

Abstract
The release of mitochondrial intermembrane space proteins to the cytosol is a key event during apoptosis. We used in situ fluorescent labeling of proteins tagged with a short tetracysteine-containing sequence to follow the release of Smac, Omi, adenylate kinase-2, cytochrome c, and apoptosis-inducing factor (AIF) during apoptosis and compared the release with that of cytochrome c tagged with GFP in individual cells observed over time. We observed a caspase-independent, simultaneous release of cytochrome c, Smac, Omi, and adenylate kinase-2. Although AIF release also was caspase-independent and commenced with that of the other proteins, it proceeded much more slowly and incompletely from mitochondria, perhaps because of a requirement for a secondary event. These results suggest that these proteins are released through the same mitochondrial pore and that apoptosis may not be regulated through a selective release of individual mitochondrial proteins. The timing and extent of AIF release makes it unlikely that it is involved in the induction of apoptosis, either upstream or downstream of mitochondrial outer membrane permeabilization.
Keywords: apoptosis-inducing factor, cytochrome c, mitochondria, Omi, Smac
Mitochondrial outer membrane permeabilization (MOMP) is a critical event during apoptosis (1, 2). This event is controlled by pro- and antiapoptotic Bcl-2-family proteins, which integrate the survival and prodeath signals leading to the decision to undergo apoptosis. Some of these proteins reside on mitochondria or translocate to mitochondria during apoptosis, where they trigger the permeabilization of the outer mitochondrial membrane and the release of several intermembrane space proteins. Among these intermembrane space proteins, cytochrome c is required for the initiation of the apoptosome and the activation of caspases, whereas Smac/DIABLO and Omi/HtrA2 enhance caspase activation through the neutralization of inhibitor of apoptosis proteins (3–5), although the role of these two proteins in apoptosis is controversial (6). Other mitochondrial proteins that are released during apoptosis have been suggested to play other roles in cell death. Endonuclease G (7), apoptosis-inducing factor (AIF) (8), and Omi/Htra2 (5, 9) each have been proposed to function in caspase-independent cell death after MOMP.
MOMP may be due to the formation of a pore formed by proapoptotic Bcl-2 family members such as Bax and Bak, alone or in combination with other proteins. Activated Bax alone can form size-indiscriminate pores in vesicles made from purified mitochondrial outer membranes or mitochondrial lipids (10). Although it has been shown that either Bax or Bak are required for MOMP in purified mitochondria and living cells (11), the nature and composition of the pore or pores remains unknown. Two other issues that require further clarification are the timing of mitochondrial permeabilization related to other apoptotic events and whether the release of individual proteins is regulated differentially. Although cytochrome c is released from virtually all of the mitochondria in a single cell within ≈5 min (12, 13), a differential release of cytochrome c, Smac, and/or AIF has been reported (14–16). A study in single cells, however, showed that the release of Smac-yellow fluorescent protein and cytochrome c-GFP commenced simultaneously, and this event preceded caspase activation (17). On the other hand, the release of Smac (18) and AIF (19–21) has been reported to occur downstream of cytochrome c release and caspase activation. The differential release of proapoptotic proteins would suggest the existence of specific pores selective for each protein, as well as the possibility of regulation of the apoptotic process at multiple levels within the mitochondrial phase.
To gain insight into the mechanism and time of release of mitochondrial intermembrane space proteins, we used a confocal microscopy-based, real-time single-cell analysis approach. This technology allowed us to directly observe the temporal relationships between the release of individual proteins and the correlation of these events with other parameters of cell death. Our results show that the release of cytochrome c, Adenylate Kinase 2, Smac, Omi, and partially, AIF, is simultaneous and does not depend on caspase activity. Any AIF release occurred slowly during the hours after the release of cytochrome c, whereas the other proteins were released in a 3- to 10-min window within a cell. These results are indicative of a mitochondrial permeabilization event that is nonprotein selective and occurs rapidly within a single cell.
Results
Smac Release Coincides with Cytochrome c Release.
To monitor the release of mitochondrial intermembrane space proteins, we used a technique that allows us to label recombinant proteins in live cells. We fused each protein to a small tag (10–15 aa residues) containing a tetracysteine motive (TC) that can be specifically recognized by a cell-permeant fluorescein- (FlAsH, fluorescein arsenical hairpin binder) or resorufin- (ReAsH, resorufin arsenical hairpin binder) based fluorescent dye (22) that binds covalently to the tag.
Recently, we compared the mitochondrial release of cytochrome c-GFP and cytochrome c-TC in two cell lines expressing both labeled proteins (13). We found that during apoptosis, these two forms of cytochrome c were released simultaneously and with identical kinetics. Based on this and other evidence (13), we concluded that the localization of cytochrome c-GFP during apoptosis accurately reflects that of cytochrome c in the cell. These studies therefore validated comparisons of the kinetics of cytochrome c-GFP release with that of TC-tagged proteins.
We generated vectors encoding TC-tagged Smac, Adenylate Kinase 2 (AK2), Omi, and AIF recombinant proteins. The proteins encoded by these vectors displayed mitochondrial localization, as detected by cell fractionation (data not shown). Localization was verified by using confocal microscopy either by costaining with the red dye ReAsH or the green dye FlAsH and mitochondrion-specific dyes (data not shown) or by transfection of cells that express cytochrome c tagged with the GFP (Fig. 1; see also Fig. 5, which is published as supporting information on the PNAS web site).
Fig. 1.

TC-tagged proteins display mitochondrial localization. HeLa-cyt.c-GFP cells were stably (Smac, Omi, and AK2) or transiently (AIF) transfected with the vector encoding the corresponding TC-tagged protein and stained with the red dye ReAsH. (Scale bars: 20 μm.) Arrow indicates a cell transfected with AIF-TC; other cells in the field did not express the protein. Full-resolution images are shown in Fig. 5.
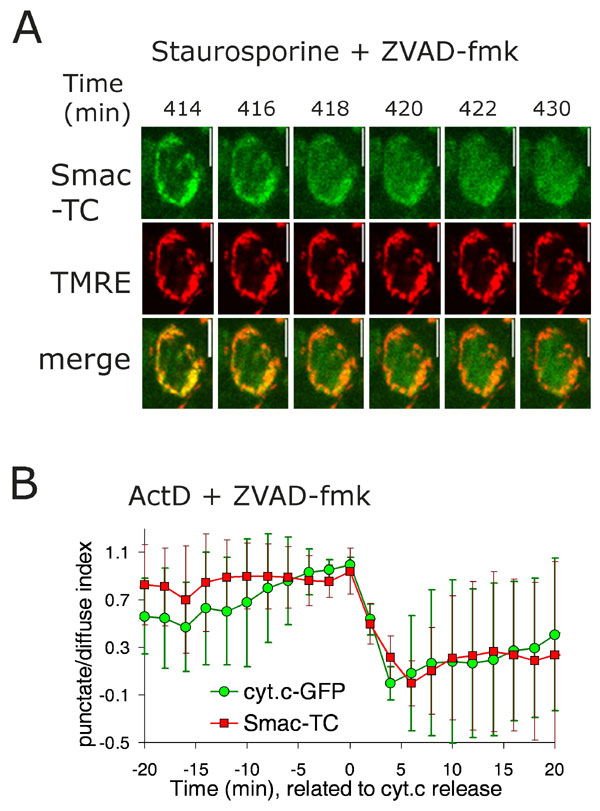
We generated cells stably expressing Smac-TC and monitored the localization of the stained protein after treatment with staurosporine to induce apoptosis. We used the pan-caspase inhibitor Z-Val-Ala-Asp(OMe)-CH2F (zVAD-fmk) to block any caspase-dependent effect on mitochondrial permeabilization. Fig. 6A, which is published as supporting information on the PNAS web site, shows the kinetics of release of Smac-TC labeled with the green dye FlAsH in one cell. Although each individual cell in the microscope field released Smac at a variable time after addition of staurosporine (between 3 and 9 h after treatment; data not shown), once a cell started releasing Smac, this release was rapid; Smac was released within single cells in 5.4 ± 1.8 min (average and SD from 10 cells from the same experiment). The kinetics of the release of Smac (Fig. 6A; see also Movie 1, which is published as supporting information on the PNAS web site) show that it is sudden, rapid, and complete, as virtually all of the protein was released from the mitochondria within a 5-min timeframe.
The mitochondrial transmembrane potential (Δψm) dissipates during apoptosis, and this event occurs around the time of MOMP. We measured Δψm by using tetramethylrhodamine ethyl ester (TMRE) and compared the timing of Δψm loss and the release of Smac. As we have observed previously for cytochrome c release (12, 13, 23, 24), Δψm is sustained throughout the initiation of Smac release, and dissipates only slowly thereafter (Fig. 2A and 6A). The rate at which Δψm dissipates after MOMP depends on caspase activation (23–25), the agent used to induce MOMP (12), and the amount of light used in assessing the cells (N. Waterhouse, unpublished data). Nevertheless, we observed changes in Δψm only after the initiation of MOMP, as assessed by Smac release. These observations support supports the idea that loss of Δψm is a result of events leading to Smac release rather than being associated with its cause.
Fig. 2.

Smac, AK2, and Omi are released with cytochrome c. (A) HeLa-cyt.c-GFP cells stably expressing Smac-TC were stained with the red dye ReAsH or the green dye FlAsH and treated with the indicated inducers. (A Upper) Scaled average and SD of the punctate/diffuse index are shown of 10 cells from the same experiment, aligned to the time of first detected Smac-FlAsH release (t = 0). Cells also were stained with the mitochondrial dye tetramethylrhodamine ethyl ester (TMRE); TMRE brightness index was calculated by averaging the values of the average intensity of the pixels within each cell. (A Lower) Scaled average of the punctate/diffuse index for Smac-ReAsH and cytochrome c aligned to the time of cytochrome c release (n = 7). Images were taken every 3 min with a ×40 objective. (B) HeLa-cyt.c-GFP cells stably transduced with AK2-TC were stained with ReAsH and treated with the indicated inducers. Graphs show the scaled average of the punctate/diffuse index from 7 (Upper) or 17 (Lower) cells. (C) HeLa-cyt.c-GFP cells were transiently transfected with a construct encoding Omi-TC. Cells were stained with ReAsH, typically 24 to 40 h after transfection, and treated with the indicated inducers. (C Upper) Graph from a representative experiment showing the scaled punctate/diffuse index of cytochrome c-GFP and Omi-TC (average and SD of five cells). (C Middle) Graph from an experiment in which cells were treated with staurosporine in the presence of 100 μM zVAD-fmk. Pictures were taken every 3 min. Images from the same experiment are shown in C Lower. Additional cells are shown in Movie 2. (Scale bar: 10 μm.)
Several reports described a selective release of Smac or cytochrome c during apoptosis (14, 18, 26). However, other experiments in single cells suggested a simultaneous release of cytochrome c and yellow fluorescent protein-tagged Smac (17, 27). We generated cells stably expressing cytochrome c-GFP and Smac-TC (Fig. 1) and monitored the fluorescence of cytochrome c-GFP and Smac-TC stained with the red dye ReAsH after treatment with staurosporine (Fig. 2A) or actinomycin D (ActD) (Fig. 6B) in the presence of caspase inhibitors. In both cases, the release of cytochrome c and Smac was simultaneous. Smac and cytochrome c were released in 6.2 ± 2.0 and 5.4 ± 2.0 min, respectively, during staurosporine-induced apoptosis and in 4.3 ± 1.3 and 3.6 ± 0.1 min, respectively, during ActD-induced apoptosis.
AK2, Omi, and Cytochrome c Are Released Simultaneously.
Although cytochrome c and Smac are proteins of special interest because they are implicated in the apoptotic process, it is possible that MOMP would allow the release of virtually any soluble protein from the mitochondrial intermembrane space (28). It has been shown by cell fractionation that a protein with adenylate kinase activity is released to the cytosol during apoptosis (29). This protein was identified as isoform II of AK2, which is a mitochondrial protein located in the intermembrane space (30). We generated cells overexpressing AK2-TC and cytochrome c-GFP, and observed that the release of AK2 and cytochrome c is simultaneous after treatment with staurosporine, in the presence or absence of zVAD-fmk (Fig. 2B). AK2 was released in 5.13 ± 1.9 and 5.11 ± 1.8 min (SD) in the presence or absence of the caspase inhibitor, respectively.
Omi/Htra2 is a serine protease that is released to the cytosol during apoptosis, where it may participate in caspase activation or caspase-independent cell death (5). Omi release has been shown to occur independently of zVAD-fmk-inhibitable caspases (20). We generated a stable cell line expressing Omi-TC and cytochrome c-GFP (Fig. 1). We observed by time-lapse experiments that Omi-TC and cytochrome c-GFP were released simultaneously when these cells were treated with staurosporine (data not shown). However, because only low levels of expression of Omi-TC were achieved in stable lines, we performed experiments after transient transfection of HeLa-cyt.c-GFP with the Omi-TC plasmid. After treatment with staurosporine, individual cells released cytochrome c and Omi at the same time (Fig. 2C). Omi-TC release proceeded with a duration of 5.9 ± 1.3 min, whereas cytochrome c-GFP release took 3.63 ± 0.89 min (eight cells analyzed). Similarly, the release of both proteins started at the same time when cells were treated with staurosporine in the presence of zVAD-fmk (Fig. 2C; see also Movie 2, which is published as supporting information on the PNAS web site), although the duration of the release of Omi-TC in this case was slightly longer: 8.7 ± 3.8 min (13 cells analyzed, difference not significant by Student t test; data not shown). In summary, these results indicate that Omi, like Smac, cytochrome c, and AK2, is released during a period of <10 min from the mitochondria within a single cell and that the release occurs at the same time as the other mitochondrial intermembrane space proteins.
AIF Release Is Slow, Incomplete, and Caspase-Independent.
We sought to determine the kinetics of AIF release by monitoring AIF and cytochrome c translocation in coexpressing, live cells. We transiently transfected HeLa-cyt.c-GFP cells with a construct encoding TC-tagged human AIF (AIF-TC) and studied the localization of both molecules. We were unable to visually detect the release of AIF-TC at the time of cytochrome c release after treatment with staurosporine (data not shown) or ActD (Fig. 3). A slow, partial release of AIF-TC was observed in individual cells between 2 and 5 h after cytochrome c release at a time close to when the plasma membrane became permeable to propidium iodide. Similar results were obtained when cells were treated with apoptotic inducers in the presence of the caspase inhibitors zVAD-fmk (data not shown) or quinoline-Val-Asp-CH2-difluorophenoxy (qVD-OPH) (Fig. 3). The software-assisted analysis of the punctate/diffuse index in four cells from the experiment shown in Fig. 3, however, indicated a slight drop in the value of this index for AIF-TC at the time of cytochrome c release. This slight drop, which we were unable to detect visually, may represent the release of a small fraction of AIF preexisting as a soluble form in the intermembrane space or being cleaved immediately after MOMP. These results are consistent with the idea that AIF is a transmembrane protein that requires a secondary event to be released during apoptosis.
Fig. 3.

AIF-TC is released slowly and in a caspase-independent manner. HeLa-cyt.c-GFP cells were transiently transfected with a construct encoding AIF-TC and stained with the red dye ReAsH typically 24 to 40 h after transfection. Images were taken every 4 min after appropriate incubation times with the inducer, as described in Methods. (A) Time shown is time of total incubation with ActD. (Scale bars: 10 μm.) (B) Average and SD of four cells from an experiment in which cells were treated with staurosporine in the presence of quinoline-Val-Asp-CH2-difluorophenoxy (qVD-OPH).
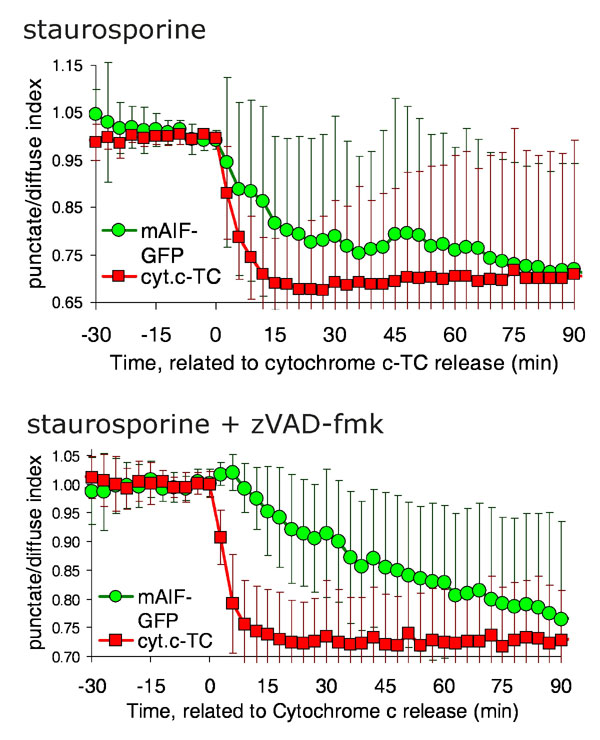
Other experiments analyzing the kinetics of AIF release had been performed by using the GFP-tagged version of the murine (31) or rat (32) AIF molecule. By analyzing cells visually at single time points, both studies concluded that AIF-GFP was released in a caspase-independent manner. Consistent with our results employing AIF-TC, Otera et al. (32) reported that at short times after treatment, Smac and cytochrome c release could be detected in many cells in which AIF was not released, suggesting that rat AIF-GFP was released after cytochrome c. We wished to track GFP-tagged AIF in live cells to confirm our previous results employing AIF-TC. We first compared the release of murine AIF-GFP (mAIF-GFP) to cytochrome c-TC by transiently transfecting HeLa cells expressing cytochrome c-TC (13) with a construct encoding GFP-tagged murine AIF (Fig. 4A). This protein, as seen with the TC-tagged AIF, was released very slowly from the mitochondria in cells treated with staurosporine, either in the presence or absence of the caspase inhibitor zVAD-fmk (Fig. 4B; see also Fig. 7, which is published as supporting information on the PNAS web site). When cells were treated with ActD in the presence of zVAD-fmk (chart in Fig. 4C), the release proceeded noticeably more slowly, and it was never complete until >10 h after cytochrome c release (data not shown).
Fig. 4.

AIF-GFP is released slowly and in a caspase-independent manner. (A–C) HeLa cells stably expressing cytochrome c-TC were transiently transfected with a construct encoding murine AIF-GFP. Pictures in A show the colocalization of AIF-GFP with the mitochondrial marker Mitotracker Red (MTR). (Scale bars: 10 μm.) (B and C) Cells were stained with the red dye ReAsH to label cytochrome c-TC (abbreviated cyt.c-TC) and treated with the indicated inducers. Images were taken every 3 min. Propidium iodide was added to the medium to monitor cell death. (D and E) HeLa cells stably expressing cyt.c-TC were transiently transfected with a construct encoding human AIF-GFP and stained with ReAsH. Graph in D shows the average and SD of punctate/diffuse index for AIF-GFP and cyt.c-TC of six cells aligned to the time of cytochrome c release. (E) Images were taken every 3 min. Numbers show time (in minutes) after drug addition. (Scale bar: 10 μm.)
The kinetics of release of human AIF-GFP showed a similar pattern. When transiently overexpressed in cells expressing cytochrome c-TC, we observed that AIF was released slowly after MOMP when cells were treated with staurosporine either in the presence (data not shown) or absence of the caspase inhibitor zVAD-fmk (Fig. 4D; see also Movie 3, which is published as supporting information on the PNAS web site). The same results were obtained when cells were treated with ActD (data not shown) or ActD and quinoline-Val-Asp-CH2-difluorophenoxy (qVD-OPH) (Fig. 4E).
Discussion
We have analyzed the release in real time of AIF, AK2, Smac, Omi, and cytochrome c from the mitochondria during apoptosis. We aimed to avoid potential artifacts arising from the study of apoptosis in asynchronous cell populations by monitoring this release in single cells. We found that the release of all these proteins commences in single cells at the same time during apoptosis triggered by two different stimuli, although the release of the transmembrane protein AIF was slow and incomplete. Our results support a model in which the permeabilization of the mitochondrial membrane is due to the rapid formation of a pore that allows the release of many soluble proteins. Previous experiments indicate that the formation of this pore requires the presence of the multidomain proapoptotic proteins Bax or Bak (11). Although the size of this pore has not been determined by direct measurements in cells or isolated mitochondria, Kuwana et al. (10) have shown that a pore formed by Bax could allow the flux of molecules as large as 2 MDa. Thus, it is reasonable to speculate that any soluble protein in the intermembrane space could be released from mitochondria through this pore. The present study and several studies reporting the release of Smac show that a protein as large as 50 kDa (monomeric Smac-yellow fluorescent protein) or 100 kDa (the predicted size of dimeric soluble Smac-yellow fluorescent protein) can be released in ≈4–20 min from all of the mitochondria within a single cell (17, 27, 33). Consistent with these results, it has been shown than tBid induces the release of many intermembrane space proteins from isolated mitochondria, of sizes up to 123 kDa (28). Although some of these proteins such as cytochrome c, Smac, and Omi play a role during apoptosis or interact with proteins involved in apoptosis, the role of many other mitochondrial proteins, such as AK2, in cell death is not well established. In the present study, we show that AK2 is coreleased in cells with cytochrome c, supporting the hypothesis that the pore is not protein-specific. Previously, we have shown that cytochrome c is completely released from essentially all of the mitochondria within a cell irrespective of cell type or stimulus used (12, 13). Our results extend these observations to other intermembrane space proteins and suggest that the pore is formed simultaneously, in ≈5 min, in virtually all of the mitochondria within a cell, because cytochrome c, Smac, Omi, and AK2 were completely released within this period.
Our results indicate that Omi, AK2, Smac, and cytochrome c are released simultaneously. However, both the murine and human form of AIF were released differently. Although the onset of the release was at the same time as that for cytochrome c and the other proteins, the duration of AIF release was longer. AIF release has been shown to be regulated differently than other proteins, because this protein is not released from purified mitochondria by Bax (21) or tBid (28). Otera et al. (32) reported that AIF spans the mitochondrial inner membrane and must be cleaved to be released. They observed that the cleavage was inhibited by overexpression of Bcl-2 and Bcl-xL, thus suggesting that the cleavage starts after the mitochondrial membrane has been permeabilized. Furthermore, the release of AIF triggered by inducers of the permeability transition pore such as atractyloside or Ca2+ is prevented by protease inhibitors (34, 35). Therefore, the requirement of a secondary event after MOMP, the proteolytic cleavage of AIF, may explain its slow kinetics. Although the kinetics of release of AIF have not been analyzed previously in real time, some reports indicate that AIF is released after cytochrome c. Consistent with our results, the authors detected cytosolic cytochrome c in cells in which AIF was not released (19, 32, 36) or showed the release of both proteins at later time points (31). Several studies, however, have reported a selective release of AIF but not cytochrome c in T lymphocytes (37, 38) and fibroblasts (16, 39), as detected by immunostaining with an antiserum directed against a mixture of AIF peptides. A similar situation has been reported for Smac where some inducers triggered the selective release of Smac but not cytochrome c, as detected by immunoblotting (14, 15). Although in our study we have focused on well established activators of the Bax/Bak-dependent mitochondrial pathway of apoptosis, we cannot presently exclude the possibility of specific mechanisms for cleavage and export of AIF and for Smac release in certain cells types and/or after treatment with certain inducers. The nature of such mechanisms requires further clarification.
It has been proposed that in certain scenarios, a partial release of cytochrome c would activate caspases, which would act back on the mitochondria through a feedback loop leading to further mitochondrial permeabilization. This hypothesis is not supported by previous studies in single cells, because the kinetics of release of cytochrome c and Smac are identical in the presence or absence of caspase inhibitors (12, 13, 17, 27, 33). Furthermore, in this study, we have not detected an effect of caspase inhibition on the kinetics of the release of any intermembrane space protein, although in our system, caspase inhibitors blocked cell rounding, blebbing, annexin V binding, and incorporation of propidium iodide for at least 12 h after cytochrome c release. It has been reported that caspase-3 and caspase-7 deficiency causes a delay in the occurrences of mitochondrial changes during apoptosis (40). These observations are consistent with the idea that MOMP can be triggered by both caspase-dependent and caspase-independent pathways that can affect when MOMP occurs but does not affect its kinetics. It has been reported that Smac release in cell populations could be blocked by caspase inhibitors under conditions in which cytochrome c release is caspase-independent (18). This result may be due to the difficulty of detecting Smac in the cytosolic fraction of apoptotic cells in the presence of caspase inhibitors, because Smac is rapidly degraded by the proteasome when caspases are inhibited (41). Similarly, some reports showed that the release of AIF, but not cytochrome c, is caspase-dependent (19–21) after treatment with the same inducers used in our studies. The authors report that a subset of cells release AIF even in the presence of caspase inhibitors. We believe the differences observed between these studies and ours could have at least two explanations: First, zVAD-fmk at high concentrations may inhibit the noncaspase cystein proteases that are presumably responsible for AIF cleavage (35), and second, the early time points used in those studies did not allow detection of AIF release that may take several hours after cytochrome c release. On the other hand, many results implying a differential release of proteins from isolated mitochondria may be due to the use of techniques that underestimate the amount of cytochrome c release that the same stimulus would trigger in cells. When assays are performed under conditions of low ionic strength, such as standard mitochondrial isolation buffers, cytochrome c, but not Smac or other soluble proteins, may remain associated with mitochondrial membranes (42). Perhaps the most common source of discrepancies between reports analyzing release of mitochondrial proteins, however, is the one we avoided by single-cell analysis: the attempt to obtain kinetic information from highly asynchronous populations observed at widely spread intervals.
Our results indicate that the permeabilization of the outer mitochondrial membrane during apoptosis is a rapid process that allows the simultaneous release of many proteins. The continuous and slow release of AIF suggests that the pore formed during MOMP remains open for many hours even in the presence of caspase inhibitors. Differences in the kinetics of release of intermembrane space proteins thus are likely to reflect sequestration of proteins that may require a secondary event to be released, either because they are transmembrane proteins or because they are tightly bound to lipids or other mitochondrial proteins. Such secondary, late events may not play major roles in apoptosis, because their occurrence probably would follow caspase activation and mitochondrial dysfunction, but roles in caspase-independent cell death are not excluded. These observations further highlight the importance of the Bcl-2 family regulated mitochondrial permeabilization as the critical point of regulation of the apoptotic process.
Methods
Cell Lines and Reagents.
HeLa cells expressing cytochrome c-GFP were generated by infection with three rounds of pBABE mouse cytochrome c-GFP virus (13) and selection with 1 mg/ml G418. An individual clone with high expression of cytochrome c-GFP (named HeLa-cyt.c-GFP) was selected for subsequent transfections. Stable cell lines were generated by infection of HeLa-cyt.c-GFP cells with Smac-TC, Omi-TC, or AK2-TC viruses and selection with 5 μg/ml puromycin for 3 days. Lipofectamine 2000 (Invitrogen) was used for transient transfections, and experiments were performed 24–48 h after transfection. zVAD-fmk (Z-Val-Ala-Asp(OMe)-CH2F, abbreviated as zVAD-fmk) and quinoline-Val-Asp-CH2-difluorophenoxy were used at 100 and 20 μM, respectively. Both caspase inhibitors were purchased from Enzyme System Products (Livermore, CA). Staurosporine and ActD were used at 1 μM and 2 μM, respectively.
Time-Lapse Confocal Microscopy and Data Analysis.
For time-lapse analysis, cells were labeled with biarsenical ligands (described in ref. 22 and Supporting Methods, which is published as supporting information on the PNAS web site), treated with the inducers, and set on the confocal microscope stage (detailed in Supporting Methods). Images were taken every 2 min unless otherwise indicated.
Images were analyzed with Metamorph 4.0 (Universal Imaging, West Chester, PA) by drawing regions around individual cells and then computing SD of the intensity of the pixels (punctate/diffuse) and integrated brightness (total brightness). The punctate/diffuse index and the total brightness index were calculated by dividing each value by the average of the five values before cytochrome c release. As a rough indication of the range of variability among cells within an experiment, error bars show SD. When noted, data were scaled by the following formula: scaled point = 1 – ((Max − x)/MaxDifference), where Max equals the maximum value in the series, x equals the point of interest, and MaxDifference equals the maximum minus the minimum value in the series. In those cases, error bars were scaled by multiplying the SD in each point by 1/MaxDifference.
Supporting Information.
Additional details are described in Supporting Methods.
Supplementary Material
Acknowledgments
Samuel Connell was invaluable for his assistance with cell sorting and microscopy. We thank J. Wilkinson and C. Duckett (both of University of Michigan, Ann Arbor, MI) for the human AIF-GFP vector. Murine AIF-GFP vector was provided by G. Kroemer (Institut Gustave Roussy, Villejuif, France). We thank members of the R. Y. Tsien and M. H. Ellisman laboratories (University of California at San Diego, La Jolla, CA) for helpful discussions, in particular G. Gaietta, T. Deerink, S. Adams, and B. Martin. All members of the D.R.G. and D.D.N. laboratories are acknowledged for continuous help and support; especially Lisa Bouchier-Hayes, who also critically read this manuscript. This work was partially supported by National Institutes of Health Grants AI40646, AI52735, and CA69381. C.M-P. was supported by the Secretaria de Estado de Universidades Investigacion and the Fondo de Investigaciones Sanitarias of Spain.
Abbreviations
- ΔΨm
mitochondrial membrane potential
- ActD
actinomycin D
- AIF
apoptosis-inducing factor
- AK2
adenylate kinase 2
- FlAsH
fluorescein arsenical helix binder
- MOMP
mitochondrial outer membrane permeabilization
- ReAsH
resorufin arsenical helix binder
- TC
tetracysteine
- zVAD-fmk
Z-Val-Ala-Asp(OMe)-CH2F.
Footnotes
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Danial N. N., Korsmeyer S. J. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 2.Green D. R. Cell. 2005;121:671–674. doi: 10.1016/j.cell.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 3.Verhagen A. M., Ekert P. G., Pakusch M., Silke J., Connolly L. M., Reid G. E., Moritz R. L., Simpson R. J., Vaux D. L. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 4.Du C., Fang M., Li Y., Li L., Wang X. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 5.Suzuki Y., Imai Y., Nakayama H., Takahashi K., Takio K., Takahashi R. Mol. Cell. 2001;8:613–621. doi: 10.1016/s1097-2765(01)00341-0. [DOI] [PubMed] [Google Scholar]
- 6.Martins L. M., Morrison A., Klupsch K., Fedele V., Moisoi N., Teismann P., Abuin A., Grau E., Geppert M., Livi G. P., et al. Mol. Cell. Biol. 2004;24:9848–9862. doi: 10.1128/MCB.24.22.9848-9862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li L. Y., Luo X., Wang X. Nature. 2001;412:95–99. doi: 10.1038/35083620. [DOI] [PubMed] [Google Scholar]
- 8.Susin S. A., Lorenzo H. K., Zamzami N., Marzo I., Snow B. E., Brothers G. M., Mangion J., Jacotot E., Costantini P., et al. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 9.van Loo G., van Gurp M., Depuydt B., Srinivasula S. M., Rodriguez I., Alnemri E. S., Gevaert K., Vandekerckhove J., Declercq W., Vandenabeele P. Cell Death Differ. 2002;9:20–26. doi: 10.1038/sj.cdd.4400970. [DOI] [PubMed] [Google Scholar]
- 10.Kuwana T., Mackey M. R., Perkins G., Ellisman M. H., Latterich M., Schneiter R., Green D. R., Newmeyer D. D. Cell. 2002;111:331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- 11.Wei M. C., Zong W.-X., Cheng E. H. Y., Lindsten T., Panoutsakopoulou V., Ross A. J., Roth K. A., MacGregor G. R., Thompson C. B., Korsmeyer S. J. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldstein J. C., Waterhouse N. J., Juin P., Evan G. I., Green D. R. Nat. Cell Biol. 2000;2:156–162. doi: 10.1038/35004029. [DOI] [PubMed] [Google Scholar]
- 13.Goldstein J. C., Munoz-Pinedo C., Ricci J. E., Adams S. R., Kelekar A., Schuler M., Tsien R. Y., Green D. R. Cell Death Differ. 2005;12:453–462. doi: 10.1038/sj.cdd.4401596. [DOI] [PubMed] [Google Scholar]
- 14.Deng Y., Ren X., Yang L., Lin Y., Wu X. Cell. 2003;115:61–70. doi: 10.1016/s0092-8674(03)00757-8. [DOI] [PubMed] [Google Scholar]
- 15.Chauhan D., Hideshima T., Rosen S., Reed J. C., Kharbanda S., Anderson K. C. J. Biol. Chem. 2001;276:24453–24456. doi: 10.1074/jbc.C100074200. [DOI] [PubMed] [Google Scholar]
- 16.Daugas E., Susin S. A., Zamzami N., Ferri K. F., Irinopoulou T., Larochette N., Prevost M.-C., Leber B., Andrews D., Penninger J., Kroemer G. FASEB J. 2000;14:729–739. [PubMed] [Google Scholar]
- 17.Rehm M., Dussmann H., Prehn J. H. J. Cell Biol. 2003;162:1031–1043. doi: 10.1083/jcb.200303123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adrain C., Creagh E. M., Martin S. J. EMBO J. 2001;20:6627–6636. doi: 10.1093/emboj/20.23.6627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arnoult D., Karbowski M., Youle R. J. Cell Death Differ. 2003;10:845–849. doi: 10.1038/sj.cdd.4401240. [DOI] [PubMed] [Google Scholar]
- 20.Arnoult D., Gaume B., Karbowski M., Sharpe J. C., Cecconi F., Youle R. J. EMBO J. 2003;22:4385–4399. doi: 10.1093/emboj/cdg423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arnoult D., Parone P., Martinou J. C., Antonsson B., Estaquier J., Ameisen J. C. J. Cell Biol. 2002;159:923–929. doi: 10.1083/jcb.200207071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adams S. R., Campbell R. E., Gross L. A., Martin B. R., Walkup G. K., Yao Y., Llopis J., Tsien R. Y. J. Am. Chem. Soc. 2002;124:6063–6076. doi: 10.1021/ja017687n. [DOI] [PubMed] [Google Scholar]
- 23.Waterhouse N. J., Goldstein J. C., von Ahsen O., Schuler M., Newmeyer D. D., Green D. R. J. Cell Biol. 2001;153:319–328. doi: 10.1083/jcb.153.2.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ricci J. E., Munoz-Pinedo C., Fitzgerald P., Bailly-Maitre B., Perkins G. A., Yadava N., Scheffler I. E., Ellisman M. H., Green D. R. Cell. 2004;117:773–786. doi: 10.1016/j.cell.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 25.Ricci J. E., Gottlieb R. A., Green D. R. J. Cell Biol. 2003;160:65–75. doi: 10.1083/jcb.200208089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pardo O. E., Lesay A., Arcaro A., Lopes R., Ng B. L., Warne P. H., McNeish I. A., Tetley T. D., Lemoine N. R., Mehmet H., et al. Mol. Cell. Biol. 2003;23:7600–7610. doi: 10.1128/MCB.23.21.7600-7610.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou L. L., Zhou L. Y., Luo K. Q., Chang D. C. Apoptosis. 2005;10:289–299. doi: 10.1007/s10495-005-0803-9. [DOI] [PubMed] [Google Scholar]
- 28.Van Loo G., Demol H., van Gurp M., Hoorelbeke B., Schotte P., Beyaert R., Zhivotovsky B., Gevaert K., Declercq W., Vandekerckhove J., Vandenabeele P. Cell Death Differ. 2002;9:301–308. doi: 10.1038/sj.cdd.4400966. [DOI] [PubMed] [Google Scholar]
- 29.Single B., Leist M., Nicotera P. Cell Death Differ. 1998;5:1001–1003. doi: 10.1038/sj.cdd.4400462. [DOI] [PubMed] [Google Scholar]
- 30.Kohler C., Gahm A., Noma T., Nakazawa A., Orrenius S., Zhivotovsky B. FEBS Lett. 1999;447:10–12. doi: 10.1016/s0014-5793(99)00251-3. [DOI] [PubMed] [Google Scholar]
- 31.Loeffler M., Daugas E., Susin S. A., Zamzami N., Metivier D., Nieminen A. L., Brothers G., Penninger J. M., Kroemer G. FASEB J. 2001;15:758–767. doi: 10.1096/fj.00-0388com. [DOI] [PubMed] [Google Scholar]
- 32.Otera H., Ohsakaya S., Nagaura Z. I., Ishihara N., Mihara K. EMBO J. 2005;24:1375–1386. doi: 10.1038/sj.emboj.7600614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Springs S. L., Diavolitsis V. M., Goodhouse J., McLendon G. L. J. Biol. Chem. 2002;277:45715–45718. doi: 10.1074/jbc.C200524200. [DOI] [PubMed] [Google Scholar]
- 34.Polster B. M., Basanez G., Etxebarria A., Hardwick J. M., Nicholls D. G. J. Biol. Chem. 2005;280:6447–6454. doi: 10.1074/jbc.M413269200. [DOI] [PubMed] [Google Scholar]
- 35.Yuste V. J., Moubarak R. S., Delettre C., Bras M., Sancho P., Robert N., d’Alayer J., Susin S. A. Cell Death Differ. 2005;12:1445–1448. doi: 10.1038/sj.cdd.4401687. [DOI] [PubMed] [Google Scholar]
- 36.Cregan S. P., Fortin A., MacLaurin J. G., Callaghan S. M., Cecconi F., Yu S.-W., Dawson T. M., Dawson V. L., Park D. S., Kroemer G., Slack R. S. J. Cell Biol. 2002;158:507–517. doi: 10.1083/jcb.200202130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bidere N., Lorenzo H. K., Carmona S., Laforge M., Harper F., Dumont C., Senik A. J. Biol. Chem. 2003;278:31401–31411. doi: 10.1074/jbc.M301911200. [DOI] [PubMed] [Google Scholar]
- 38.Dumont C., Durrbach A., Bidere N., Rouleau M., Kroemer G., Bernard G., Hirsch F., Charpentier B., Susin S. A., Senik A. Blood. 2000;96:1030–1038. [PubMed] [Google Scholar]
- 39.Yu S.-W., Wang H., Poitras M. F., Coombs C., Bowers W. J., Federoff H. J., Poirier G. G., Dawson T. M., Dawson V. L. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- 40.Lakhani S. A., Masud A., Kuida K., Porter G. A., Jr., Booth C. J., Mehal W. Z., Inayat I., Flavell R. A. Science. 2006;311:847–851. doi: 10.1126/science.1115035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun X. M., Butterworth M., MacFarlane M., Dubiel W., Ciechanover A., Cohen G. M. Mol. Cell. 2004;14:81–93. doi: 10.1016/s1097-2765(04)00156-x. [DOI] [PubMed] [Google Scholar]
- 42.Uren R. T., Dewson G., Bonzon C., Lithgow T., Newmeyer D. D., Kluck R. M. J. Biol. Chem. 2004;280:2266–2274. doi: 10.1074/jbc.M411106200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}