scholar]
- 5.Sahashi K, Engel AG, Lambert EH, Howard FM., Jr Ultrastructural localization of the terminal and lytic ninth complement component (C9) at the motor end-plate in myasthenia gravis. J Neuropathol Exp Neurol. 1980;39:160–172. doi: 10.1097/00005072-198003000-00005. [DOI] [PubMed] [Google Scholar]
- 6.Fazekas A, Komoly S, Bozsik B, Szobor A. Myasthenia gravis: demonstration of membrane attack complex in muscle end-plates. Clin Neuropathol. 1986;5:78–83. [PubMed] [Google Scholar]
- 7.Nakano S, Engel AG. Myasthenia gravis: quantitative immunocytochemical analysis of inflammatory cells and detection of complement membrane attack complex at the end-plate in 30 patients. Neurology. 1993;43:1167–1172. doi: 10.1212/wnl.43.6.1167. [DOI] [PubMed] [Google Scholar]
- 8.Lennon VA, Seybold ME, Lindstrom JM, Cochrane C, Ulevitch R. Role of complement in the pathogenesis of experimental autoimmune myasthenia gravis. J Exp Med. 1978;147:973–983. doi: 10.1084/jem.147.4.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lennon VA, Lambert EH. Monoclonal autoantibodies to acetylcholine receptors: evidence for a dominant idiotype and requirement of complement for pathogenicity. Ann NY Acad Sci. 1981;377:77–96. doi: 10.1111/j.1749-6632.1981.tb33725.x. [DOI] [PubMed] [Google Scholar]
- 10.Corey AL, Richman DP, Agius MA, Wollmann RL. Refractoriness to a second episode of experimental myasthenia gravis. Correlation with AChR concentration and morphologic appearance of the postsynaptic membrane. J Immunol. 1987;138:3269–3275. [PubMed] [Google Scholar]
- 11.Corey AL, Richman DP, Shuman CA, Gomez CM, Arnason BG. Use of monoclonal antiacetylcholine receptor antibodies to investigate the macrophage inflammation of acute experimental myasthenia gravis: refractoriness to a second episode of acute disease. Neurology. 1985;35:1455–1460. doi: 10.1212/wnl.35.10.1455. [DOI] [PubMed] [Google Scholar]
- 12.Biesecker G, Gomez CM. Inhibition of acute passive transfer experimental autoimmune myasthenia gravis with Fab antibody to complement C6. J Immunol. 1989;142:2654–2659. [PubMed] [Google Scholar]
- 13.Christadoss P. C5 gene influences the development of murine myasthenia gravis. J Immunol. 1988;140:2589–2592. [PubMed] [Google Scholar]
- 14.Piddlesden SJ, Jiang S, Levin JL, Vincent A, Morgan BP. Soluble complement receptor 1 (sCR1) protects against experimental autoimmune myasthenia gravis. J Neuroimmunol. 1996;71:173–177. doi: 10.1016/s0165-5728(96)00144-0. [DOI] [PubMed] [Google Scholar]
- 15.Janeway, C.A., Jr., Travers, P., Walport, M., and Shlomchik, M. 2001. Innate immunity. In Immunobiology. The immune system in health and disease. C.A. Janeway, Jr., P. Travers, M. Walport, and M. Shlomchik, editors. Garland Publishing. New York, New York, USA. 35–91.
- 16.Medof ME, Kinoshita T, Nussenzweig V. Inhibition of complement activation on the surface of cells after incorporation of decay-accelerating factor (DAF) into their membranes. J Exp Med. 1984;160:1558–1578. doi: 10.1084/jem.160.5.1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seya T, Turner JR, Atkinson JP. Purification and characterization of a membrane protein (gp45-70) that is a cofactor for cleavage of C3b and C4b. J Exp Med. 1986;163:837–855. doi: 10.1084/jem.163.4.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davies A, et al. CD59, an LY-6-like protein expressed in human lymphoid cells, regulates the action of the complement membrane attack complex on homologous cells. J Exp Med. 1989;170:637–654. doi: 10.1084/jem.170.3.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holguin MH, Fredrick LR, Bernshaw NJ, Wilcox LA, Parker CJ. Isolation and characterization of a membrane protein from normal human erythrocytes that inhibits reactive lysis of the erythrocytes of paroxysmal nocturnal hemoglobinuria. J Clin Invest. 1989;84:7–17. doi: 10.1172/JCI114172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harada R, Okada N, Fujita T, Okada H. Purification of 1F5 antigen that prevents complement attack on homologous cell membranes. J Immunol. 1990;144:1823–1828. [PubMed] [Google Scholar]
- 21.Lin F, et al. Tissue distribution of products of the mouse decay-accelerating factor (DAF) genes. Exploitation of a Daf1 knock-out mouse and site-specific monoclonal antibodies. Immunology. 2001;104:215–225. doi: 10.1046/j.0019-2805.2001.01287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spicer AP, Seldin MF, Gendler SJ. Molecular cloning and chromosomal localization of the mouse decay-accelerating factor genes. Duplicated genes encode glycosylphosphatidylinositol-anchored and transmembrane forms. J Immunol. 1995;155:3079–3091. [PubMed] [Google Scholar]
- 23.Lennon VA, Lambert EH. Myasthenia gravis induced by monoclonal antibodies to acetylcholine receptors. Nature. 1980;285:238–240. doi: 10.1038/285238a0. [DOI] [PubMed] [Google Scholar]
- 24.Karachunski PI, Ostlie N, Bellone M, Infante AJ, Conti-Fine BM. Mechanisms by which the I-ABM12 mutation influences susceptibility to experimental myasthenia gravis: a study in homozygous and heterozygous mice. Scand J Immunol. 1995;42:215–225. doi: 10.1111/j.1365-3083.1995.tb03648.x. [DOI] [PubMed] [Google Scholar]
- 25.Conti-Fine, B., Bellone, M., Howard, J.J., and Protti, M. 1997. Myasthenia gravis. The immunobiology of an autoimmune disease. Neuroscience Intelligence Unit, R.G. Landes Co. Austin, Texas, USA. 230 pp.
- 26.Conroy WG, Saedi MS, Lindstrom J. TE671 cells express an abundance of a partially mature acetylcholine receptor alpha subunit which has characteristics of an assembly intermediate. J Biol Chem. 1990;265:21642–21651. [PubMed] [Google Scholar]
- 27.Karachunski PI, Ostlie NS, Okita DK, Conti-Fine BM. Interleukin-4 deficiency facilitates development of experimental myasthenia gravis and precludes its prevention by nasal administration of CD4+ epitope sequences of the acetylcholine receptor. J Neuroimmunol. 1999;95:73–84. doi: 10.1016/s0165-5728(98)00262-8. [DOI] [PubMed] [Google Scholar]
- 28.Karachunski PI, Ostlie NS, Monfardini C, Conti-Fine BM. Absence of IFN-gamma or IL-12 has different effects on experimental myasthenia gravis in C57BL/6 mice. J Immunol. 2000;164:5236–5244. doi: 10.4049/jimmunol.164.10.5236. [DOI] [PubMed] [Google Scholar]
- 29.Schneiter R, et al. A yeast acetyl-coenzyme A carboxylase mutant links very long chain fatty acid synthesis to structure and function of the nuclear membrane-pore complex. Mol Cell Biol. 1996;16:7161–7172. doi: 10.1128/mcb.16.12.7161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wood SJ, Slater CR. Safety factor at the neuromuscular junction. Prog Neurobiol. 2001;64:393–429. doi: 10.1016/s0301-0082(00)00055-1. [DOI] [PubMed] [Google Scholar]
- 31.Engel AG, Lindstrom JM, Lambert EH, Lennon VA. Ultrastructural localization of the acetylcholine receptors in myasthenia gravis and in its experimental autoimmune model. Neurology. 1977;27:307–315. doi: 10.1212/wnl.27.4.307. [DOI] [PubMed] [Google Scholar]
- 32.Engel, A. 1994. Myasthenic syndromes. In Myology. A. Engel and C. Franzini-Armstrong, editors. McGraw-Hill Professional. New York, New York, USA. 1798–1835.
- 33.Boonyapisit K, Kaminski HJ, Ruff RL. Disorders of neuromuscular junction ion channels. Am J Med. 1999;106:97–113. doi: 10.1016/s0002-9343(98)00374-x. [DOI] [PubMed] [Google Scholar]
- 34.Ubogu U, Kaminski HJ. Preferential involvement of extraocular muscle by myasthenia gravis. Neuroophthalmology. 2001;25:219–228. [Google Scholar]
- 35.Porter JD, et al. Extraocular muscle is defined by a fundamentally distinct gene expression profile. Proc Natl Acad Sci USA. 2001;98:12062–12067. doi: 10.1073/pnas.211257298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matsuo S, et al. In vivoeffects of monoclonal antibodies that functionally inhibit complement regulatory proteins in rats. J Exp Med. 1994;180:1619–1627. doi: 10.1084/jem.180.5.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu C, et al. A critical role for murine complement regulator Crry in fetomaternal tolerance. Science. 2000;287:498–501. doi: 10.1126/science.287.5452.498. [DOI] [PubMed] [Google Scholar]
- 38.Lin F, Emancipator SN, Salant DJ, Medof ME. Decay accelerating factor confers protection against complement-mediated podocyte injury in acute nephrotoxic nephritis. Lab Invest. 2002;82:563–569. doi: 10.1038/labinvest.3780451. [DOI] [PubMed] [Google Scholar]