

Abstract
The gating pathways of mechanosensitive channels of large conductance (MscL) in two bacteria (Mycobacterium tuberculosis and Escherichia coli) are studied using the finite element method. The phenomenological model treats transmembrane helices as elastic rods and the lipid membrane as an elastic sheet of finite thickness; the model is inspired by the crystal structure of MscL. The interactions between various continuum components are derived from molecular-mechanics energy calculations using the CHARMM all-atom force field. Both bacterial MscLs open fully upon in-plane tension in the membrane and the variation of pore diameter with membrane tension is found to be essentially linear. The estimated gating tension is close to the experimental value. The structural variations along the gating pathway are consistent with previous analyses based on structural models with experimental constraints and biased atomistic molecular-dynamics simulations. Upon membrane bending, neither MscL opens substantially, although there is notable and nonmonotonic variation in the pore radius. This emphasizes that the gating behavior of MscL depends critically on the form of the mechanical perturbation and reinforces the idea that the crucial gating parameter is lateral tension in the membrane rather than the curvature of the membrane. Compared to popular all-atom-based techniques such as targeted or steered molecular-dynamics simulations, the finite element method-based continuum-mechanics framework offers a unique alternative to bridge detailed intermolecular interactions and biological processes occurring at large spatial scales and long timescales. It is envisioned that such a hierarchical multiscale framework will find great value in the study of a variety of biological processes involving complex mechanical deformations such as muscle contraction and mechanotransduction.
INTRODUCTION
Many fundamentally important biological processes rely on the mechanical response of biomolecules and their assemblies. A set of well-known examples includes actions that implicate molecular motors (1). For instance, muscle contraction involves the cooperative mechanical response of a large number of myosin molecules, the actin filaments, and the elastic titin assembly; (2,3) cytokinesis also depends critically on the mechanical properties of the cortex (4–6). Another class of remarkable biomechanical processes is mechanosensation (7), which converts mechanical force exerted on the cell membrane into biochemical or electrical signals through cytoskeleton molecules (8,9) and/or mechanosensitive channels (10).
An important aspect of many biomechanical processes is that phenomena on multiple length-scales play a key role. For example, mechanotransduction may involve nanometer-scale conformational changes in one protein but much larger scale (up to μm) variations in the cell membrane or cytoskeleton. Similarly, muscle contraction occurs at the macroscopic scale but originates from nanometer-scale conformational transitions in muscle proteins. Although direct mechanical measurements of single biomolecules or cells are possible (8,11,12), a mechanical testing at multiple length-scales is difficult. Advanced computer simulations that can bridge these length-scales are therefore a powerful technique for exploring fundamental principles associated with the production, transduction, and regulation of mechanical response in biological systems.
The computational study of systems on multiple length-scales and timescales is a significant challenge because it requires the development of a framework and computational model that is sufficiently coarse-grained to treat large length- and timescales while, at the same time, including sufficient detail to faithfully capture the characteristics of the specific system. This is particularly important in biological systems where features on an atomistic scale are crucial to structure and function. The challenge is to develop a framework that complements the traditional all-atom simulations, which are most appropriate for studying nanometer-scale biological processes (13,14), with continuum simulations that can treat large length-scales and timescales.
In this work, we make a useful step toward this direction by establishing a phenomenological continuum-mechanics framework based on the finite element method (FEM) for studying the conformational response of a macromolecule to external mechanical perturbations. The FEM analysis is widely used in the engineering field for solving mechanical and transport problems in systems with complex geometries and boundary conditions, and FEM-based continuum models are therefore more versatile than those based on highly idealized geometries and mechanical properties (15,16). We develop a new FEM framework specifically for the gating behavior of mechanosensitive (MS) channels.
We develop FEM models for a specific type of MS channel from two bacteria, Mycobacterium tuberculosis (Tb) and Escherichia coli (E. coli), and parameterize these models using molecular-mechanics energy calculations. Even with this minimal quasi-atomistic description, it is shown that reasonable gating behaviors are observed for both channels studied, when compared to experimental measurements and previous all-atom MD simulations. The unique versatility of the FEM model is demonstrated by studying the response of the MS channels to two different types of membrane deformations, i.e., in-plane stretching and out-of-the-plane bending. We conclude that FEM-based models have tremendous potential in the study of biological systems, although much work remains to be done.
The mechanosensitive (MS) channels
Although certain processes such as tissue remodeling involve cell adhesion molecules like integrin, most cellular responses to force are due to mechanosensitive (MS) channels (7,10,17). In response to load perturbation applied to the cell membrane or other membrane-associated components, MS channels undergo significant conformational transitions to change their conductive state, which can lead to depolarization or hyperpolarization, Ca2+ entry with a subsequent cascade of intracellular biochemical events, or release of osmolytes. Mechanosensitive channels have been identified in more than 30 cell types (7), and abnormality in their functions may contribute to serious health problems such as neuronal degeneration, hypertension, and glaucoma.
Despite extensive research efforts over the last few decades (7,10,18–20), detailed molecular mechanisms by which MS channels sense and convert the mechanical deformation into biological signals remain unclear. The challenge is evident considering the diverse forms of mechanical stimuli potentially exerted to cells (1,7), which include steady-state contacts, high-frequency vibrations, osmotic pressure gradients, hemodynamic pressure, and fluid-shear stresses. All these external stimuli are present in the background of internally generated forces such as those arising from hydrostatic pressure and cytoskeletal polymerization. Thus the mechanotransduction pathways have to be designed through evolution to filter out irrelevant ones while extracting specifically relevant stimulus. In certain cases, the function of MS channels involve intracellular cytoskeleton and molecular motors (8,9), while many MS channels also function only through interacting with the membrane (7,10,18,21); the specific system that we focus on here, the MS channel of large conductance (MscL) in bacteria, belongs to the latter class.
Overall, how different modes of membrane deformation (see discussion below and Fig. 1) regulate the function and sensitivity of MS channels is poorly understood (7,19,22,23). Major challenge arises for both experiment and simulation due to the involvement of multiple length- and temporal scales in mechanotransduction; e.g., it is technically difficult to experimentally characterize the conformational changes in both the protein and the membrane at the quantitative level. The relationship between the channel opening probability and membrane tension was studied by Sukharev et al. (24) using the patch-clamp technique on MscL in E. coli. A simple five-subconductance-states model was established, which showed that the tension-dependent conformational transition is primarily attributed to the pore-area variation that occurs between the closed state and the open state. The structural rearrangements in the MscL in E. coli have been proposed by Sukharev et al. (25,26) based on Cys cross-linking experiments, and were confirmed by Perozo et al. (19,23,27) using electron paramagnetic resonance spectroscopy (EPR) and site-directed spin labeling, which suggested a plausible molecular mechanisms of gating in MscL.
FIGURE 1.

The four basic deformation modes of a membrane: (a) tension, (b) shear, (c) torsion, and (d) bending. Both tension and bending are investigated in this work.
Previous analytical and simulation studies of MS channels: insights and limitations
Several groups have attempted to develop analytical models for the gating transition in MscL. By considering possible deformation mechanisms (e.g., membrane tension and bending), Markin and Sachs (28) presented a general formulation for the thermodynamics of mechanotransduction, which relates the probability of the channel opening to membrane properties such as stiffness, thickness, and curvature. Wiggins and Phillips (29) developed an analytic model to characterize the free energy of the protein-bilayer system and suggested that the competition of hydrophobic mismatch could be a physical mechanism that governs gating. This model was further improved by adding triggers (besides the channel radius change) in the transition from the closed to the open state (30). Finally, Turner and Sens (31) proposed a gating-by-tilt model as an alternative to dilatational gating, where the gate opening is due to the swinging of the lipids near the channel with respect to a pivot. These theoretical models provide insight into common features in MS channels. They lack sufficient structural detail, however, and their validity to any specific system is difficult to judge.
There have also been simulation studies of various MS channels using different approaches. Most simulations have relied on the single MS channel x-ray structure for an MscL from Mycobacterium tuberculosis (Tb) solved by Chang et al. (32), which is generally believed to mimic the fully closed state of the channel (33,34). As shown in Fig. 2, the Tb-MscL is a homopentamer with each monomer containing two transmembrane helices (TM1,TM2) and a cytoplasmic helix. By retaining the main structural features of the Tb-MscL and taking related experimental data as constraints, Sukharev et al. (25,26) developed a series of structural models for the gating transition in both Tb-MscL and E. coli-MscL; 13 conformational states were constructed that ranged from the most closed state to an open conformation in which the narrowest part of the pore has a diameter of ∼36 Å. In an attempt to construct a gating pathway with more detailed energetic considerations, Gulligsrud et al. (35) carried out all-atom MD simulations for Tb-MscL with explicit solvent and lipid molecules. During the rather short (∼3 ns) simulation, however, the lipid membrane maintained a constant volume well before the conformation of MscL could be affected, and the increase in the pore radius h, with a surface tension of 60 dyne/cm, was merely 4 Å. In a subsequent steered MD simulation (20), the gating (closed→open) transition in E. coli-MscL was studied by the same authors using the structural models of Sukharev et al. (26). Instead of simulating the stretched bilayer explicitly, the lateral and normal pressure profiles exerted by the deformed bilayer (due to equi-biaxial tension) on the protein was estimated and then applied to the protein atoms in the form of an external steering force. Unfortunately, even with such a bias, after 12 ns of simulation the channel opened to a pore radius of only 9.4 Å, which is significantly smaller than the fully opened state proposed by Sukharev et al. (25). In a complementary study, Kong et al. (36) applied target molecular dynamics (TMD) (37) to study the same gating transition. Since TMD applies a holonomic constraint during the simulation, reach of the final target is guaranteed; the constraining force on the protein atoms in this type of simulation, however, can be extremely large compared to the realistic gating force exerted by the deformed membrane, which makes TMD simulations useful as a qualitative structural biology tool but inappropriate for the purpose of analyzing the membrane-mediated gating mechanism. In fact, the lipid membrane was entirely ignored in the TMD study (36). Colombo et al. (38) have also studied the gating process using equilibrium MD simulations with different pressure conditions. The tilting of a subset of transmembrane helices was observed as a consequence of the applied lateral tension, although a full opening was not observed due to the nanosecond timescale of the simulations. Even though the gating/steering forces are sometimes unrealistic, these atomistic simulations yield helpful insights on the mechanisms of mechanotransduction and they will be compared with the FEM model in this study.
FIGURE 2.

Structural models and FEM models of the Tb and E. coli MscL channels. (a,b) Top and side views of the structural model of Tb. (c,d) Top and side views of the FEM model of Tb. (e,f) Top and side views of the structural model of E. coli. (g,h) Top and side views of the FEM model of E. coli. In the FEM model, the TM1 helices are in orange and TM2 helices are in blue.
A continuum-mechanics model with the finite element representation
It is widely acknowledged that the short timescales (∼10–100 ns) accessible to all-atom simulations (with explicit solvent and lipid molecules) hamper the possibility of observing the gating transition in MscL under realistic conditions. A less emphasized fact is that the conformational response of both model lipid vesicles and cells to mechanical stimulation can be rather complex, and it is difficult to introduce complex membrane deformations in typical nanometer-scale all-atom simulations. As shown in Fig. 1, any membrane deformation can be decomposed into a combination of several basic deformation modes: stretching (or dilatation and thinning), shearing, bending, and twisting. The first two are in-plane distortions and the last two are out-of-plane distortions. The out-of-plane modes are capable of initiating both stretching and shearing stresses, which lead to complicated stress fields that may result in different gating thresholds in MS channels. In addition, deformation due to osmotic pressure variation inside the membrane will be superimposed onto those caused by external forces. Therefore, mechanotransduction through MS channels is likely to be sensitive to both intrinsic (e.g., membrane curvature, thickness, osmotic pressure, vesicle bilayer structure, and material property) and extrinsic (e.g., types of loads, mechanochemical environment) factors, and both need to be considered explicitly in a simulation. Most importantly, the local stress-strain field surrounding a MS channel is inevitably governed by the remote mechanical load/stimuli applied to the entire system. The remote load can be complex (such as adhesion between cells), leading to intricate local deformation fields. Even in cases where the remote load is relatively simple, due to the large rotation caused by the flexible shell-like geometry and orientation of each individual cell, the local deformation is still a complicated combination of the four basic deformation modes described above. To conduct a complete investigation and to simulate mechanotransduction in a more realistic manner, it is critical to understand how MS channels respond to different types of local stress-strain field, and how such local deformation is derived from remotely applied loads. This is clearly beyond the capability of conventional MD simulation techniques.
Ideally, a concurrent multiscale model that treats the lipid and solvent molecules near the protein differently from those far away is most suited to study the hierarchical mechanochemical coupling phenomenon. Although such an idea has been pursued effectively in the context of hybrid quantum mechanical and classical mechanical simulations (39–41) as well as in the hybrid atomic/continuum framework for material simulations (42), developing the appropriate boundary condition for soft-matter systems is not a trivial task (43). Instead, inspired by the successful applications of simple continuum mechanics models in molecular motor studies (15,16,44,45), we establish a new phenomenological continuum framework for studying MS channel gating using the FEM analysis, which is a technique that finds a broad range of applications in engineering. The major difference compared to previous mechanical models of biomolecules is that the FEM model can adopt complex geometry and highly heterogeneous mechanical properties specific to the system under study; the parameterization process for the FEM model makes a natural connection between continuum mechanics and previous all-atom simulations. In the current work, as a proof of concept, we develop rather simple FEM models for both the Tb- and E. coli-MscLs, which include only the transmembrane helices as homogeneous elastic rods embedded into an elastic membrane. Nevertheless, simulations using these simple models generate gating transitions that are remarkably consistent with available experimental data and all-atom simulations. More importantly, it is straightforward to incorporate different deformation modes into the elastic membrane and to study the corresponding response of the MscL; here we only illustrate this point with equi-biaxial membrane tension and axisymmetric bending.
THEORY AND METHODS
In this section, we first present the finite element model and the relevant parameterization procedures, and then describe the detailed simulation protocols for studying the mechanical response of the MscL channels. Here we restrict ourselves to comparing them to in vitro experimental studies of MscL in lipid vesicles, which means that the model includes a single MscL in a homogeneous lipid bilayer.
The finite element (FEM) model
We restrict ourselves to a minimalist mechanical model that includes only the transmembrane domain of the MscL; it is commonly assumed that the gating behavior is largely determined by the interaction between the transmembrane domain and the surrounding membrane (17,23,33,46). Although the cytoplasmic helices and the loops that connect TM1 and TM2 helices (Fig. 2) may also play an important role (26,47,48), they were ignored in the present proof-of-concept model; they can be included in a straightforward manner in a more complete model. For simplicity, the TM1 and TM2 helices are treated as homogeneous elastic rods (see below for details) and are embedded into a homogeneous elastic membrane (49). The model is parameterized using molecular-mechanics energy calculations. Although hydration of polar groups upon channel opening has been proposed to make an important contribution to the gating process (22), to be consistent with the simple description of the helices, solvent molecules are not included.
The helices
The structural and FEM models for the Tb and E. coli MscL channels are shown in Fig. 2. The five TM1 helices form the inner boundary for the pore with limited contact with the lipid while the shorter TM2 helices form the outer boundary that interact extensively with the lipid membrane. The TM1 and TM2 helix bundles share the same fivefold symmetry axis, denoted as the z axis here, which is also the direction of the membrane norm. In Tb–MscL, the TM1 helix contains residues Val-15–Thr-40, and TM2 includes residues Val-71–Val-90; more precisely, the first few residues of TM1 adopt 310 rather than α-helix. In the E. coli MscL, both helices are longer; TM1 and TM2 helices correspond to residues Asn-15–Gly-50 and Val-77–Glu-107, respectively. We note that there is a break in TM1 due to Pro-43, which (for simplicity) is not taken into account in our model. Within the continuum-mechanics framework, each helix is modeled as a cylindrical elastic rod of 5 Å diameter with spherical caps at both ends (see Fig. 2, c and d, and Fig. 2, g and h, for the top/side views of Tb and E. coli models, respectively); spherical caps were employed to ensure a smooth surface of the elastic rod and to obtain converged numerical results (see below). (Special Note: The diameter of 5 Å was chosen based on the main-chain structure of an ideal α-helix. The volume of side chain is not considered explicitly, which is not expected to be a major problem in the context of the current work; the effect was included implicitly through nonbonded parameters between helices.) Since the elastic rods are assumed to be homogeneous in this work, the only system-specific (Tb versus E. coli) property is the geometry of the TM1 (orange) and TM2 (blue) helices, including length, radius, and orientation. Under this assumption, the mechanical property of the elastic rod is homogeneous and isotropic, and the only relevant constitutive parameters are the Young's modulus and the Poisson's ratio, values of which are taken from the previous MD study of Sun et al. (51). They found that the mechanical properties of α-helices are not strong functions of the sequence; the effective Young's moduli of various helices were found to be in the range of 60–180 GPa. In this study, the Young's modulus of TM1 and TM2 helices is taken to be 100 GPa and a Poisson's ratio of 0.3 is adopted; these are tabulated along with the relevant geometrical parameters of the helices in Table 1. Under external loads, these elastic rods may stretch, bend and/or twist, and they interact with each other as well as with the lipid membrane as elaborated below.
TABLE 1.
Material properties of MscL transmembrane helices and lipid membrane used in the finite element model
| Transmembrane
|
Helices
|
||||
|---|---|---|---|---|---|
| Properties | Lipid membrane | Tb TM1 | MscL TM2 | E. coli TM1 | MscL TM2 |
| Length/diameter* (Å) | 400.0 | 42.0 | 29.0 | 55.6 | 45.6 |
| Thickness/diameter† (Å) | 35.0 | 5.0 | 5.0 | 5.0 | 5.0 |
| Young's modulus E (GPa) | 0.1 | 100.0 | 100.0 | 100.0 | 100.0 |
| Poisson's ratio ν | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 |
The lengths of helices for Tb-MscL are based on the x-ray structure (PDB code 1MSL); those for E. coli-MscL are based on the structural model of Sukharev et al. (26).
The length is for the square lipid sheet used for stretching (Fig. 3 a); the diameter is for the spherical lipid sheet used for bending (Fig. 3 b).
The thickness is for the lipid membrane; the diameter is for the helices.
The lipid bilayer membrane
The lipid bilayer is modeled as an elastic sheet of thickness 35 Å, which is close to that of a dipalmitoylphosphatidylcholine (DPPC) bilayer and spans the transmembrane domains of the Tb x-ray structure (32). No distinction is made between the hydrophobic regions and the polar heads. For the simplest case of in-plane membrane stretching, a flat square membrane (within the x,y plane) with a size of 400 × 400Å is employed (Fig. 3 a). The equi-biaxial membrane tension is most likely induced by osmotic pressure: assuming the cell is spherical with a typical diameter of 1 μm, a patch of membrane with the size of 400 × 400 Å corresponds to a center angle of ∼5°, which suggests that the curvature of the patch is negligible.
FIGURE 3.

(a) Schematic for the equi-biaxial tension of the lipid membrane, and a zoomed-in view of the 10-petal lipid hole that encompasses the protein. (b) Schematic for the axisymmetric bending deformation of the lipid membrane. The bending is realized by a four-point bending flexure. (c) The finite element mesh for membrane and protein (E. coli-MscL) during equi-biaxial tension.
The lipid membrane is modeled as homogeneous and isotropic and the Young's modulus is estimated from the area compressibility. The mechanical failure of the lipid membrane is not considered although this might be important in some patch-clamp studies (24). The elastic properties of the lipid bilayer are assumed to be pressure-independent and estimated as follows. The stress-strain relationship of a thin membrane subjecting to equi-biaxial tension is σ = Eε/(1 – ν), where E is the Young's modulus, ν is the Poisson's ratio (which is ∼0.3 for most materials) (52), ε is the in-plane membrane strain, and σ is the in-plane tensile stress. The value σ can be estimated from the surface tension, γ, which in turn can be obtained from the bilayer area compressibility, Ka. In particular, σ = γ/h, where h is the thickness of the membrane (20,53,54) and, under equi-biaxial tension (since the normal pressure is always much smaller than the lateral pressure (20,53,54)), γ = KaΔA/A (53), where A is the undeformed membrane area and ΔA is the area increment. For small deformations, ΔA/A = (1 + ε)2 – 1 ∼ 2ε, and therefore E = (1 − ν)Ka2ε/hε = 2(1 – ν)Ka/h. Based on MD simulations (53) and experiments (54) on the stretching of DPPC membrane at ambient temperature, Ka is ∼230–350 dyne/cm, which results in a Young's modulus of ∼92–140 MPa for the DPPC membrane. Therefore the Young's modulus of the lipid is taken to be 100 MPa in this study.
To embed the channel into the continuum membrane, a cavity (hole) with the shape of a 10-petal flower is created in the elastic sheet (Fig. 3 a), with the size and shape of the cavity determined using the geometrical parameters of the elastic rods (Table 1) and the corresponding structure of the closed channel. The specific shape of the hole conforms to the embedded protein (e.g., Tb or E. coli MscL), such that the initial distance between the surface of TM1 (or TM2) helices and the surface of lipid cavity is set to be their equilibrium distance of ∼5.5 Å, which was measured from the trajectories in previous all-atom simulations (20) (based on main-chain atoms in the helices to lipid molecules, to be consistent with the helical radius used; see Special Note in text above). The interactions between the lipid and helices are described in the next subsection. Calculations were performed to verify that the system maintained mechanical equilibrium in the absence of external force.
Interaction among continuum components
The nonbonded interaction among the transmembrane helices, and that between helix and lipid, are represented by a simple pairwise effective potential of the familiar Lennard-Jones form,
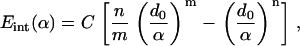 |
(1) |
where Eint(α) is the effective interaction (per surface area) between the surfaces of two continuum components. For any given pair of interactions, d0 is the equilibrium distance between the two surfaces, and α is the distance between the two surfaces. Taking the first derivative of Eint with respect to α leads to the pressure-distance relationship between two surfaces (adopting the sign convention that repulsive pressure is positive),
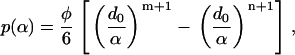 |
(2) |
where p is the interaction pressure between two surfaces, and φ = 6Cn/d0. This nonbonded interaction model has been successfully applied to study the deformation and buckling of double-walled carbon nanotubes (55) and radial elastic properties of multiwalled carbon nanotubes (56) as well as nano-indentation of nanotubes (57). In this study, nonbonded interaction pairs are included between the lipid hole and each TM1 helix, between the lipid hole and each TM2 helix, between each pair of TM1 helices, and between each TM1 helix and each TM2 helix. The interactions between the TM2 helices were neglected because they are far apart (see Fig. 2). Based on the structural models for the closed state, d0 for each interaction pair is taken as the shortest distance between the surfaces of the two corresponding continuum components. For instance, there are 10 interaction pairs among TM1 helices, which lead to one unique d0 value based on the shortest distance between TM1 helices in the closed-state structure. For other types of pairwise combinations, i.e., lipid–TM1, lipid–TM2, and TM1–TM2, d0 takes on different values (see Table 2). Since the Tb and E. coli MscLs have different structures, the values of d0s are also different.
TABLE 2.
Parameters for the nonbonded interactions between helices and between helix and lipid membrane
| Interaction pair | Tb d0 (Å) | MscL φ (GPa) | m | n | E. coli d0 (Å) | MscL φ (GPa) | m | n |
|---|---|---|---|---|---|---|---|---|
| Lipid–TM1 | 5.5 | 2.0 | 9 | 3 | 5.5 | 2.0 | 9 | 3 |
| Lipid–TM2 | 5.5 | 2.0 | 7 | 3 | 5.5 | 2.0 | 7 | 3 |
| TM1–TM1 | 0.5 | 7.0 | 2 | 1 | 1.5 | 3.3 | 2 | 1 |
| TM1–TM2 | 2.5 | 3.0 | 9 | 3 | 5.0 | 1.7 | 9 | 3 |
The interactions between TM2 helices are ignored because they are generally far apart (>17 Å). The value d0 is measured based on the atomic structures using a diameter of 5 Å for all helices.
The well-depth, C, and the exponents (n, m) are determined from molecular-mechanics calculations using the program CHARMM (58). For each pair of helices (TM1–TM1 or TM1–TM2), the interaction energy in the vacuum is calculated using the polar-hydrogen set of CHARMM force field (CHARMM 19 (58,59)) with the internal structure of the helices fixed to that in the x-ray structure (for Tb) or the homology model (for E. coli). The calculations are done for different combinations of helix pairs, which effectively sample different relative orientations; for each pair, the position of one helix varies between −20 Å and 20 Å along the direction of the center-of-mass separation vector projected onto the membrane. The nonbonded (electrostatic and van der Waals) interactions are calculated without a cutoff. To estimate the helix-lipid interactions, the insertion energy profile of a single helix (TM1 or TM2) is calculated with an implicit membrane model; i.e., the helix is gradually transferred from the implicit membrane to the implicit bulk solution along the membrane norm. Several implicit membrane models available in CHARMM including the EEF1 (60), GBIM (61), and GBSW (62) models are tested and the results are rather similar at the semiquantitative level; to be consistent with the parameterization of these models, the CHARMM 19 force field (59) is used for EEF1 (with the corresponding cutoff scheme) and GBIM, while the CHARMM 22 set (63) is used for GBSW. The EEF1 results are used in the model parameterization. In these implicit membrane calculations, the membrane thickness is taken to be 23.5 Å, which corresponds to the hydrophobic part of the membrane. The implicit membrane model is used to avoid the need to sample a large set of lipid configurations, although this can be done for a more sophisticated parameterization (see Future Directions).
To illustrate the fitted parameters, the nonbonded interaction energies (per unit area) between a pair of nearest TM1–TM2 helices and those between a pair of nearest TM1–TM1 helices of E. coli-MscL are shown in Fig. 4, as a function of the normalized separation between the two components (i.e., the deformed distance between the two surfaces normalized by d0). The symbols indicate data computed from molecular mechanics (MM) calculations and the lines are fits based on Eq. 1 with the parameters listed in Table 2. A number of representative nonbonded interaction curves obtained from MM analyses are shown, which correspond to the configurations (orientations) of helices in the close, intermediate, and open models of Sukharev et al. (26). Although the parameters are fitted based on the intermediate structural state only, the agreement between FEM and MM results is rather good for other structural states as well, which indicates that the parameters are fairly transferable.
FIGURE 4.

The fitting of nonbonded interaction between helices of E. coli-MscL. The x axis is the normalized separation between the helices (with 1.0 being the equilibrium spacing). The parameters (shown in Table 2) were fitted based on molecular mechanics (MM) calculations for the helical pairs in the intermediate structural model in Sukharev et al. (26). This set of parameters is fairly transferable to other structural states, as shown by the comparison between FEM and MM calculations for helical pairs in the open and closed structural states.
Finite element analysis and simulation procedures
Theoretical estimates of stress and strain
The averaged radius of the cavity (hole) is measured to be ∼22 Å in the closed state, which is much smaller than the size of the membrane (∼400 Å) and suggests that the deformation of the hole is dominated by the external load while the effect of the channel is much smaller (since most of the strain energy is stored in the membrane). This allows an analytical analysis of membrane deformation under mechanical stress and estimates for the magnitude of tension and bending moments to be used in the FEM simulations.
For a flat membrane containing a circular hole, when the membrane is under equi-biaxial in-plane tension, the increment of the hole radius can be derived based on plane stress elasticity theory (52). The closed form solution of an annulus (with an outer radius b and inner radius a) under uniform boundary pressure is
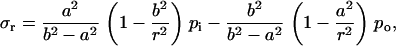 |
(3) |
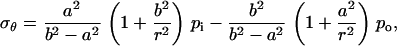 |
(4) |
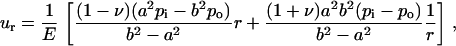 |
(5) |
where r is the radial distance from the center of the hole, ur is the radial displacement, and σr and σθ are the radial and hoop stress components, respectively, and po and pi are the pressures applied on the outer and inner boundaries, respectively. Since the hole is much smaller than the membrane span, then a ≪ b. Moreover, and since the nonbonded interaction between the helix bundle and lipid bilayer is small compared with the external load, then pi ≪ po. These considerations lead to a set of simplified solutions:
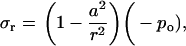 |
(6) |
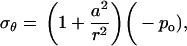 |
(7) |
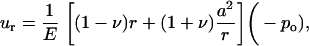 |
(8) |
where −po = σ is the equi-biaxial membrane tensile stress. Accordingly, the increment in the hole inner radius is
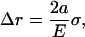 |
(9) |
which provides an upper bound for the channel pore in the open state. Using this result, if the desired increment in the lipid hole diameter is ∼30 Å (an estimate compatible with the fully opened MscL, see below), with a = 22 Å and E = 100 MPa, the required tension for fully opening the channel is ∼35 MPa. The strain in the direction normal to the membrane is
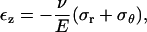 |
(10) |
which leads to a reduction in the membrane thickness of
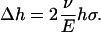 |
(11) |
With the membrane tension of 35 MPa, the thickness is predicted to reduce from 35 Å to ∼27 Å, which is an ∼24% change. This estimate is in close agreement with the 20% reduction in the thickness of membrane spanning part of MscL measured by Perozo et al. (27), which suggests that the current continuum model is at least qualitatively correct. This analysis also suggests that the membrane deformation is dominated by the external load applied to the lipid bilayer, i.e., the lipid-helix interactions (discussed below) play a minor role in lipid deformation, although these interactions are clearly crucial to the gating behavior of MscL (64).
For axisymmetric bending, a four-point bend flexure of a circular membrane is employed as sketched in Fig. 3 b. The normal of the membrane is z axis and a cylindrical coordinate system (z, r, θ) is employed. The outer radius of the membrane is 250 Å, and the 10-petal flower-shaped cavity, whose detailed geometry depends on the embedded protein, is located at the center. A circular ring of roller with a radius of 200 Å is placed on top of the membrane, and a uniform line load is imposed on the outer rim to bend the circular disk upwards. At equilibrium, the resulting bending moment within the roller is a constant (denoted by Mo); thus, this flexure simulates the axisymmetric pure bending of the lipid membrane. The amount of load needed for gating can be estimated from elastic plate theory (65). For a circular plate with an outer radius b (200 Å in the present case) containing a circular hole of radius a (∼22 Å for both Tb and E. coli MscLs), the radial and circumferential bending moments per unit length are, respectively,
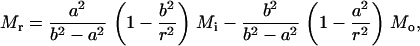 |
(12) |
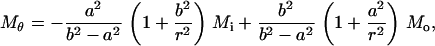 |
(13) |
where Mi and Mo are the uniformly distributed bending moments applied on the inner and outer boundaries of the annulus, respectively. Moreover, the deflection of the membrane (which is counted negative) can be solved as
 |
(14) |
where D = Eh3/[12(1 – ν2)] and h is the membrane thickness. Once again, we assume that the interaction moment between helices and lipid is much smaller than the external bending moment (i.e., Mi < < Mo); when a ≪ b, we have
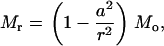 |
(15) |
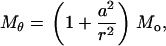 |
(16) |
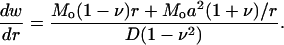 |
(17) |
The curvature along the r-direction is
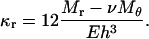 |
(18) |
Thus, the membrane curvature at the cavity is
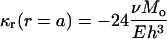 |
(19) |
and the angle of rotation in the cavity wall is
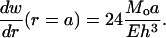 |
(20) |
Based on the structural models of E. coli-MscL (Fig. 2 h), the tilting angle of the TM1 helices in the closed state is ∼10°. Upon axisymmetric bending, if the cavity wall rotates by the same angle in the opposite direction the helices may become upright and gating-by-tilt (31) becomes possible. With h = 35 Å, the distributed bending moment Mo needed for gating is estimated to be ∼14.3 pN; i.e., the total bending moment exerted on the circular membrane with a radius of 200 Å is ∼1800 pN·nm. For Tb-MscL, the initial tilting angle of TM1 is almost twice as large (Fig. 2 d), leading to a doubled gating moment.
Simulation protocols
Four-node tetrahedron finite elements are used to mesh the helices and the membrane (Fig. 3 c) with the commercial package ABAQUS (66). Each helix contains ∼1800 nodes and ∼7000 elements, with all nodes roughly equally spaced. The lipid bilayer consists of ∼23,000 nodes and ∼118,000 elements, with the mesh more refined toward the boundary of the inner hole. Finite element calculations are performed using ABAQUS (66) with the option for finite deformation and strain employed. The nonbonded interactions described above are implemented as a user interaction subroutine (UINTER), which enables the user to specify the constitutive interactions between a master and a slave surface. Within the current formulation, both master and slave surfaces contribute equally in the interaction algorithm. The subroutine is called for each slave node at each time-increment during the numerical analysis to compute the tractions at these nodes based on their relative positions with respect to the master surface. The normal tractions (i.e., p in Eq. 2) can be either positive or negative, which indicate surface repulsion and attraction, respectively. In addition to computing the nodal traction, the UINTER subroutine was used to calculate the corresponding Jacobian to help accelerate convergence of the computations. The typical computational time for a gating process (under equi-biaxial tension) is ∼3 h on a Dell workstation with 3.2 GHz Intel Xeon CPU and 2 Gb of memory.
Since the membrane deformation is dominated by the external load, this allows us to simplify the gating simulation. We divide the simulation into two stages: the first focuses on the membrane deformation due to the external load, and the second focuses on the channel conformational transition due to the displacement field of the lipid-helix interface. We illustrate this two-stage procedure for the case of equi-biaxial membrane tension below.
During the first stage, the protein is not included and the membrane, with an embedded cavity (hole), is stretched by applying equi-biaxial displacement on its outer boundary (Fig. 3 a). The displacement components are applied uniformly on all finite element nodes on the four external edges, as illustrated in Fig. 3 c, and rigid body motion is eliminated during this process. The quasi-stretching process is divided into 20 steps to a maximum membrane tension of σ = 35 MPa, which is our estimate for what is needed to fully open the channel. At the end of each step, the nodal displacements of the inner hole surface are recorded and transferred to the next stage. The reaction forces acting on the external boundary nodes are also calculated, from which the membrane tension (stress) is calculated as a function of the prescribed displacement.
During the second stage, only the protein deformation is followed explicitly. In each step, the displacement field of the lipid hole surface recorded in the first stage is employed as a displacement boundary condition, and the nonbonded interactions between the lipid and the helix bundle gradually pull the channel open. There are multiple increments within each step, and the MscL structure is updated after each increment. The simulation is advanced by explicit time integration, although the timescale in such quasi-static simulations (i.e., the MscL configuration was at mechanical equilibrium at a fixed load) does not correspond to the physical timescale associated with the gating process, which is on the order of 10–30 μs (17,67).
Similarly, for axisymmetric bending, a total bending moment of 1800 (3600) pN·nm is exerted on the circular membrane for E. coli (Tb) in the first stage, where the sequential deformed profiles of the lipid hole are computed during the explicit-lipid simulation. The results are then imposed as the displacement boundary condition in the second stage, which governs the conformational transition of the channel.
RESULTS AND DISCUSSION
In this section, we first describe and compare the gating behaviors of MscL from Tb and E. coli in response to membrane stretch (tension), and then present similar analysis for the gating response to membrane bending.
Gating of MscL in response to membrane stretch
The gating pathway of MscL
The structural variations during the gating transition observed in the FEM model (at different fractional times) are in qualitative agreement with the structural models developed by Sukharev et al. (26) based on experimental constraints. Figs. 5 and 6 depict the gating pathways predicted by these two approaches for Tb-MscL and E. coli-MscL, respectively. As the membrane tension is increased, the lipid hole expands and, through the nonbonded attraction, the TM2 bundle expands radially. This is clearly seen in the top view. As a consequence of the TM2–TM1 and lipid–TM1 interactions, the TM1 bundle follows the changes in the lipid and TM2 helices, which leads to increase in the pore size. The arrangement of the helices can be examined from the side view in Figs. 5 and 6. In the undeformed configuration of Tb-MscL (Fig. 5), the projection of the transmembrane helix bundle in the direction of the fivefold symmetry axis (the z-axis) is roughly equal to the membrane thickness. As the membrane tension is increased the helix bundles tilt, and the projected length of the helix bundle is shortened by ∼30% when fully opened. The helix titling is largely due to the reduction in the membrane thickness during dilatation. In E. coli-MscL (Fig. 6), the helix tilting is more striking than in Tb-MscL: in the closed state, part of the TM1 and TM2 helices protrudes out of the lipid bilayer, but with increasing membrane tension the projection of the helix bundle on the z axis becomes shorter than the membrane thickness. For both MscLs, the tilting of TM1 helix bundle is more significant than that of the TM2 helix bundle. At the quantitative level, the TM1 titling angle predicted from the FEM model agrees well with the structural models derived based on experimental constraints for E. coli (Fig. 6 c) (26). In our model, the helices are highly elastic and flexible, and they are significantly stretched and bent to maintain mechanical equilibrium during the gating process; this has not been shown in any experimental studies so far due to the limitation in resolution.
FIGURE 5.

Comparison between gating pathways of Tb-MscL under equi-biaxial tension: (a) the structural model in Sukharev et al. (26) and (b) the present FEM model. The x axis of the FEM model is a virtual time-variable, with 0.0 being the closed state (with an effective radius of 6.5 Å) and 1.0 being the fully opened state (with an effective radius of ∼20 Å (24)); the TM1 helices are in orange and TM2 helices are in blue). The dashed lines indicate the approximate location of membrane/water interface.
FIGURE 6.

Comparison between gating pathways of E. coli-MscL under equi-biaxial tension: (a) the structural model in Sukharev et al. (26) and (b) the present FEM model. The TM1 helices are in orange and TM2 helices are in blue. (c) Comparison of the TM1 helix tilting angle between the structural model and FEM model. The dashed lines indicate the approximate location of membrane/water interface. Note that both TM1 and TM2 helices bend significantly in the FEM model; whether these structural changes are realistic remains to be clarified with further investigation. The model developed in Sukharev et al. (26) also indicates some degree of bending of these helices, which is not apparent in the rod representation used here; note that TM1 has an intrinsic break at Pro-43.
Evolution of the pore radius
A critical parameter in the context of studying MS channel gating is the size of the channel pore. When only the transmembrane helices are considered, the five TM1 helices form a pore (see Figs. 5 and 6) with a pentagon-shaped projection onto the x,y (membrane) plane. The area enclosed by the pentagon can be calculated based on the FEM analysis, and we define an effective pore radius as the radius of a circle with the same area as this hexagon-shaped pore. We define the effective radial strain (Fig. 7 b) as the increase of the effective pore radius normalized by that in the undeformed state. For the purpose of making qualitative comparisons with previous simulation studies (e.g., steered or targeted MD, see below), a fractional channel radius (Fig. 7 a) is also defined as the unitized ratio of the effective radial strain, with 0.0 denoting the fully closed state and 1.0 denoting the maximum opening in a specific simulation, regardless of the actual value of the maximum channel radius computed in that simulation.
FIGURE 7.

Results for equi-biaxial tension. (a) The comparison between the fractional channel radii computed from the present FEM model and those from the steered (20) and targeted (36) MD simulations. (b) The comparison between the effective radial strain versus membrane strain of the FEM model and that from the steered MD simulation.
In the FEM simulation, the pore radius increases monotonically with increasing membrane tension, as can be seen in Fig. 7 a where the fractional (virtual) time is proportional to the membrane stress. Although there are oscillatory features due to the equilibrium iterations of many-body interactions in the FEM model, the overall trend is close to a linear behavior. Recall that in the continuum model, the relationship between membrane tension and the lipid hole radius is strictly linear (Eq. 9). Thus, the results show that the evolution of the pore radius and the membrane hole are tightly coupled. Note that the increment of pore radius and radial strain have stepwise features, which are related to the channel stretching-relaxing cycles as the helices iterate to their equilibrium positions. Interestingly, such features were also found in all-atom simulations (see below).
We note that the maximum load of 35 MPa imposed in the FEM simulation might be slightly excessive and this indicates that the evolution of the pore, which is enclosed by the five TM1 helices, does not quite follow that of the lipid hole (in our simulations). The maximum pore radius in the FEM model is ∼10–20% larger than in the structural models (26) and good agreement between the fully opened states is found at the fractional time (in the FEM simulation) of t = 0.9 for E. coli and t = 0.8 for Tb. When the lipid is under tension, the deformed lipid hole surface is kept normal to the flat surface of the membrane, whose opening radius can be determined as a function of stress (Eq. 9) and this relationship is used to estimate the load needed to gate the MscL. However, the actual radius of MscL is governed by the five tilted TM1 helices enclosing the channel. It is the tilting angle that accounts for the difference between the MscL pore radius and lipid hole radius. Nevertheless, the closed form solution in Theoretical Estimates of Stress and Strain provides a good estimate for the gating process (see below) and it is straightforward to increase or decrease the tension stress σ in the FEM model to achieve a desired maximal pore radius.
Comparison with experiment and previous simulations
The FEM simulations for the membrane strain required for gating are in good agreement with experimental data. In the FEM simulations, a complete opening of MscL is achieved when the applied membrane tension is ∼28 MPa and 32 MPa for Tb- and E. coli-MscLs, respectively, which corresponds to a dimensionless membrane strain of ε = σ(1 – ν)/E = 22% for the full gating of E. coli-MscL. The corresponding strain for 50% probability for gating in the patch-clamp experiments can be estimated (see below) to be 13%, which is in good agreement with the prediction of the current work considering the simplicity of the model.
The strain in the patch-clamp experiments of Sukharev et al. (24) on the E. coli MscL can be estimated from the membrane tension and the Young's modulus. The membrane tension has been measured, and is ∼3.4 MPa. The Young's modulus used in the patch-clamp experiment can be estimated as follows: the radius of the pipette was c ∼3.2 μm; with a constant pressure of pp = 42 mmHg, a circular patch of membrane that was initially flat became a bulged spherical cap with height w ∼1.5 μm. In the bulge test, the pressure is related with the deflection by pp = 8Ehw3/3c4 (68), from which E ∼ 18 MPa. Therefore, the membrane strain encountered in the patch-clamp experiment is about ε ∼ 13%.
The monotonic behavior found for the pore radius as a function of membrane tension suggests that the effective energy surface is downhill toward the open state in the presence of tension. This is inconsistent with the energy profile estimated in Sukharev et al. (24), which involves five states (closed, open, and three intermediates) separated by sizable barriers, even in the presence of tension. To capture the realistic behavior of the channel with such an energy landscape, more refined continuum mechanics models need to be developed and the effect of temperature (thermal fluctuation) also needs to be considered.
The evolution of the pore radius during the gating process as found in the FEM simulations is comparable to that in all-atom MD studies (20,36). As shown in Fig. 7 a, once the simulation time and pore radii in the steered (20) and targeted MD (36) simulations are converted into the fractional time and the fractional channel radius with respect to the maximum degree of channel opening in these simulations, the behavior is very similar in the three very different studies. The fractional channel radius varies essentially linearly with the fractional time; the zigzag feature is present in both FEM and steered MD simulations although the magnitude is much larger in the latter, which might be due, in part, to the larger scaling factor used for steered MD simulations (where the channel opening is much smaller) in Fig. 7 a. The evolution of the pore size is almost monotonic in the targeted MD simulation, which could be due to the strong monotonic constraint (though in RMSD, not in the pore radius per se) used in such simulations (37).
Regarding the effective radial strain as a function of the membrane strain, a quantitative comparison can be made only between FEM and the steered MD because no lipid was present in the targeted MD study. In the steered MD study, the lipid used by Schulten et al. (20) was dilauroylphosphatidylethanolamine, for which Young's modulus could not be obtained from the literature; moreover, both the membrane surface tension and the steering force acting on selected protein residues coexist in the atomic system, which makes it very difficult to estimate the membrane stress. Instead, we estimated the dimensionless membrane strain from Eq. 9, that is, ε = σ(1 – ν)/E = Δr(1 – ν)/2a, where a ∼21.4 Å is the average undeformed radius of the lipid hole and Δr ∼ 3.3 Å is the increment of the hole radius by the end of the simulation, both measured from the trajectories of the steered MD simulation (20). Therefore, the maximum membrane strain is ∼5.4% in the steered MD study.
As shown in Fig. 7 b, although the channel was far from fully opened in the nanosecond steered MD simulation, the relationship between the effective radial strain and membrane strain in the steered MD analyses is in good agreement with the current FEM model at small strain, which nicely illustrates that the FEM model has a very reasonable description for the forces involved in the gating process, yet is capable to overcome the length- and timescale limits and achieve a much larger MscL gating profile.
Gating of MscL in response to membrane bending
The gating pathway of MscL
The out-of-plane bending deformation of the membrane depends critically on the remote mechanical load applied to the system, which exceeds the capability of typical all-atom simulations. Simulating the response of MscL to axisymmetric bending nicely illustrates the unique power of the present FEM model. Based on the discussions in Theoretical Estimates of Stress and Strain, if a maximum total bending moment of ∼1800 (3600) pN·nm were applied uniformly on the circular membrane (Fig. 3 b) for E. coli (Tb) MscL, the cavity wall would rotate for ∼10° (20°) to fully reduce the tilting angle of the helices. The tilting angle of the transmembrane helix bundle, α, is defined in terms of the effective radii of the five TM1 helical bundle at the locations that correspond to the surfaces of the lipid membrane (see Fig. 8 a for E. coli MscL).
FIGURE 8.

Results of the present FEM model as the membrane was subject to axisymmetric bending. (a) The TM1 helix bundles at fractional time 0.0 and 1.0, and the definition of the tilting angle α for the TM1 helix bundle; (b) the gating pathways of E. coli-MscL; and (c) the gating pathways of Tb-MscL. The TM1 helices are in orange and TM2 helices are in blue. The dashed lines indicate the approximate location of membrane/water interface.
With bending deformation, the wall of the lipid hole rotates whereas the averaged radius of the lipid cavity throughout the thickness remains essentially unchanged. Through the lipid-helix interactions, both TM1 and TM2 helices become upright and the pore radius is only moderately increased. Similar trends are found in the E. coli (Fig. 8 b) and Tb MscLs (Fig. 8 c); the gate-by-tilting is more obvious in the latter case, leading to a more expanded pore as quantified below.
Evolution of the pore radius
As shown in Fig. 9 a for E. coli MscL, with increasing bending curvature, the effective radial strain of the pore shows interesting nonmonotonic behavior: it first increases for ∼4%, then decreases to ∼−8%, and then increases monotonically to ∼8%. Such a trend is related to the moving pattern of the TM1 helices: upon bending, the TM1 helices not only become more upright (which increases the pore radius) but also slide toward the interior of the membrane (which decreases the pore radius). As a result, the evolution of the channel radius and the radial strain exhibit nonmonotonic behavior as shown in Fig. 9; note that the effective radial strain εR in Fig. 9 a (or that in Fig. 7 b) can be converted to the channel radius with the relation R = R0(εR + 1), where R0 = 6.5 Å is the undeformed radius for E. coli-MscL. As shown in Fig. 9 b, the tilting angle of TM1 helices reduces gradually with the fractional time. At the maximum loading, the remnant tilting angle is ∼1° for E. coli and the TM1 helices have not yet become upright, indicating that we have slightly underestimated the bending moment required for rotating the helices to achieve maximum gating-by-tilt. Recall that the bending moment is estimated (see Theoretical Estimates of Stress and Strain) based on the tilting angle of the lipid hole whereas the result plotted in Fig. 9 b is that of the TM1 helices.
FIGURE 9.

Results for axisymmetric bending of the present FEM model. (a) The evolution of the effective radial strain as a function of the fractional time. (b) The variation of the tilting angle of the TM1 helix bundle as a function of the fractional time.
CONCLUDING REMARKS
The value of continuum-mechanics models
Given the importance of mechanically driven processes in biology (1,3,4,7,9), effective theoretical and computational methods for their analysis are of considerable significance. The development of such methods is not a straightforward task because of the multiscale nature of typical biomechanical problems. Here we take an important step forward by adopting a continuum-mechanics model in the finite element (FEM) framework. Although simple continuum-mechanics models have been used to describe highly simplified models of biomolecules (15,16), adopting a flexible implementation using FEM makes it possible to study biological systems with more realistic representation of their irregular shapes, heterogeneous properties, and complex structural changes. The parameterization process of the FEM model also makes a natural connection to all-atom simulations.
As a first illustration, we apply the simulation model to study the gating behavior of mechanosensitive channels (7,10,17) of large conductance (MscL) in bacteria; the MscL is chosen based on its relatively simple structural topology and striking structural response to external mechanical perturbation, which have been rather well-characterized in a series of experiments (17,21). Even with the simple parameterization as chosen in the current study, the FEM-based model captures the major physical properties of MscL.
The model makes some predictions that can be tested experimentally. For example, the FEM model predicts that the MscL responds in very different fashion to equi-biaxial (in-plane) tension and axisymmetric (out-of-plane) bending of the lipid membrane: With the equi-biaxial tension, the channel opens fully at a maximal tension; however, with axisymmetric bending, the channel opens only slightly. This sensitivity of MscL to the form of the mechanical perturbation has been implicitly assumed in previous work but has never been shown explicitly using either experiment or theory (20,21,31,42). Another prediction is that both TM1 and TM2 helices bend substantially during the gating process; although it is possible that these are artifacts of the current FEM model due to the highly simplified description of helices, the result makes physical sense.
It is encouraging to see that the simple model developed here shows characteristic differences in the behavior of the MscL in the two different bacteria. With longer TM helices, the E. coli-MscL exhibits more striking tilting for the helices during gating, which was also featured in the structural model of Guy et al. (26) based on experimental constraints. Moreover, the gating pathway and pore radius evolution in the FEM simulations are in quite close agreement with the all-atom steered (20) and targeted (36) MD simulations. Given that the steered simulations were too short to observe the entire opening process and the targeted simulations employed unphysically large forces, the agreement between FEM results and these simulations has limited quantitative significance. It does indicate, however, that the FEM framework captures the essential forces for the gating process. Application of the FEM model to study the structural response of MscL to membrane bending clearly illustrated the unique value of continuum-mechanical models.
Future directions
The FEM model of this work is highly simplified, the encouraging results for gating notwithstanding, and is only meant to illustrate the value of FEM-based continuum-mechanics models in biology. The FEM-based model complements all-atom simulations and purely phenomenological models in two ways:
First, the FEM model overcomes the length- and timescale limits of atomistic simulations, yet it is not intended to reproduce all relevant atomistic details or to replace all-atom simulations; e.g., even large-scale conformational transitions in biomolecules typically involve important local structural rearrangements (69,70), which need to be treated implicitly in a continuum model.
On the other hand, based on useful insights obtained from atomistic simulations and/or experiments, the FEM model can be improved or decorated to capture the most important features that dictate the mechanical response of biomolecules, thus making it sufficiently detailed and less phenomenological. Specifically in the context of mechanosensation, further studies are being carried out in our research groups to either make the model more quantitative to fully take advantage of the flexibility offered by FEM, or to go beyond the quasi-static approach adopted here for the study of more complex biomechanical processes.
Regarding the quantitative improvements of the continuum-mechanical model, the helix mechanical properties could be readily made to be inhomogeneous and/or anisotropic. For example, the presence of hinge regions in a helix (e.g., at the proline in TM1 or around glycine in general) can be taken into consideration by modifying the mechanical properties or surface area of such regions. Moreover, the surface of a helix may be divided into hydrophilic and hydrophobic sections with different Lennard-Jones interaction potentials with the lipid; this can be a crucial quantitative improvement considering the postulated importance of hydrophobic mismatch to the function of membrane proteins (17,23,33,46,71). Solvation of the hydrophilic regions, which has been proposed to make a crucial contribution to the gating process (22), can be included in a similar fashion to implicit solvent models in all-atom simulations (72). The relevant parameters can be obtained from more sophisticated all-atom simulations compared to those performed here; e.g., potential of mean force at a relevant temperature instead of potential energy can be computed. The cytoplasmic S1 helices and the loops linking TM1 and TM2 helices, which may also play an important role in determining the quantitative gating behavior (17,26,47,48), should also be included. Only with all these improvements considered is a model likely to capture the more complex features of the MscL energy landscape as estimated by patch-clamp studies (24). For biomolecules with more complex secondary and tertiary structures, constructing an effective continuum model is more challenging although certainly possible and does not introduce additional difficulties into the FEM simulation.
The membrane can be made more heterogeneous to mimic realistic cellular membranes that include many components. (1) For example, experimental measurements of the population response of E. coli MscLs in the native membrane reveal that there is ∼5% variation in the energy cost of the opening among the channels, or ∼5% variation in tension (therefore, effective expansion area) between the individual channels due to the environment (73). Although 5% seems to be small, for such a system with steep sigmoidal dependence of the opening probability on tension it leads to the dramatic 50% decrease in the apparent gating energy cost and expansion area measured over the whole population (∼27 kBT, 12 nm2). This compares to the measurements of individual channels (∼50 kBT, ∼20 nm2). There are other subtle but potentially important issues such as the peristaltic deformation of the membrane and/or the asymmetric motion of the membrane leaflets that may influence the gating behavior and therefore need to be considered (74). Recent advances in developing effective coarse-grained models for multicomponent lipids (75,76) will greatly facilitate the efforts in constructing continuum models for heterogeneous membrane systems. In addition to studying individual biomolecules, the availability of effective continuum mechanical models will also raise the exciting possibility of studying cooperativity between many biomolecules, such as in muscle contraction and signal transduction across the cellular membrane (3,77–80).
Regarding the FEM simulation, it is fairly straightforward to go beyond the quasi-static approach adopted here and include time- as well as temperature-dependent features. Simulating the lipid and channel in a fully coupled fashion makes it possible to treat lipid-protein interactions on a more explicit and realistic level. Both time- and temperature-dependent physical properties of the helix and membrane, as well as the interactions among them, can be readily incorporated into the FEM model. For example, it is possible to include a dynamic (oscillatory) mechanical load, which can be very interesting to the study of phenomena such as desensitization of ion channels (81). Temperature is also an interesting variable because some biomechanical processes can also be triggered or regulated by change in temperature (82); how to include the effect of thermal fluctuation in a continuum simulation, however, is a challenging issue. It is straightforward to vary the intrinsic variables (e.g., membrane curvature, thickness, and physical properties of the protein or macromolecules) and extrinsic parameters (e.g., osmotic pressure and other environment-related variables), and to explore their effects. With its flexible length- and timescale formulation, the FEM model can be used to simulate the conformational transitions of proteins or other macromolecules when the system is under very complex mechanical load such as cell adhesion. Moreover, low-resolution intermediate structures accessible in FEM simulations can be further refined by subsequent all-atom simulations. Evidently, combining recent advances in all-atom simulations and solid mechanics will greatly expand the limitations in the spatial and timescales for the biological problems that can be analyzed computationally.
Acknowledgments
We thank Dr. J. Gullingsrud for sending us the trajectories reported in their published work (20) and Dr. R. H. Guy for sending us the coordinates for the models developed in Sukharev et al. (26). Discussions with Prof. C. Kung and Dr. A. Anishkin are greatly appreciated. We also thank Prof. N. Baker for a critical reading of the manuscript. Computational resources from the National Center for Supercomputing Applications at the University of Illinois are greatly appreciated.
The work of Y.T., G.C., and X.C. is supported in part by the National Science Foundation grant No. CMS-0407743 and in part by the Academic Quality Fund of Columbia University. J.Y. is partially supported by a grant from the National Institutes of Health (No. R01-GM071428 to Q.C.). Q.C. is an Alfred P. Sloan Research Fellow.
References
- 1.Alberts, B., D. Bray, J. Lewis, M. Raff, K. Roberts, and J. D. Watson. 1994. Molecular Biology of the Cell. Garland Publishing, New York.
- 2.Holmes, K. C., and M. A. Geeves. 1999. Structural mechanism of muscle contraction. Annu. Rev. Biochem. 68:687–728. [DOI] [PubMed] [Google Scholar]
- 3.Geeves, M., and K. C. Holmes. 2005. The molecular mechanism of muscle contraction. Adv. Protein Chem. 71:161–193. [DOI] [PubMed] [Google Scholar]
- 4.Robinson, D. N., and J. A. Spudich. 2000. Towards a molecular understanding of cytokinesis. Trends Cell Biol. 10:228–237. [DOI] [PubMed] [Google Scholar]
- 5.Robinson, D. N., and J. A. Spudich. 2004. Mechanics and regulation of cytokinesis. Curr. Opin. Cell Biol. 16:182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scholey, J. M., I. Brust-Mascher, and A. Mogilner. 2003. Cell division. Nature. 422:746–752. [DOI] [PubMed] [Google Scholar]
- 7.Hamill, O. P., and B. Martinac. 2001. Molecular basis of mechanotransduction in living cells. Physiol. Rev. 81:685–740. [DOI] [PubMed] [Google Scholar]
- 8.Huang, H., R. D. Kamm, and R. T. Lee. 2004. Cell mechanics and mechanotransduction: pathway, probes and physiology. Am. J. Physiol. Cell Physiol. 287:C1–C11. [DOI] [PubMed] [Google Scholar]
- 9.Janmey, P. A. 1998. The cytoskeleton and cell signaling: component localization and mechanical coupling. Physiol. Rev. 78:763–781. [DOI] [PubMed] [Google Scholar]
- 10.Martinac, B. 2004. Mechanosensitive ion channels: molecules of mechanotransduction. J. Cell Sci. 117:2449–2460. [DOI] [PubMed] [Google Scholar]
- 11.Vliet, K. J. V., G. Bao, and S. Suresh. 2003. The biomechanics toolbox: experimental approaches for living cells and biomolecules. Acta Mater. 51:5881–5905. [Google Scholar]
- 12.Bustamante, C., Y. R. Chemla, N. R. Forde, and D. Izhaky. 2004. Mechanical processes in biochemistry. Annu. Rev. Biochem. 73:705–748. [DOI] [PubMed] [Google Scholar]
- 13.Karplus, M., and J. Kuriyan. 2005. Molecular dynamics and protein function. Proc. Natl. Acad. Sci. USA. 102:6679–6685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Snow, C. D., E. J. Sorin, Y. M. Rhee, and V. S. Pande. 2005. How well can simulation predict protein folding kinetics and thermodynamics? Annu. Rev. Biophys. Biomol. Struct. 34:43–69. [DOI] [PubMed] [Google Scholar]
- 15.Wang, H. Y., and G. Oster. 1998. Energy transduction in the F1 motor of ATP synthase. Nature. 396:279–282. [DOI] [PubMed] [Google Scholar]
- 16.Zumdieck, A., M. C. Lagomarsino, C. Tanase, K. Kruse, B. Mulder, M. Dogterom, and F. Julicher. 2005. Continuum description of the cytoskeleton: ring formation in the cell cortex. Phys. Rev. Lett. 95:258103. [DOI] [PubMed] [Google Scholar]
- 17.Perozo, E. 2006. Gating prokaryotic mechanosensitive channels. Nat. Rev. Mol. Cell Biol. 7:109–119. [DOI] [PubMed] [Google Scholar]
- 18.Sukharev, S., P. Blount, B. Martinac, and C. Kung. 1997. Mechanosensitive channels of Escherichia coli: the MscL gene, protein and activities. Annu. Rev. Physiol. 59:633–657. [DOI] [PubMed] [Google Scholar]
- 19.Perozo, E., and D. Rees. 2003. Structure and mechanism in prokaryotic mechanosensitive channels. Curr. Opin. Struct. Biol. 13:432–442. [DOI] [PubMed] [Google Scholar]
- 20.Gullingsrud, J., and K. Schulten. 2003. Gating of MscL studied by steered molecular dynamics. Biophys. J. 85:2087–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kung, C. 2005. A possible unifying principle for mechanosensation. Nature. 436:647–654. [DOI] [PubMed] [Google Scholar]
- 22.Sukharev, S., and A. Anishkin. 2004. Mechanosensitive channels: what can we learn from “simple” model systems? Trends Neurosci. 27:345–351. [DOI] [PubMed] [Google Scholar]
- 23.Perozo, E., A. Kloda, D. M. Cortes, and B. Martinac. 2002. Physical principles underlying the transduction of bilayer deformation forces during mechanosensitive channel gating. Nat. Struct. Biol. 9:696–703. [DOI] [PubMed] [Google Scholar]
- 24.Sukharev, S. I., W. J. Sigurdson, C. Kung, and F. Sachs. 1999. Energetic and spatial parameters for gating of the bacterial large conductance mechanosensitive channel, MscL. J. Gen. Physiol. 113:529–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sukharev, S., M. Betanzos, C.-S. Chiang, and H. R. Guy. 2001. The gating mechanism of the large mechanosensitive channel MscL. Nature. 409:720–724. [DOI] [PubMed] [Google Scholar]
- 26.Sukharev, S. I., S. R. Durell, and H. R. Guy. 2001. Structural models of the MscL gating mechanism. Biophys. J. 61:917–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perozo, E., D. M. Cortes, P. Sompornpisut, A. Kloda, and B. Martinac. 2002. Open channel structure of MscL and the gating mechanism of mechanosensitive channels. Nature. 418:942–948. [DOI] [PubMed] [Google Scholar]
- 28.Markin, V. S., and F. Sachs. 2004. Thermodynamics of mechanosensitivity. Phys. Biol. 1:110–124. [DOI] [PubMed] [Google Scholar]
- 29.Wiggins, P., and R. Philips. 2003. Analytical models for mechanotransduction: gating a mechanosensitive channel. Proc. Natl. Acad. Sci. USA. 101:4071–4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wiggins, P., and R. Philips. 2005. Membrane-protein interactions in mechanosensitive channels. Biophys. J. 88:880–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Turner, M. S., and P. Sens. 2004. Gating-by-tilt of mechanically sensitive membrane channels. Phys. Rev. Lett. 93:118103. [DOI] [PubMed] [Google Scholar]
- 32.Chang, G., R. H. Spencer, A. T. Lee, M. T. Barclay, and D. Rees. 1998. Structure of the MscL homolog from Mycobacterium tuberculosis: a gated mechanosensitive ion channel. Science. 282:2220–2226. [DOI] [PubMed] [Google Scholar]
- 33.Anishkin, A., and C. Kung. 2005. Microbial mechanosensation. Curr. Opin. Neurol. 15:397–405. [DOI] [PubMed] [Google Scholar]
- 34.Anishkin, A., C. S. Chiang, and S. Sukharev. 2005. Gain of function mutations reveal expanded intermediate states and a sequential action of two gates in MscL. J. Gen. Physiol. 125:155–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gullingsrud, J., D. Kosztin, and K. Schulten. 2001. Structural determinants of MscL gating studied by molecular dynamics simulations. Biophys. J. 80:2074–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kong, Y., Y. Shen, T. E. Warth, and J. Ma. 2002. Conformational pathways in the gating of Escherichia coli mechanosensitive channel. Proc. Natl. Acad. Sci. USA. 99:5999–6004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schlitter, J., M. Engels, P. Krüger, E. Jacoby, and A. Wollmer. 1993. Targeted molecular dynamics simulation of conformational change—application to the T ↔ R transition in insulin. Mol. Simul. 10:291–308. [Google Scholar]
- 38.Colombo, G., S. J. Marrink, and A. E. Mark. 2003. Simulation of MscL gating in a bilayer under stress. Biophys. J. 84:2331–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Field, M. J., P. A. Bash, and M. Karplus. 1990. A combined quantum mechanical and molecular mechanical potential for molecular dynamics simulations. J. Comput. Chem. 11:700–733. [Google Scholar]
- 40.Friesner, R. A., and V. Guallar. 2005. Ab intio QM and QM/MM methods for studying enzyme catalysis. Annu. Rev. Phys. Chem. 56:389–427. [DOI] [PubMed] [Google Scholar]
- 41.Riccardi, D., P. Schaefer, Y. Yang, H. Yu, N. Ghosh, X. Prat-Resina, P. Konig, G. Li, D. Xu, H. Guo, M. Elstner, and Q. Cui. 2006. Development of effective quantum mechanical/molecular mechanical (QM/MM) methods for complex biological processes. J. Phys. Chem. B. 110:6458–6469. [DOI] [PubMed] [Google Scholar]
- 42.Phillips, R., M. Dittrich, and K. Schulten. 2002. Quasicontinuum representations of atomic-scale mechanics: from proteins to dislocations. Annu. Rev. Mater. Res. 32:219–233. [Google Scholar]
- 43.Villa, E., A. Balaeff, and K. Schulten. 2005. Structural dynamics of the lac repressor-DNA complex revealed by a multi-scale simulation. Proc. Natl. Acad. Sci. USA. 102:6783–6788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bustamante, C., D. Keller, and G. Oster. 2001. The physics of molecular motors. Acc. Chem. Res. 34:412–420. [DOI] [PubMed] [Google Scholar]
- 45.Julicher, F., A. Ajdari, and J. Prost. 1997. Modeling molecular motors. Rev. Mod. Phys. 69:1269–1281. [Google Scholar]
- 46.Moe, P., and P. Blount. 2005. Assessment of potential stimuli for mechano-dependent gating of MscL: effects of pressure, tension and lipid headgroups. Biochemistry. 44:12239–12244. [DOI] [PubMed] [Google Scholar]
- 47.Blount, P., M. J. Schroeder, and C. Kung. 1997. Mutations in a bacterial mechanosensitive channel change the cellular response to osmotic stress. J. Biol. Chem. 272:32150–32157. [DOI] [PubMed] [Google Scholar]
- 48.Hase, C. C., A. C. L. Dain, and B. Martinac. 1997. Molecular dissection of the large mechanosensitive ion channel (MscL) of E. coli: mutants with altered channel gating and pressure sensitivity. J. Membr. Biol. 157:17–25. [DOI] [PubMed] [Google Scholar]
- 49.Markin, V. S., and J. P. Albanesi. 2002. Membrane fusion: stalk model revisited. Biophys. J. 82:693–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reference deleted in proof.
- 51.Choe, S., and S. X. Sun. 2005. The elasticity of α-helices. J. Chem. Phys. 122:244912. [DOI] [PubMed] [Google Scholar]
- 52.Sadd, M. H. 2005. Elasticity: Theory, Applications and Numerics. Elsevier, Dordrecht, The Netherlands.
- 53.Lindahl, E., and O. Edholm. 2000. Spatial and energetic-entropic decomposition of surface tension in lipid bilayers from molecular dynamics simulations. J. Chem. Phys. 113:3882–3893. [Google Scholar]
- 54.Evans, E., and W. Rawicz. 1990. Entropy-driven tension and bending elasticity in condensed-fluid membranes. Phys. Rev. Lett. 64:2094–2097. [DOI] [PubMed] [Google Scholar]
- 55.Pantano, A. B. M.C., and D. Parks. 2003. Nonlinear structural mechanics based modeling of carbon nanotube deformation. Phys. Rev. Lett. 91:145504–1–4. [DOI] [PubMed] [Google Scholar]
- 56.Cao, G., Y. Tang, and X. Chen. 2006. Elastic properties of carbon nanotubes in radial direction. J. Nanoeng. Nanosys. In press.
- 57.Cao, G., and X. Chen. 2006. Mechanisms of nanoindentation on single-walled carbon nanotubes: the effect of nanotube length. 2006. J. Mater. Res. 21:1048–1070. [Google Scholar]
- 58.Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM: a program for macromolecular energy, minimization and dynamics calculations. J. Comput. Chem. 4:187–217. [Google Scholar]
- 59.Neria, E., S. Fischer, and M. Karplus. 1996. Simulation of activation free energies in molecular systems. J. Chem. Phys. 105:1902–1921. [Google Scholar]
- 60.Lazaridis, T. 2003. Effective energy function for proteins in lipid membranes. Proteins Struct. Funct. Gen. 52:176–192. [DOI] [PubMed] [Google Scholar]
- 61.Spassov, V. Z., L. Yan, and S. Szalma. 2002. Introducing an implicit membrane in generalized-Born/solvent accessibility continuum solvent models. J. Phys. Chem. B. 106:8726–8738. [Google Scholar]
- 62.Im, W., M. Feig, and C. L. Brooks. 2003. An implicit membrane generalized-Born theory for the study of structure, stability, and interactions of membrane proteins. Biophys. J. 85:2900–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.MacKerell, A. D. Jr., D. Bashford, M. Bellott, R. L. Dunbrack, Jr., J. D. Evenseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-Mc, L. Carthy, K. Kuchnir, F. T. K. Kuczera, C. Lau, S. Mattos, T. Michnick, D. T. Ngo, B. Nguyen, W. E. R. I. I. I. Prodhom, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiórkiewicz-Kuczera, D. Yin, and M. Karplus. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- 64.Yoshimura, K., T. Nomura, and M. Sokabe. 2004. Loss of function mutations at the rim of the funnel of mechanosensitive channel MscL. Biophys. J. 86:2113–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Timoshenko, S., and S. Krieger. 1987. Theory of Plates and Shells, 2nd Ed. McGraw-Hill, New York.
- 66.ABAQUS. 2004. ABAQUS 6.4 User's Manual. ABAQUS Inc., Pawtucket, RI.
- 67.Shapovalov, G., and H. A. Lester. 2004. Gating transitions in bacterial ion channels measured at 3 μs resolution. J. Gen. Physiol. 124:151–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vinci, R. P., and J. J. Vlassak. 1996. Thin film mechanical properties. Annu. Rev. Mater. Res. 26:431–462. [Google Scholar]
- 69.Ma, J., T. C. Flynn, Q. Cui, A. G. W. Leslie, J. E. Walker, and M. Karplus. 2002. A dynamic analysis of the rotation mechanism for conformational change in F1-ATPase. Structure. 10:921–931. [DOI] [PubMed] [Google Scholar]
- 70.Formaneck, M., L. Ma, and Q. Cui. 2006. Reconciling the “old” and “new” views of protein allostery. A molecular simulation study of chemotaxis Y protein (Che Y). Proteins. 63:846–867. [DOI] [PubMed] [Google Scholar]
- 71.Suchyna, T. M., S. E. Tape, R. E. Koeppe, O. S. Andersen, F. Sachs, and P. A. Gottlieb. 2004. Bilayer-dependent inhibition of mechanosensitive channels by neuroactive peptide enantiomers. Nature. 430:235–240. [DOI] [PubMed] [Google Scholar]
- 72.Feig, M., and C. L. Brooks. 2004. Recent advances in the development and application of implicit solvent models in biomolecular simulations. Curr. Opin. Struct. Biol. 14:217–224. [DOI] [PubMed] [Google Scholar]
- 73.Chiang, C. S., A. Anishkin, and S. Sukharev. 2004. Gating of the large mechanosensitive channel in situ: estimation of the spatial scale of the transition from channel population responses. Biophys. J. 86:2846–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brannigan, G., and F. Brown. 2006. A consistent model for thermal fluctuations and protein-induced deformations in lipid bilayers. Biophys. J. 90:1501–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Marrink, S. J., A. H. de Vries, and A. E. Mark. 2004. Coarse-grained model for semiquantitative lipid simulations. J. Phys. Chem. B. 108:750–760. [Google Scholar]
- 76.Nielsen, S. O., C. F. Lopez, G. Srinivas, and M. L. Klein. 2004. Coarse-grained models and the computer simulation of soft materials. J. Phys. Cond. Matt. 16:R481–R512. [Google Scholar]
- 77.Bray, D. 1998. Signaling complexes: biophysical constraints on intracellular communication. Annu. Rev. Biophys. Biomol. Struct. 27:59–75. [DOI] [PubMed] [Google Scholar]
- 78.Bray, D., and T. Duke. 2004. Conformational spread: the propagation of allosteric states in large multiprotein complexes. Annu. Rev. Biophys. Biomol. Struct. 33:53–73. [DOI] [PubMed] [Google Scholar]
- 79.Marx, S. O., K. Ondrias, and A. R. Marks. 1998. Coupled gating between individual skeletal muscle Ca2+ release channels (ryanodine receptors). Science. 281:818–821. [DOI] [PubMed] [Google Scholar]
- 80.Vaknin, A., and H. C. Berg. 2006. Osmotic stress mechanically perturbs chemoreceptors in E. coli. Proc. Natl. Acad. Sci. USA. 103:592–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Auerbach, A. 2005. Gating of acetylcholine receptor channels: Brownian motion across a broad transition state. Proc. Natl. Acad. Sci. USA. 102:1408–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Clapham, D. E. 2003. TRP channels as cellular sensors. Nature. 426:517–524. [DOI] [PubMed] [Google Scholar]